
Abstract
A cypermethrin-degrading bacterium (SG4) was isolated from the pesticide-contaminated soil in the agricultural field of the crop research centre of the University, and characterized as Bacillus thuringiensis strain, SG4. The bacterium degraded 78.9 % of cypermethrin (50 ppm) in 15 days when grown in a minimal medium. To understand the functional proteins of cypermethrin degradation in Bacillus thuringiensis strain SG4, a comparative proteomic analysis was performed in the presence/absence of cypermethrin after 5 days of incubation in minimal medium. More than 450 spots corresponding to different proteins were recorded by 2D electrophoresis. We report expression of 223 and 250 unique proteins under normal and induced conditions (cypermethrin stress), respectively. Identified proteins were categorized into different functional groups on the basis of their biological functions, viz., catabolic enzymes, translational and stress proteins, etc. Characterization of cypermethrin-specific proteins in a bacterial strain will help in biodegradation practices in situ.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cypermethrin belongs to a synthetic pyrethroid group of insecticides and is used to control insects of cotton and lettuce. It is a broad-spectrum insecticide and kills beneficial insects along with target insects. Fish are particularly more susceptible to cypermethrin. Resistance to cypermethrin (developed quickly in insects exposed frequently to this chemical) renders it ineffective in insects. It readily crosses the blood-brain barrier, enters the brain and eventually leads to nigrostriatal dopaminergic neurodegeneration in rats after prolonged exposure (Tiwari et al. 2010; Singh and Singh 2011; Pankaj et al. 2016). Cypermethrin induces the progressive degeneration of dopaminergic neurons in adults. Degree of neuronal loss is considerably increased when rats are preexposed with low doses of cypermethrin during the critical period of brain development (Singh et al. 2012). Cypermethrin induced nigrostriatal dopaminergic neurodegeneration altering the mitochondrial function was studied using proteomics by Agrawal et al. (2015).
Proteomics has emerged as an excellent approach for gaining insight of physiological changes at cellular level (Jin et al. 2010; Meng et al. 2010) but relatively few attempts have been made to apply this technique to characterize resistance-related proteins in bacteria under pesticide stress. Till date no reports are available analyzing the physiological protein expression in cypermethrin-resistant bacteria in relation to susceptible strain, using proteomics. Proteome-based approaches are effective in making direct links with the pesticide degradation in natural environment. As compared to other biomolecules, proteins are promising markers as they reflect actual functionality with respect to metabolic reactions and regulatory cascades and give direct information about microbial activity than the functional genes and even the corresponding messenger RNAs (Wilmes and Bond 2006). Functional metaproteome from contaminated soil and ground water was analyzed by Benndorf et al. (2007) using 2D electrophoresis. Differential protein expression in response to imidacloprid in Myzus persicae (Sulzer) was reported by Meng et al. (2014).
In the present study, combination of two-dimensional gel electrophoresis (2-DE), matrix-assisted laser/desorption ionization time of flight (MALDI–TOF) and mass spectroscopy (MS) were used to identify cypermethrin-resistant proteins in Bacillus thuringiensis strain, SG4. A majority of changes in proteins expression are related to signal transduction, RNA processing, protein processing and cypermethrin-degrading catabolic proteins. This study reveals that resistance towards cypermethrin in Bacillus thuringiensis strain is a combination of complex responses that are displayed by proteomic expression profiles of resistant bacteria.
Materials and methods
Bacterial strain used in the study
Soil samples, collected from pesticide-contaminated agriculture fields of Udham Singh Nagar, Uttarakhand, India, were suspended in minimal medium (MM) containing 50 ppm of cypermethrin in 50 ml medium. After 5 days of incubation, 10 % from the medium was transferred into fresh MM containing cypermethrin@ 50 ppm. Bacterial colonies appeared were transferred individually to 50 ml of minimal medium, supplemented with cypermethrin which acted as a carbon and nitrogen source. Residual concentration of cypermethrin in the presence of bacterial culture was determined by HPLC (Dionex) at different time interval. A pure bacterial isolate showing highest degradation activity was selected for the proteomic study.
Identification of bacterial strain
Cypermethrin-degrading bacterial isolate SG4 was identified on the basis of morphological, biochemical, and molecular characters (based on 16S rDNA gene sequence). Cell morphology was observed using scanning electron microscopy. Genomic DNA of the bacterial strain was extracted according to Bazzicalupo and Fani (1995). 16S rDNA gene was amplified as described by Negi et al. (2014). PCR product was purified using Genei gel purification kit and sequenced by Biotech Centre, Delhi University, South Campus. Resulting 16S rDNA gene sequences were compared with the sequences in the GenBank nucleotide library using BLAST program (Pankaj et al. 2016). Multiple sequence alignment was carried out using Clustal-W and phylogeny was analyzed using MEGA 5.0 software Pankaj (2015).
Sample preparation for 2D gel electrophoresis
Whole cell extracellular differential proteome with and without cypermethrin was extracted from an actively growing bacterial isolate (SG4) after 5 days of incubation in minimal medium. The concentration of the pesticide was 200 ppm. Extraction procedure and methodology for PAGE analysis were followed as described by Laemmli (1970). Protein samples were lyophilized at Department of Molecular Biology and Genetic Engineering. GBPUAT, Pantnagar. Lyophilized protein samples were sent to Sandor life Science Pvt. Ltd., Hyderabad for 2D gel electrophoresis.
Gel image analysis
In silico analysis of 2D gel was performed on the basis of isoelectric point (pI) and molecular weight of separated protein(s) as reported by Jain et al. (2010). ExPASy is a SIB Bioinformatics Resource Portal which provides access to scientific databases and software tools in proteomics, genomics, phylogeny, systems biology and transcriptomics, etc. We have used TagIdent (a software tool) to analyze stress proteins that appeared as prominent spots in two-dimensional gel electrophoresis in SG4 exposed to pesticide.
MALDI–TOF–MS analysis and MASCOT database searches
MALDI–TOF analysis of match ID 2 and 42 was done at Sandor Proteomics Pvt Ltd, Hyderabad using Bruker Daltonics—UltraflexTM III Mass Spectrometer with spectra internally calibrated using trypsin auto-digestion products. Obtained peptide masses were searched on MascotTM Peptide Mass Fingerprint database (Matrix Science). Obtained data was used to identity the proteins using Mascot search tool (http://www.matrixscience.com).
Results
Isolation and identification of cypermethrin-degrading strain
A highly efficient, cypermethrin-degrading bacterial strain, designated as SG4, was isolated from pesticide-contaminated agriculture fields of Udham Singh Nagar using enrichment method. The organism utilized cypermethrin as a carbon and nitrogen source when grown in minimal medium and degraded the compound by 78.9 % within 15 days (Fig. 3). Bacterial isolate was Gram-positive and rod-shaped (Fig. 2). Phylogenetic analysis of 16S rDNA gene sequences revealed that the strain showed 99–100 % homology with Bacillus thuringiensis (Fig. 1). 16S rDNA sequences of Bacillus thuringiensis strain SG4 were deposited in GenBank under the nucleotide accession number, KT186610.

Phylogenetic analysis of SG4 strain on the basis of 16S rDNA using MEGA 5.0 software. The numbers in parentheses represent the sequence accession number in GenBank. Bar represents sequence divergence

Electron micrograph of strain SG4 with cypermethrin

Growth-linked biodegradation of cypermethrin by SG4
2-DE analysis of proteins in Bacillus thuringiensis strain SG4
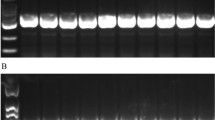
Difference in protein expression in any kind of stress (susceptible and resistant) in bacterial strains is observed during physiological adaptations. Protein samples of SG4 (extracted from SG4, exposed to cypermethrin and one control) were subjected to two-dimensional gel electrophoresis (Fig. 4a, b). Representative 2D maps are shown in Figs. 5, 6, 7 and 8. Separated proteins spots are evident in both the dimensions. Gel maps were of good quality and could be used as reference 2-D maps for Bacillus thuringiensis strain SG4 proteins. More than 450 protein spots were reproducibly detected by Image analysis software. Quantitative image analysis revealed the presence of 51 protein spots that showed significant and reproducible changes in protein expression under pesticide stress (Figs. 6, 7). Some proteins were overexpressed and others were underexpressed. Magnified view of differentially expressed proteins is given in Fig. 8.

Two-dimensional gel electrophoresis of cypermethrin-induced proteins in SG4. a Normal protein profile without cypermethrin. b Cypermethrin-induced protein

Comparative 2-D profiling on the basis of observed spots

Comparative 2-D profiling of expressed proteins on the basis of observed spots and identification of unique spots

Analysis of matched spots in 2D gel (normal and under stressed conditions)

Analysis of matched spots in 2D gel (down-regulation)
Identification of differentially expressed proteins
Differentially expressed proteins in SG4 were identified on the basis of their PI values and molecular weight using ExPASy protein database. Highly up-regulated proteins were identified using MALDI–TOF mass spectrometer. Out of 51 differentially expressed proteins, 39 protein spots showed apparent similarities with database, including 21 down-regulated spots and 18 up-regulated spots in resistant Bacillus thuringiensis strain SG4 (Table 1). Remaining 12 proteins did not show major changes statistically. We report the expression of 223 unique proteins in SG4 under normal conditions and 250 unique proteins under cypermethrin-induced conditions. Majority of the spots in 2D gels displayed a reasonable correlation of theoretical molecular weight (MW) values with the experimental data. Some of spots showed significant deviation from the experimentally measured MW from the expected theoretical values, implying that PTMs (post translational modifications) such as fragmentation might had occured in these proteins (Liu et al. 2008). Most of the theoretical values were based on proteins originating from other species (rather than Bacillus thuringiensis), rendering the exact MW of these proteins unknown. Based on the biological functions, identified proteins were classified into several functional categories including Stress response, hypothetical/uncharacterized protein, catabolic/cypermethrin-degrading proteins, protein synthesis/modifications, gene regulation/transcription, energy production/chemotaxis etc. Hypothetical proteins and some proteins with unknown function were also identified in the database (Fig. 9a–c).

a Distribution of proteins expressed during normal metabolism in SG4. b Distribution of proteins expressed during cypermethrin-induced metabolism in SG4. c Overexpressed and underexpressed proteins in cypermethrin-induced condition in strain SG4
Discussion
Cypermethrin is used widely against serious pests of most of the crops worldwide. Extensive use of number of pesticides has resulted in rapid development of insecticide resistance in naturally occurring microbiota (Meng et al. 2014; Foster et al. 2003; Slater et al. 2011). Development of resistance against an insecticide in a bacterial system is an universal phenomena. A comparative proteomic analysis of susceptible and resistant bacteria is helpful to elucidate innate responses and adaptive physiological changes contributing towards cypermethrin resistance. The goal of the present study was to identify and characterize cypermethrin responsive proteins in a cypermethrin-resistant Bacillus thuringiensis strain SG4 using proteomic approach. Differentially expressed protein (51 spots in gel) were observed in resistant Bacillus thuringiensis strain SG4 in comparison to the same strain growing without cypermethrin stress. Total number of protein spots was 223 in normal condition whereas 250 in cypermethrin induced conditions. Most of proteins were found to have important biological functions, providing a basis for understanding and annotating their role in response to cypermethrin exposure. Identified proteins could be classified into six groups: stress response, hypothetical/uncharacterized protein, catabolic/cypermethrin-degrading proteins, protein synthesis/modifications, gene regulation/transcription, energy production and chemotaxis-related proteins.
Stress response related proteins
Expression of functional proteins through signal transduction and regulation of gene expression might work cooperatively to maintain cellular homeostasis under various external conditions. In this study some chaperones were identified, including molecular chaperone DnaK (spot 2868), DNA repair protein MutL (spot 34), chaperone proteinHsc A (spot 2869), Grp E (spot 6559),with one homolog (spot 6421) and chaperone protein HtpG (spot 6401). HscA is a heat shock protein whose synthesis is stimulated under stress (Pumirat et al. 2009). It is known that oxidative stress can be induced by pesticides either by overproduction of free radicals or by alterations in antioxidant defence mechanism (Abdollahi et al. 2004). Catalase (CAT) is one of the essential enzymes in the process of reactive oxygen species (ROS) detoxification (Gao et al. 2008). Our results showed that predicted catalase activity was inhibited in cypermethrin-resistant bacteria. Inhibition of catalase activity could be explained by level of toxicity of cypermethrin or by the presence of O2 radicals during resistance development. Several studies have demonstrated that higher quantity of O2− radicals acts as catalase inhibitor, so it is possible that exposure to cypermethrin during the development of resistance, primarily induced the generation of O2− radicals which caused inhibition of the expression of CAT. Other studies have also reported inhibition of CAT activity after exposure to some pesticides (Ferrari et al. 2011; Velki and Hackenberger 2012).
Cypermethrin-degrading proteins
Metabolic pathways produce energy for the maintenance of bacterial growth under stress condition. Primary metabolic pathways (metabolism of carbohydrate, fatty acid for energy production) are needed to stabilize a new homeostasis in the resistant bacteria. Some enzymes like malate dehydrogenase, α-ketoglutarate semialdehyde dehydrogenase, isocitrate dehydrogenase kinase/phosphatase, glycerol-3- phosphate dehydrogenase and formate dehydrogenase were down-regulated in cypermethrin-resistant bacteria. All the mentioned enzymes have significant role in tri-carboxylic acid cycle suggesting that bacteria might have taken up cypermethrin through novel metabolites for its growth. Such findings were not reported earlier in the similar studies conducted by Mishra and Shukla (2003). Reduction in malate dehydrogenase activity in Clarias batrachus under endosulfan stress was reported by Mishra and Shukla (2003) and it could be due to binding of endosulfan or its metabolites with enzyme molecules and/or by blocking the enzyme synthesis.
Protein synthesis/modifications
Protein synthesis can be regulated by modifications in various enzymes at post transcriptional, translational and post translational level. Present study reports the involvement of different up and down-regulated proteins in these processes and mentions the up-regulation of polynucleotide nucleotidyltransferase (Match ID39), Threonine-tRNA ligase (Match ID 36), Glutaryl-tRNA (Gln) amidotransferase subunit A, (match ID 0), and RNA polymerase II subunit A C-terminal domain phosphatise (Match ID 2), whereas the DNA-directed RNA polymerase (β 1 subunit, Match ID 51) was down-regulated.
Gene regulation/transcription
Gene expression can be regulated at replication, transcriptional and translational level. Proteins involved in all these processes were identified in control and cypermethrin-resistant bacteria. Overexpressed proteins were DNA mismatch repair protein MutL (Match ID 34) and type 2 DNA topoisomerase 6 subunit B (Match ID 6). Besides these, several unique proteins were also expressed positively. Some important proteins were: elongation factor 4 (spot 2833), protein RecA (spot 2739), transcription factor TBF1 (spot 2852), elongation factor Ts (spot 2767), peptide chain release factor 1 (spot 2736) and cysteine ligase BshC (spot 2712) (Table 1).
Energy production/chemotaxis
Chemotaxis, a chemical interaction between the chemicals and bacterial cell surface receptors plays important role in energy production. A number of proteins take part in energy production and chemotaxis. In the present study, Phosphatise CheZ (Match ID 12) and putative glycine rich membrane protein (Match ID 41) were overexpressed in cypermethrin-resistant bacteria. Whereas translocase subunit Sec A protein (match ID 45) was down-regulated under cypermethrin induction in Bacillus thuringiensis strain SG4. Other prominent proteins in resistant bacteria were NAD kinase (6572), putative arsenical pump driving ATPase (6598), chemotaxis response regulator glutamate methyl transferase (6570), ATPase synthase (subunit α 6437) and ATPase synthase subunit β (6449), etc.
Conclusion
The present study offers an example of application of proteomic approach to screen cypermethrin resistance proteins in Bacillus thuringiensis strain, SG4. Response of Bacillus thuringiensis strain, SG4 towards cypermethrin is a complex phenomenon, as differentially expressed proteins are involved in multiple functional categories. Most of the altered proteins were closely associated with stress response, hypothetical/uncharacterized proteins, catabolic/cypermethrin-degrading proteins, protein synthesis/modifications, gene regulation/transcription, energy production and chemotaxis-related proteins. But identifying all the proteins in insecticide-resistant bacteria is not sufficient to reveal the complexity underlying the engineered object. The proteomic resources and knowledge developed by this study will contribute towards better understanding of the mechanisms which govern development of insecticide resistance in bacterial systems.
References
Abdollahi M, Ranjbar A, Shadnia S, Nikfar S, Rezaiee A (2004) Pesticides and oxidative stress: a review. Med Sci Monit 10:144–147
Agrawal S, Singh A, Tripathi P, Mishra M, Singh PK, Singh MP (2015) Cypermethrin-induced nigrostriatal dopaminergic neurodegeneration alters the mitochondrial function: a proteomics study. Mol Neurobiol 51(2):448–465
Bazzicalupo M, Fani R (1995) The use of RAPD for generating specific DNA probes for microorganisms. In: Clapp JP (ed) Methods in molecular biology. Humana, Totowa, pp 155–175
Benndorf D, Balcke GU, Harms H, Von Bergen M (2007) Functional metaproteome analysis of protein extracts from contaminated soil and groundwater. ISME J 1:224–234
Ferrari A, Lascano C, Pechen AM, de Angelo D, Venturino A (2011) Effects of azinphos methyl and carbaryl on Rhinella arenarum larvae esterases and antioxidant enzymes. Comp Biochem Phys 153:34–39
Foster S, Denholm L, Thompson R (2003) Variation in response to neonicotinoid insecticides in peach-potato aphids, Myzus persicae (Homoptera: Aphididae). Pest Manag Sci 59:166–173
Gao Y, Sun X, Sun Z, Zhao N, Li Y (2008) Toxic effects of enrofloxacin on growth rate and catalase activity in Eisenia fetida. Environ Toxicol Pharmacol 2:177–180
Jain A, Bacolla A, Chakraborty P, Grosse F, Vasquez KM (2010) Human DHX9 helicase unwinds triple-helical DNA structures. Biochemistry 49:6992–6999
Jin T, Zeng L, Lu YY, Xu YJ, Liang GW (2010) Identification of resistance responsive proteins in larvae of Bactrocera dorsalis (Hendel), for pyrethroid toxicity by a proteomic approach. Pestic Biochem Physiol 96:1–7
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685
Liu N, Song WJ, Wang P, Lee KC, Chan W, Chen HL, Cai ZW (2008) Proteomics analysis of differential expression of cellular proteins in response to avian H9N2 virus infection in human cells. Proteomics 8:1851–1858
Meng JY, Zhang CY, Lei CL (2010) A proteomic analysis of Helicoverpa armigera adults after exposure to UV light irradiation. J Insect Physiol 56:405–411
Meng JY, Zhang CY, Chen XJ, Cao Y, Shang SH (2014) Differential protein expression in the susceptible and resistant Myrus persicae (Sulzer) to imidacloprid. Pest Biochem Physiol 115:1–8
Mishra R, Shukla SP (2003) Endosulfan effects on muscle malate dehydrogenase of the freshwater catfish Clarias batrachus. Ecotoxicol Environ Saf 56:425–433
Negi G, Pankaj, Srivastava A, Sharma A (2014) In situ biodegradation of endosulfan, imidacloprid, and carbendazim using indigenous bacterial cultures of agriculture fields of Uttarakhand, India. Int J Biol Biomol Agric Food Biotechnol Eng 8(9):973–981
Pankaj (2015) Microbiological and molecular analysis of biodegradation potential of indigenous bacterial cultures against cypermethrin, fipronil and sulfosulfuron pesticides. Ph.D thesis, Department of Microbiology, CBSH, GBPUAT Pantnagar
Pankaj, Sharma A, Gangola S, Khati P, Kumar G, Srivastava A (2016) Novel pathway of cypermethrin biodegradation in a Bacillus sp. strain SG2 isolated from cypermethrin-contaminated agriculture field. 3 Biotech 6:45. doi:10.1007/s13205-016-0372-3
Pumirat P, Saetun P, Sinchaikul S, Chen ST, Korbsrisate S, Thongboonkerd V (2009) Altered secretome of Burkholderia pseudomallei induced by salt stress. Biochim Biophys Acta 1794:898–904
Singh NS, Singh DK (2011) Biodegradation of endosulfan and endosulfan sulfate by Achromobacter xylosoxidans strain C8B in broth medium. Biodegradation 22:845–857
Singh AK, Tiwari MN, Prakash O, Singh MP (2012) A current review of cypermethrin-induced neurotoxicity and nigrostriatal dopaminergic neurodegeneration. Curr Neuropharmacol 10:64–71
Slater R, Paul VL, Andrews M, Garbay M, Camblin P (2011) Identifying the presence of neonicotinoidresistant peach–potato aphid (Myzus persicae) in the peach-growing regions of southern France and northern Spain. Pest Manag Sci 68:634–638
Tiwari MN, Singh AK, Ahmad I, Upadhyay G, Singh D, Patel DK, Singh C, Prakash O, Singh MP (2010) Effects of cypermethrin on monoamine transporters, xenobiotic metabolizing enzymes and lipid peroxidation in the rat nigrostriatal system. Free Radic Res 44:1416–1424
Velki M, Hackenberger BK (2012) Species-specific differences in biomarker responses in two ecologically different earthworms exposed to the insecticide dimethoate. Comp Biochem Physiol Part C 156:104–112
Wilmes P, Bond PL (2006) Metaproteomics: studying functional gene expression in microbial ecosystems. Trends Microbiol 14:92–97
Acknowledgments
The authors are thankful to the Ministry of Environment for providing financial support in the form of a project. First author, Dr. Pankaj, thankfully acknowledges the Junior Research fellowship in the project during the course of this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Pankaj, Negi, G., Gangola, S. et al. Differential expression and characterization of cypermethrin-degrading potential proteins in Bacillus thuringiensis strain, SG4. 3 Biotech 6, 225 (2016). https://doi.org/10.1007/s13205-016-0541-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-016-0541-4