
Abstract
Surface derivatized magnetic nanoparticles have been commonly used for magnetic separation. Facile mechanisms are needed to be developed for the design of bio-functionalized magnetic hybrid materials, where the surfaces can be re-generated for the re-use of the developed platforms. Superparamagnetic iron oxide nanoparticles with a diameter below 10 nm were synthesized via a novel microwave-assisted hydrothermal method in the presence of citrate ions, which allowed to obtain uniform and negatively charged nanoparticles. These were then coupled with Poly-l-lysine (PLL), forming micrometer-sized self-assembled spherical entities. Cross-linking the PLL within these microspheres with glutaraldehyde stabilized them chemically and mechanically. The active bio-functionality was introduced by a protein grafting methodology, using m-maleimidobenzoyl-N-hydroxysulfosuccinimide ester (SMBS). The Moringa oleifera Coagulant Protein (MOCP) from a seed extract was employed for its characteristic coagulation activity. The performance of the MOCP functionalized microspheres was evaluated as a function of turbidity removal of problematic colloidal clay from water via magnetic separation, resulting in over 80% of activity within 15 min. Surface of these hybrid materials can be re-generated by treatment with alcohol, allowing their easy magnetic separation and re-use. The rapid and strong response with tunable magnetic property makes these hybrid microspheres a powerful tool for many potential applications, due to the general applicability of the developed methodology.
Similar content being viewed by others

Introduction
Nanoparticles (NPs) have widely been applied to develop novel environment-friendly methods and techniques. These constitute an advancement to traditional manufacturing, dramatically reducing the contamination or negative impact, to the environment (Cui et al. 2019; Wang et al. 2018). Chemical mechanical polishing slurries (Zhang et al. 2019), diamond wheels (Zhang et al. 2015) and novel machining methods (Zhang et al. 2013) have been employed in a wide spectrum of fields, such as aerospace, ocean, nuclear, and power engineering (Zhang et al. 2012).
Furthermore, magnetic NPs are nowadays used in almost every area of application from food (McClements and Xiao 2017) to contrast agents for imaging-magnetic resonance imaging (MRI) (Busquets et al. 2015), X-ray imaging (Justin et al. 2017), sensors and detection, water remediation (preconcentration, separation, and capture of analytes) (Beveridge et al. 2011), cancer treatments (Storozhuk and Iukhymenko 2019; Mody et al. 2014), etc. The high surface-to-volume ratio of nanoparticles indeed enables novel properties which can be tuned by their size control.
Superparamagnetism (SPM) is a desired characteristic of magnetic NPs, where colloidal stability and magnetic field responsive particles are deemed essential, especially in biological (Neuberger et al. 2005; Lu et al. 2017) and environment-related applications (Yantasee et al. 2007). The synthesis methods can be tuned to obtain NPs with size and surface properties adjusted to a specific application. Superparamagnetic iron-oxide NPs (SPIONs) have been previously synthesized using solution precipitation, thermolysis and hydrothermal reactions (Daou et al. 2006; Yoffe et al. 2012; Patsula et al. 2016). Microwave (MW) assisted hydrothermal synthesis is a flexible, highly controllable, reproducible and energy-efficient method for the synthesis in nanoscale (Gawande et al. 2014; Saxena et al. 2019). MW assisted hydrothermal method was recently used for a high-yield synthesis of high quality and water dispersible SPIONs (Carenza et al. 2014).
In recent years, there has been a considerable interest in the development and employment of hybrid nanomaterials for energy harvesting (Wang et al. 2010; Tsai et al. 2017), storage (Zhou et al. 2013), medical imaging (Cheng et al. 2014) and relevant environmental issues, including water treatment (Okoli et al. 2012). Specifically, surface derivatized magnetic NPs are developed for separation and pre-concentration of common pollutants from water, where coating with inorganic components (e.g., silica); or modification with organic molecules [e.g., octadecylsilane (ODS), polymer or surfactant] are commonly utilized (Chen et al. 2011). The bottom-up approach to synthesize hybrid materials utilizes NPs as core constituents, and polymers or complementary entities to form stable and bio-friendly material platforms with a higher complexity for specific applications (Zhang et al. 2008). The controlled assembly of SPIONs has been demonstrated to be a successful route to synthesize stable SPM microspheres (Ramanujan and Chong 2004). Several methods as emulsion (Kondo et al. 1994) and dispersion (Horák et al. 2003) polymerization were proposed for nanoparticle aggregation. “Click Chemistry” is a term that was introduced by Sharpless in 2001 to describe reactions that are high-yield, wide in scope, create only byproducts that can be removed without chromatography, are simple to perform, and can be conducted in easily removable or benign solvents (Kolb et al. 2001). In recent years, a self-assembly, or click-chemical, method was reported combining polyelectrolyte complexation with a cross-linking process (Toprak et al. 2007a). The stability (Toprak et al. 2007b) of the resultant spherical particles constitutes an important characteristics for protein or other type of biomolecule functionalization.
This work focuses on the assembly of bio-functionalized SPM microspheres, using the Moringa oleifera Coagulant Protein (MOCP), as a model functional protein, to obtain a hybrid magnetic platform with coagulation properties (Jahn 1988). The platform has a backbone composed of NPs and amino acid, held together by covalent bond. A green chemical MW assisted synthesis was used for the synthesis of SPIONs capped with citrate. The bio-functionalized microspheres would constitute a green alternative to common coagulants associated with risks (Gauthier et al. 2000) and, due to the magnetic separation possibility, they are environmentally friendly and ready-to-use tools for separation purposes, for example in turbidity removal from water. This methodology represents an alternative to centrifugation, gravitational separation, and filtration processes, for both laboratory and industrial scales. Finally, the developed strategy to synthesize a stable, magnetically separable, bio-functionalized platform has a wide range of potential applications, depending on the chosen target protein, and thus featuring the generality of the developed methodology.
Results and discussion
The experimental methods follow a bottom-up approach: after the synthesis of SPIONs, their morphological and magnetic properties were investigated. The characterization of the cross-linked coacervates was performed to study their stability and functional properties. A wide variety of analytical techniques was used to evaluate the crosslinking with the target protein and a possible biological and promising application in water treatment for turbidity removal was demonstrated.
To show the predominant crystalline phase of the synthetized particles, X-ray powder diffraction analysis (XRPD) was performed (Fig. 1). The diffraction pattern presents the characteristic peaks of cubic Fe3O4—magnetite (ICDD card no: 88-0315; COD card no: 9005837). Using the Scherrer equation a crystallite size of 6.2 nm was obtained (see Supplementary information). This value is in a good agreement with the average value of 6.1 nm obtained from AFM (Figure S1) indicating the single-crystalline nature of the Fe3O4 NPs.

XRPD pattern for as-made iron oxide NPs. The main crystalline planes are indexed with corresponding Miller indices of cubic Fe3O4—magnetite (ICDD card no:88-0315; COD card no: 9005837)
Magnetic characterisation was performed on Fe3O4 NPs to investigate their response to an external applied magnetic field (H) (Fig. 2). The absence of coercivity would indicate the SPM character of the as-made iron oxide NPs. The obtained SPIONs have very low coercivity (\(\Delta H_{\text{C}} = 2\) Oe) due to their small size, and a saturation magnetization of 72 emu/g, slightly lower than the saturation magnetisation of bulk magnetite (92 emu/g). The calculated magnetic diameter is 6.1 nm (see Figures S2, S3 for details on the plot of the estimated probability density function and cumulative distribution function). The experimental data were fitted with a theoretical model, described by the following function:
where \(M\) is the magnetisation, \(M_{\text{S}}\) is the saturation magnetization, \(L\) is the Langevin function and \(f(y)\) is the log-normal distribution function with the reduced diameter \(y\) as independent variable (Chantrell et al. 1978). By numerically integrating, the magnetic diameter (\(D_{\text{M}}\)), the coercivity (\(\Delta H_{\text{C}} )\) the saturation magnetization of the sample was computed (Saladino 2019).

Magnetization curve for as-made iron oxide NPs
The coating with citrate ions on the NPs surface was investigated by the Fourier Transform Infrared Spectroscopy (FTIR); the resultant spectra for citrate alone (TSC) and citrate coated SPIONs are shown in Fig. 3. The vibrational bands of citrate are clearly present in the SPIONs spectrum, the band at 1606 cm−1 (Band D) being attributed to asymmetric stretching of COO. The band at 572 cm−1 (Band A) is attributed to vibrational mode of Fe–O in the SPIONs lattice (Ahn et al. 2001). A detailed spectral identification is presented in Table S1.

FTIR spectra of a citrate coated SPIONs and b TSC
The organic content of the SPIONs was assessed by thermal gravimetric analysis (TGA) (Fig. 4). The TGA thermogram shows three different decomposition steps, until at about 660 °C. The first step until 180 °C can be attributed to water desorption, with a 2% weight loss; the second and third step (180–660 °C) is attributed to the partial decomposition of citrate in two steps, with CO2 evolution (Marcilla et al. 2018). The citrate coating of SPIONs constituted 7% of the total mass.

TGA thermogram of the SPION sample. Four decomposition stages are identified: (I) water desorption (25–180 °C), (II, III) citrate decomposition in two steps (180–660 °C) and (IV) steady-state (> 660 °C)
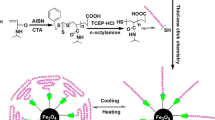
The SPIONs suspension with additional citrate (C-SPIONs) was used for the coacervation process, to form micrometre-sized hybrid entities constituted by C-SPIONs and poly-l-lysine (PLL). The process was completed by intermolecular cross-linking part of amine groups of PLL macromolecules by glutaraldehyde (GA), which formed stable double-bond interactions, i.e. the Schiff base (Toprak et al. 2007a). The coacervation and cross-linking processes resulted in the formation of self-assembled poly-electrolytic spheres (SAPES). The cross-linking process is shown in Scheme 1, where a GA molecule constitutes the bridge between two PLL macromolecules.

Cross-linking PLL macromolecules with GA within SAPES
The coacervation and cross-linking processes were confirmed by changes in the FTIR spectrum (Fig. 5). The bands at 798 cm−1 and 1030 cm−1 correspond to C–H bending and C–N stretching, respectively. The band at 1655 cm−1 is associated with the C=N stretching mode, which is a strong indication of the success of the cross-linking process and subsequent formation of the Schiff-bases.

FTIR spectra of SPIONs and SAPES
Further, we investigated the morphology and size of the SAPES using SEM (Fig. 6), highlighting some NPs on the surface, while most are contained inside the polymeric entity. The size measurements from SEM micrographs (Figure S4) were fit to a log-normal distribution accordingly to a model (Söderlund et al. 1998) predicting the size distribution of SAPES, shown in Figure S5. The average size obtained was 1.5 ± 0.5 µm, in agreement with the earlier reports (Toprak et al. 2007a).

A typical SEM micrograph of a cross-linked coacervate, SAPES
The bio-functionalization was introduced by coating the SAPES surface with MOCP. The grafting methodology was designed by studying the amino acid sequence of an equivalent recombinant protein, MO2.1, already present in UniProtKB, central hub containing functional information on proteins, with the identification code P24303. An online suite of tools, Phyre2, was employed to predict and analyse protein structure, function and mutations, based on a homology/analogy recognition engine (Kelley et al. 2015). The platform provided the results with a confidence in the model of 99.9%, where 53 residues, constituting the 88% of the protein sequence, were modelled by the sweet protein mabinlin-2 chain b, coherently to previous studies (Gassenschmidt et al. 1995). The generated PDB file was analysed via the software Chimera. In Fig. 7, two cysteine residues, Cys12 and Cys25 are shown and highlighted on the protein ribbon structure of MO2.1; each of them presents a sulfhydryl group, which can be targeted by the heterobifunctional cross-linker SMBS, presenting two functional groups, NHS ester and maleimide.

MO2.1 Ribbon Structure highlighting cysteine residues with the ball-and-stick structures
The maleimide group can, hence, be used to covalently bind the sulfhydryl group of the two cysteine residues of MO2.1, and, equivalently, of MOCP. Whilst, the NHS ester allowed the binding with free amine groups remained on the SAPES. The grafting procedure was confirmed with \(\zeta\)-potential analyses on SAPES and on protein-grafted sample (MO-SAPES) at neutral pH. The charge on SPIONs was negative (− 36 mV), due to the presence of citrate molecules. Upon coacervation and GA cross-linking the surface charge changes to positive values, (+ 6.7 mV), due to the amine groups on the PLL entities in SAPES. Upon MOCP conjugation, a dramatic increase in the surface potential, from + 6.7 mV of SAPES to + 30.2 mV for MO-SAPES, was observed, confirming the presence of the protein on the surface (Fig. 8).

\(\zeta\)-Potentials for SPIONs, SAPES and MO-SAPES at neutral pH
Furthermore, the hydrodynamic size of MO-SAPES entities was estimated using DLS, which showed an increase in the size of more than 1 µm for MO-SAPES, from 2.5 µm of SAPES to 3.9 µm. This is ascribed to the large protein molecules bound to SAPES surface and its surface-bound water molecules. A comparison of their distribution is shown in Figure S6.
To verify the correct covalent grafting by SMBS, the UV–vis spectroscopy was utilized to identify the absorption peak from the benzyl groups present in the cross-linker. The absorption peak at 270 nm appears in the protein-grafted microspheres spectrum (MO-SAPES), confirming the presence of covalently bound protein (Fig. 9). This is corroborated by an organic layer observed on the spherical entities in the SEM micrograph, attributable to the grafted protein (Figure S7).

UV–vis spectra of SAPES and MO-SAPES
The active bio-functionalization of the MO-SAPES was demonstrated via the characteristic coagulation property of MOCP, for turbidity removal from polluted water. The activities of SAPES and MO-SAPES (containing 170 µg SPIONs), MOCP (1 µg) and only clay solution (\(I_{0} = 1.14\)) in 1 mL sample after 15 min were compared. To exclude the passive sedimentation effect, all the samples were mixed before applying a magnetic field and separating the collected entities. In the sample with only clay, a 30% of activity was recorded, which is dominantly due to passive sedimentation. By the addition of MOCP, the activity increased only by 1%, reaching to 31%, due to the limited interaction time of 15 min. The activity was increased to 47% when using SAPES, due to a combined effect of magnetic separation and their strong positive surface charge. The coagulation functionality of MO-SAPES was confirmed as 83% of activity, which is dramatically improved owing to the covalently surface bound MOCP (Fig. 10).

Activities for only clay, MOCP, SAPES and MO-SAPES samples for turbidity removal via magnetic separation
The characteristic morphology of kaolin clay (Asma Muhmed 2013) is clearly identifiable as electrostatically attached microstructures on the surface of MO-SAPES after magnetic separation, shown in the SEM micrograph in Fig. 11.

SEM Micrograph of a hybrid microsphere MO-SAPES covered with clay
Chemicals commonly used for the various treatment units, as synthetic organic and inorganic substances, to remove pollutants—such as clays causing turbidity in water, and chemical residues—are also associated with human health and environmental problems (Gauthier et al. 2000). Safer alternatives are natural products as specific proteins extracted from plants that possess coagulation properties and can be used for the collection and separation of water pollutants, such as clays (Yin 2010). The designed hybrid material in this respect can be a useful coagulation and separation platform for a variety of applications, not to mention the validation for removal of turbidity from water containing clays. Furthermore, their surface can be re-generated by treatment with alcohol, making them re-cyclable and re-usable.
Conclusions
We successfully demonstrated the use of green-chemical synthesis in connection with the self-assembly process and click chemistry, to fabricate multifunctional complex hybrid materials with a bio-active surface. The presented synthetic route resulted in bio-functionalized and environmentally friendly microspheres, with efficient applicability in pollutant removal. By combining the magnetic properties of SPIONs, embedded in the PLL matrix, and coagulation properties of the grafted MOCP, we showed significant activity values in the turbidity removal test, reaching over 80% for MO-SAPES. The stability provided by covalent bonds constitutes the essential requirement for a future regeneration process. The tunability of the whole process for the assembly of the bio-functionalized hybrid material platform opens avenues to a wide variety of environmental and biomedical applications, where the non-permanent magnetic characteristics and functional surface would play a key role.
Methods
Ferric chloride hexahydrate (FeCl3·6H2O, 97%), ferrous sulfate heptahydrate (FeSO4·7H2O, 99%), sodium hydroxide (NaOH, 98%), sodium citrate dihydrate (Na3C6H5O7·2H2O, TSC, 99.5%), Poly-l-lysine hydrobromide (70 − 150 kDa, PLL), Glutaraldehyde solution (25% in H2O, GA), Glycine (Gly, 99%) and PBS tablets were all purchased from Sigma Aldrich. The cross-linker m-maleimidobenzoyl-N-hydroxysulfosuccinimide ester (SMBS) was bought from Thermo Fisher.
Nanoparticle synthesis 3.622 g of ferric chloride was dissolved in 130 mL of deionized (DI) H2O, while 1.825 g of ferrous sulfate in 130 mL of DI H2O, to get a stock solution with a concentration of 0.1 M and 0.05 M, respectively. Furthermore, stock solutions of TSC (0.5 M) and sodium hydroxide (1 M) were prepared. To a mixture of 16 mL of ferric chloride and 16 mL of ferrous sulphate, 6.8 mL of NaOH was added, followed by the dissolution of 16 mL of TSC. The sample was treated under microwave irradiation of flexi-WAVE (Milestone SRL), according to the following recipe: a temperature of 150 °C was reached with a 5-min ramp and kept constant for a duration of 25 min. A frequency of 2.45 GHz was employed, under continuous magnetic stirring (70% mode). The sample was then washed three times, via centrifugation, to remove the residuals of the reaction.
Coacervation process 20 mg of PLL was dissolved in 10 mL of DI water, while a 2.5 M solution was made for Gly; finally, a 2.5% GA stock solution was prepared. From the washed sample of SPIONs, 375 μL aliquot was diluted to a total volume of 15 mL with DI water. To this sample, 165 μL of 0.5 M TSC was added, followed by 3 s-pulsed sonication for 30 min. Thus, 120 μL of C-SPIONs were mixed with 20 μL of PLL and incubated for 10 min. Then, 120 μL of GA was added (vortex for 15 s) and let it react for 5 min to accomplish the crosslinking of the amine groups from different PLL molecules, via Schiff base formation (Tong et al. 2005). Finally, the extra GA was quenched by adding 40 μL of Gly (Toprak et al. 2007a). Following this procedure, 10 samples were prepared in parallel and collected together in a 4 mL cuvette after quenching. Magnetic separation was employed for the washing procedure to remove all the non-magnetic entities present in solution. Finally, the sample, called SAPES, was diluted to 2 mL of DI water and stored for further uses.
Protein extraction The protein from M. oleifera seeds was extracted and purified as previously described (Ghebremichael et al. 2005). Oil was removed from the seed kernel using ethanol followed by extracting protein with ammonium acetate buffer. The protein was purified by Ion exchange beads by batch purification method. The coagulation activity of the protein has been examined in synthetic turbid water (clay solution) by mixing protein and clay using UV–Vis spectrophotometer, as detailed below.
Protein grafting A standard sample was obtained using 3 mL dispersion of SAPES, where DI water was substituted by PBS via magnetic separation. Thus, 3 mg of SMBS was added to the sample and followed by an incubation time of 30 min. Therefore, 300 μL of MOCP (3 mg/mL) was quickly added in the dispersion, letting the thiol groups be crosslinked for 1 h. The sample was washed and refilled with 3 mL of DI water. Called MO-SAPES, it was stored at 4 °C.
Characterization techniques
The hydrodynamic size, based on dynamic light scattering (DLS), and \(\zeta\)-potential distributions were measured at room temperature with Zetasizer Nano ZS90 at an incident angle of 90°. All samples were diluted in DI H2O at neutral pH. The measurements were performed in triplicates.
Thermal gravimetric analysis (TGA) was done using TGA 55 (TA Instruments). The dried samples were heated from 25 °C to 750 °C with 20 °C/min heating rate and a nitrogen flow rate of 60 mL/min.
Fourier-transform infrared spectroscopy (FTIR) was used to obtain infrared spectra in transmission mode (KBr Mini-Pellet Press, Specac) with a Thermo Scientific Nicolet iS20 FTIR spectrometer in the 4000–450 cm−1 range.
The magnetization curve of samples was measured on dried sample with Vibrating-Sample Magnetometry (VSM, Lake Shore 7300-series). The field intensity maximum was fixed at 5 kOe and the instrument scanned the range [− 5; 5] kOe twice.
X-ray powder diffraction (XRPD) was used to identify the crystalline phase of the nanoparticles on a PANalytical X’Pert PRO Alpha-1, equipped with a Copper anode (Cu-K1 radiation) with a step size of 0.24° in continuous mode and a scan speed of 0.04°/s.
Ultraviolet–visible spectroscopy (UV–Vis), was performed using Nanophotometer NP80 (IMPLEN) in 2 mL quartz cuvettes with a path length of 1 cm. A quantitative estimation of the microsphere functionality was given by the activity \(\alpha\), related to the relative amount of turbidity removed by magnetic separation:
where \(I_{0}\) is the initial absorbance of the sample and \(I_{\text{f}}\) the absorbance after magnetic separation. The standard turbidity was quantified using a Clay Solution (Kaolin, 1% by wt) which approximately possesses a unitary absorbance for light with wavelength \(\lambda = 500\) nm.
Scanning electron microscopy (SEM) was done on a FEI Nova 200 system at 10 keV acceleration voltage. The samples were prepared by dropping 10 µL of dispersion on a graphite-coated aluminium holder and air dried at room temperature.
The iron concentration in nanoparticle suspension was measured with Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES), using Thermo Scientific ICAP 6500 system.
Atomic force microscopy (AFM) was performed using an AFM Bruker Dimension FastScan Bio for the imaging of 10 µL of dried sample on a silica wafer, setting the scan rate to 1 Hz and the tip velocity to 0.303 m/s. Size distribution analysis was attained by measuring the size of approximately 150 entities.
Abbreviations
- DLS:
-
Dynamic light scattering
- TGA:
-
Thermal gravimetric analysis
- FTIR:
-
Fourier-transform infrared spectroscopy
- VSM:
-
Vibrating-sample magnetometry
- XRD:
-
X-ray powder diffraction
- UV–Vis:
-
Ultraviolet–visible spectroscopy
- NPs:
-
Nanoparticles
- MW:
-
Microwave
- SPIONs:
-
Superparamagnetic iron oxide nanoparticles
- C-SPIONs:
-
Citrate-enriched SPIONs
- SAPES:
-
Self-assembled polyelectrolytic spheres
- MOCP:
-
Moringa oleifera coagulant protein
- MO-SAPES:
-
MOCP-functionalized self-assembled polyelectrolytic spheres
- GA:
-
Glutaraldehyde
- Gly:
-
Glycine
- TSC:
-
Tri-sodium citrate
- PLL:
-
Poly-l-lysine
- SMBS:
-
m-Maleimidobenzoyl-N-hydroxysulfosuccinimide ester
References
Ahn Y, Choi EJ, Kim S, Ok HN (2001) Magnetization and Mössbauer study of cobalt ferrite particles from nanophase cobalt iron carbonate. Mater Lett 50(1):47–52. https://doi.org/10.1016/S0167-577X(00)00412-2
Asma Muhmed AM (2013) Effect of lime stabilisation on the strength and microstructure of clay. IOSR J Mech Civ Eng 6(3):87–94. https://doi.org/10.9790/1684-638794
Beveridge JS, Stephens JR, Williams ME (2011) The use of magnetic nanoparticles in analytical chemistry. Annu Rev Anal Chem 4(1):251–273. https://doi.org/10.1146/annurev-anchem-061010-114041
Busquets MA, Estelrich J, Sánchez-Martín MJ (2015) Nanoparticles in magnetic resonance imaging: from simple to dual contrast agents. Int J Nanomed 10(1):1727. https://doi.org/10.2147/IJN.S76501
Carenza E, Barceló V, Morancho A, Montaner J, Rosell A, Roig A (2014) Rapid synthesis of water-dispersible superparamagnetic iron oxide nanoparticles by a microwave-assisted route for safe labeling of endothelial progenitor cells. Acta Biomater 10(8):3775–3785. https://doi.org/10.1016/J.ACTBIO.2014.04.010
Chantrell R, Popplewell J, Charles S (1978) Measurements of particle size distribution parameters in ferrofluids. IEEE Trans Magn 14(5):975–977. https://doi.org/10.1109/TMAG.1978.1059918
Chen L, Wang T, Tong J (2011) Application of derivatized magnetic materials to the separation and the preconcentration of pollutants in water samples. TrAC Trends Anal Chem. https://doi.org/10.1016/j.trac.2011.02.013
Cheng K, Yang M, Zhang R, Qin C, Su X, Cheng Z (2014) Hybrid nanotrimers for dual T1 and T2-weighted magnetic resonance imaging. ACS Nano 8(10):9884–9896. https://doi.org/10.1021/nn500188y
Cui J, Zhang Z, Jiang H, Liu D, Zou L, Guo X, Lu Y, Parkin IP, Guo D (2019) Ultrahigh recovery of fracture strength on mismatched fractured amorphous surfaces of silicon carbide. ACS Nano 13(7):7483–7492. https://doi.org/10.1021/acsnano.9b02658
Daou TJ, Pourroy G, Bégin-Colin S, Grenèche JM, Ulhaq-Bouillet C, Legaré P, Bernhardt P, Leuvrey C, Rogez G (2006) Hydrothermal synthesis of monodisperse magnetite nanoparticles. Chem Mater 18(18):4399–4404. https://doi.org/10.1021/cm060805r
Gassenschmidt U, Jany KD, Tauscher B, Niebergall H (1995) Isolation and characterization of a flocculating protein from Moringa oleifera Lam. Biochim Biophys Acta 1243(3):477–481. https://doi.org/10.1016/0304-4165(94)00176-x
Gauthier E, Fortier I, Courchesne F, Pepin P, Mortimer J, Gauvreau D (2000) Aluminum forms in drinking water and risk of Alzheimer’s disease. Environ Res 84:234–246. https://doi.org/10.1006/enrs.2000.4101
Gawande MB, Shelke SN, Zboril R, Varma RS (2014) Microwave-assisted chemistry: synthetic applications for rapid assembly of nanomaterials and organics. Acc Chem Res 47(4):1338–1348. https://doi.org/10.1021/ar400309b
Ghebremichael KA, Gunaratna KR, Henriksson H, Brumer H, Dalhammar G (2005) A simple purification and activity assay of the coagulant protein from Moringa oleifera seed. Water Res 39(11):2338–2344. https://doi.org/10.1016/j.watres.2005.04.012
Horák D, Semenyuk N, Lednický F (2003) Effect of the reaction parameters on the particle size in the dispersion polymerization of 2-hydroxyethyl and glycidyl methacrylate in the presence of a ferrofluid. J Polym Sci Part A Polym Chem 41(12):1848–1863. https://doi.org/10.1002/pola.10728
Jahn SAA (1988) Using Moringa seeds as coagulants in developing countries. J Am Water Works Assoc 80(6):43–50. https://doi.org/10.1002/j.1551-8833.1988.tb03052.x
Justin C, Philip SA, Samrot AV (2017) Method of synthesis of SPIONs. Appl Nanosci 7(7):463–475. https://doi.org/10.1007/s13204-017-0583-x
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10(6):845–858. https://doi.org/10.1038/nprot.2015.053
Kolb HC, Finn MG, Sharpless KB (2001) Click chemistry: diverse chemical function from a few good reactions. Angew Chemie Int Ed 40(11):2004–2021. https://doi.org/10.1002/1521-3773(20010601)40:11%3c2004:aid-anie2004%3e3.3.co;2-x
Kondo A, Kamura H, Higashitani K (1994) Development and application of thermo-sensitive magnetic immunomicrospheres for antibody purification. Appl Microbiol Biotechnol 41(1):99–105. https://doi.org/10.1007/BF00166089
Lu HD, Yang SS, Wilson BK, McManus SA, Chen CVHH, Prud’homme RK (2017) Nanoparticle targeting of gram-positive and gram-negative bacteria for magnetic-based separations of bacterial pathogens. Appl Nanosci 7(3–4):83–93. https://doi.org/10.1007/s13204-017-0548-0
Marcilla A, Gómez-Siurana A, Beltrán M, Martínez-Castellanos I, Blasco I, Berenguer D (2018) TGA-FTIR study of the pyrolysis of sodium citrate and its effect on the pyrolysis of tobacco and tobacco/SBA-15 mixtures under N 2 and air atmospheres. J Sci Food Agric 98(15):5916–5931. https://doi.org/10.1002/jsfa.9121
McClements DJ, Xiao H (2017) Is nano safe in foods? Establishing the factors impacting the gastrointestinal fate and toxicity of organic and inorganic food-grade nanoparticles NPJ Sci Food 1(1):6. https://doi.org/10.1038/s41538-017-0005-1
Mody VV, Cox A, Shah S, Singh A, Bevins W, Parihar H (2014) Magnetic nanoparticle drug delivery systems for targeting tumor. Appl Nanosci (Switzerland). https://doi.org/10.1007/s13204-013-0216-y
Neuberger T, Schöpf B, Hofmann H, Hofmann M, von Rechenberg B (2005) Superparamagnetic nanoparticles for biomedical applications: possibilities and limitations of a new drug delivery system. J Magn Magn Mater 293(1):483–496. https://doi.org/10.1016/J.JMMM.2005.01.064
Okoli C, Boutonnet M, Järås S, Rajarao-Kuttuva G (2012) Protein-functionalized magnetic iron oxide nanoparticles: time efficient potential-water treatment. J Nanopart Res 14(10):1194. https://doi.org/10.1007/s11051-012-1194-9
Patsula V, Kosinová L, Lovrić M, Ferhatovic Hamzić L, Rabyk M, Konefal R, Paruzel A, Šlouf M, Herynek V, Gajović S et al (2016) Superparamagnetic Fe3O4 nanoparticles: synthesis by thermal decomposition of Iron(III) glucuronate and application in magnetic resonance imaging. ACS Appl Mater Interfaces 8(11):7238–7247. https://doi.org/10.1021/acsami.5b12720
Ramanujan RV, Chong WT (2004) The synthesis and characterization of polymer coated iron oxide microspheres. J Mater Sci Mater Med 15(8):901–908. https://doi.org/10.1023/B:JMSM.0000036278.09272.97
Saladino GM (2019) SPfit-superparamagnetic fit. Zenodo. https://doi.org/10.5281/ZENODO.3244439
Saxena N, Dholia N, Akkireddy S, Singh A, Yadav UCS, Dube CL (2019) Efficient microwave synthesis, functionalisation and biocompatibility studies of spion based potential nano-drug carriers. Appl Nanosci. https://doi.org/10.1007/s13204-019-01153-8
Söderlund J, Kiss LB, Niklasson GA, Granqvist CG (1998) Lognormal size distributions in particle growth processes without coagulation. Phys Rev Lett 80(11):2386–2388. https://doi.org/10.1103/PhysRevLett.80.2386
Storozhuk L, Iukhymenko N (2019) Iron oxide nanoparticles modified with silanes for hyperthermia applications. Appl Nanosci 9(5):889–898. https://doi.org/10.1007/s13204-018-0777-x
Tong W, Gao C, Möhwald H (2005) Manipulating the properties of polyelectrolyte microcapsules by glutaraldehyde cross-linking. Chem Mater 17(18):4610–4616. https://doi.org/10.1021/cm0507516
Toprak MS, McKenna BJ, Mikhaylova M, Waite JH, Stucky GD (2007a) Spontaneous assembly of magnetic microspheres. Adv Mater 19(10):1362–1368. https://doi.org/10.1002/adma.200602114
Toprak MS, McKenna BJ, Waite JH, Stucky GD (2007b) Control of size and permeability of nanocomposite microspheres. Chem Mater 19(17):4263–4269. https://doi.org/10.1021/cm071215b
Tsai M-L, Tsai D-S, Tang L, Chen L-J, Lau SP, He J-H (2017) Omnidirectional harvesting of weak light using a graphene quantum dot-modified organic/silicon hybrid device. ACS Nano 11(5):4564–4570. https://doi.org/10.1021/acsnano.6b08567
Wang S, Goh BM, Manga KK, Bao Q, Yang P, Loh KP (2010) Graphene as atomic template and structural scaffold in the synthesis of graphene–organic hybrid wire with photovoltaic properties. ACS Nano 4(10):6180–6186. https://doi.org/10.1021/nn101800n
Wang B, Zhang Z, Chang K, Cui J, Rosenkranz A, Yu J, Lin C-T, Chen G, Zang K, Luo J et al (2018) New deformation-induced nanostructure in silicon. Nano Lett 18(7):4611–4617. https://doi.org/10.1021/acs.nanolett.8b01910
Yantasee W, Warner CL, Sangvanich T, Addleman RS, Carter TG, Wiacek RJ, Fryxell GE, Timchalk C, Warner MG (2007) Removal of heavy metals from aqueous systems with thiol functionalized superparamagnetic nanoparticles. Environ Sci Technol 41(14):5114–5119. https://doi.org/10.1021/es0705238
Yin C-Y (2010) Emerging usage of plant-based coagulants for water and wastewater treatment. Process Biochem 45(9):1437–1444. https://doi.org/10.1016/J.PROCBIO.2010.05.030
Yoffe S, Leshuk T, Everett P, Gu F (2012) Superparamagnetic iron oxide nanoparticles (SPIONs): synthesis and surface modification techniques for use with MRI and other biomedical applications. Curr Pharm Des 19(3):493–509. https://doi.org/10.2174/1381612811306030493
Zhang L, Chan JM, Gu FX, Rhee J-W, Wang AZ, Radovic-Moreno AF, Alexis F, Langer R, Farokhzad OC (2008) Self-assembled lipid–polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano 2(8):1696–1702. https://doi.org/10.1021/nn800275r
Zhang Z, Huo F, Zhang X, Guo D (2012) Fabrication and size prediction of crystalline nanoparticles of silicon induced by nanogrinding with ultrafine diamond grits. Scr Mater 67(7–8):657–660. https://doi.org/10.1016/j.scriptamat.2012.07.016
Zhang Z, Huo Y, Guo D (2013) A model for nanogrinding based on direct evidence of ground chips of silicon wafers. Sci China Technol Sci 56(9):2099–2108. https://doi.org/10.1007/s11431-013-5286-2
Zhang Z, Guo D, Wang B, Kang R, Zhang B (2015) A novel approach of high speed scratching on silicon wafers at nanoscale depths of cut. Sci Rep. https://doi.org/10.1038/srep16395
Zhang Z, Cui J, Zhang J, Liu D, Yu Z, Guo D (2019) Environment friendly chemical mechanical polishing of copper. Appl Surf Sci 467–468:5–11. https://doi.org/10.1016/j.apsusc.2018.10.133
Zhou G, Yin L-C, Wang D-W, Li L, Pei S, Gentle IR, Li F, Cheng H-M (2013) Fibrous hybrid of graphene and sulfur nanocrystals for high-performance lithium-sulfur batteries. ACS Nano 7(6):5367–5375. https://doi.org/10.1021/nn401228t
Acknowledgements
Open access funding provided by Royal Institute of Technology. VSM analysis was made possible by E. Holmgren and D. M. Polishchuk from the Nanostructure Physics division (Department of Applied Physics, KTH), through the courtesy of V. Korenivski.
Author information
Authors and Affiliations
Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saladino, G.M., Hamawandi, B., Vogt, C. et al. Click chemical assembly and validation of bio-functionalized superparamagnetic hybrid microspheres. Appl Nanosci 10, 1861–1869 (2020). https://doi.org/10.1007/s13204-020-01274-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13204-020-01274-5