
Abstract
A hydroperoxidized polypropylene (HPP) was obtained by oxidizing PP porous particle in solid phase, followed by impregnating dual polar monomers of pentaerythritol triacrylate (PETA) and styrene (St) into the HPP with the aid of supercritical carbon dioxide (scCO2). Then, HPP was used as polymerization initiators and PETA/St were grafted onto microporous PP backbone in water medium. Effects of hydroperoxidation temperature, time, oxidant dosage and PP diameter on hydroperoxides concentration and G p and G e were illustrated systematically. Besides, effects of scCO2 swell time, pressure, monomers concentration and ratio, grafting reaction time and temperature on G p and G e were also examined. Results showed that G p can be easily controlled by changing process conditions; G e was observed to be greater than 90% in most of the cases. Gel content of grafted samples was also determined. The structures and thermal properties of grafted copolymers were characterized through FTIR, SEM, TGA and DSC.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
As one of the most important general purpose polymers, polypropylene (PP) is widely used in various fields. However, its low surface energy and poor compatibility with other polar polymers impose a limit on certain applications. In the same sense, their adhesion to materials like wood, metals or reinforcing fibers requires special attention [1,2,3,4,5,6,7,8]. Grafting various monofunctional and multifunctional polar monomers onto its backbone offers an effective approach to overcome these disadvantages. A number of modifying approaches have been explored [9, 10]. Among them, scCO2 grafting has been investigated extensively in recent years [11,12,13,14,15,16,17,18].
The advantages of scCO2 include that high diffusivities, low viscosity, and near zero surface tension, mild critical parameters (T c = 31.1 °C, P c = 7.38 MPa), and nontoxic, nonflammable and inexpensive. At properly chosen densities, scCO2 can dissolve nonpolar or not exceedingly polar compounds to a manageable size. Most of all, scCO2 swells polymer but not dissolve it. In this way, the diffusion resistance is reduced and the solute may be dissolved and distributed more homogeneous in the amorphous domains of the bulk polymer [14,15,16,17,18,19].The density of scCO2 is continuously tunable from gas to liquid by changing its temperature or pressure. This provides the ability to control the degree of swell of a polymer [15, 18]. The low viscosity of a supercritical fluid allows for rapid mass transfer of penetrate into a swollen polymer. Moreover, because CO2 is a gas at ambient conditions, removal of the solvent from product is facilitated [18]. According to this special property, researchers have reported extensively the use of scCO2 to diffuse some polar substances into a polymer matrix such as PP to modify its physical and chemical properties [20,21,22].
The methods reported above included one-step and two-step processes. In one-step process, polar monomers and initiator dissolved in scCO2 are carried into PP matrix, simultaneously thermal free-radical grafting polymerization, resulting in composites [14, 16, 18,19,20,21]. This is a simple technical art, but in this course CO2 is retained in the system from beginning to end. The pressure in reactor increases rapidly with temperature, which is not favorable to product in large scale. Two-step process is reported as follows [10, 17,18,19]: first, monomers and initiator dissolved in scCO2 are carried into PP matrix; thereafter, CO2 is discharged and the monomers and initiator would be left in the polymer; at last, thermal free-radical grafting polymerization is achieved in mild condition. However, grafting reaction and auto-polymerization of monomers occur at the same time. There is little literature about a high grafting percentage (G p) and grafting efficiency (G e).
A hydroperoxidized polypropylene (HPP) can be easily obtained by oxidizing PP porous particle in solid phase and further used for the grafting of polar monomers onto its backbone. Most of the works report grafting through cleavage of hydroperoxide group using metal chelates. However, requirement of surfactant as interfacial agent, metal salts and other ingredients, makes grafting dependent on more parameters. Surekha Devi [23] has reported a novel method of cleavage hydroperoxide group in water medium and gained a high grafting percentage (G p) and grafting efficiency (G e). The schematic mechanism of grafting polar monomers on the surface of HPP was also proposed. However, due to the diffusion limitation resulted from small pore size of PP, grafting chains most showed in surfaces of PP and led to heterogeneous composition. Herein, we describe grafting of St and PETA onto PP backbone using scCO2 as a solvent and swell agent, HPP as initiator. Effects of various factors, such as the hydroperoxidation time and temperature, oxidant dosage, scCO2 swell time and pressure, monomers concentration and ratio, grafting reaction time and temperature on grafting level were studied. The chemical and thermal properties of the composites were also examined. In this work, grafting level and homogeneity can be easily improved by combining hydroperoxidation of PP with the swell ability of scCO2. To our knowledge, the combination of these two methods has not been reported so far, and this work should be interesting to researchers in polymer academy and industry.
Experimental section
Materials
Polypropylene: Isotactic polypropylene (iPP) powder, average particle diameter of 0.9–2.5 mm, was supplied by Daqing Petrochemicals Company and washed several times with acetone and dried under vacuum at 40 °C before use; Monomers: Styrene (St) was made free from inhibitor by washing with 10% potassium hydroxide solution and then drying over anhydrous calcium chloride. It was further purified by vacuum distillation and stored at 0 °C. Pentaerythritol Triacrylate (PETA) analytical grade, from Tianjin Tianjiao Chemical Reagent Plant (Tianjin, China), used as received; Oxidant: Benzoyl peroxide (BPO), Beijing Chemical Plant, was recrystallized twice from chloroform and methanol; Other Reagents: xylene, methyl alcohol, acetone, and chloroform, analytical grade; Nitrogen with a purity of 99.9%; CO2 with a purity of 99.95% was supplied by Daqing Xuelong and used as received. Distilled water was used throughout the work.
Grafting procedures
The grafting process is followed by three individual steps:
Hydroperoxidation of PP
Hydroperoxidation process was performed in a three-neck flask of 250 ml equipped with a twin-blade mechanical stirrer operated at 100 rpm. The temperature of the flask was maintained with an accuracy of ± 1.0 °C. First, the reactor was flushed with nitrogen for 3–5 min at room temperature; 30 g PP granules were then added, followed by a mixed solution of oxidant BPO and 3 ml xylene. The mixture was stirred for 15 min. Then, the flask was placed in an oil bath at the given temperature and dry air prepared by passing through anhydrous calcium chloride was passed at a constant rate of one bubble per second from a 2 mm diameter pipe with mild stirring. The reaction mixture was then cooled to room temperature under nitrogen atmosphere, and the hydroperoxidation of PP product (HPP) in powder form was soaked in acetone for 4 h to remove any unreacted BPO. This was followed by filtration, drying under vacuum, storage at 0 °C before use [9].
Swell PP using supercritical CO2
A high-pressure kettle reactor of 110 ml made from stainless steel was used. The HPP from first step and a mixture of PETA and St were added into the reactor and then air was replaced by CO2. The reactor was sealed and placed into an electric-heated thermostatic water bath. The temperature was measured with a calibrated mercury thermometer and was controlled at an accuracy of 0.5 °C. After the system had reached thermal equilibrium, CO2 was pumped in until the desired pressure was reached. The HPP particles were swollen and PETA/St was diffused into the HPP particle reactors. After the desired soaking time, the reactor was cooled to room temperature and the fluid in the system was released.
Thermal-induced grafting on hydroperoxidized polypropylene
The samples swelled from the second step were transferred into another three-neck flask of 250 ml and suspended in proper amount of distilled water. The air in the flask was replaced by nitrogen, and then the flask was placed in a constant temperature oil bath at desired temperature. After a suitable reaction time, the flask was taken out and cooled to room temperature; this was followed by common purification process as details described elsewhere [16, 17].
To investigate the grafting percentage of PETA and St onto the PP substrate, the resulting composites were Soxhlet-extracted using hot acetone for 10 h to remove ungrafted monomers. After dried, the extracted composites were weighed, and then were re-extracted. These procedures were repeated until the weight of the composites was constant. It was assumed that the monomers were removed completely after the extraction process. The grafting percentage (G p) and grafting efficiency of monomers (G e) were determined gravimetrically, and calculated from the following relation:
where w 0 stands for the weight of original PP, w 1 for the weight of grafted PP after removal of unreacted PETA and St and w 2 for the weight of monomers.
Determination of the concentration of active oxygen sites
The overall concentration of “active oxygen sites (OPP)” (peroxide groups initiating radical polymerization) in oxidized polypropylene powder was measured by iodometric titration. Two types of hydroperoxidic groups are present on the surface of the oxidized polypropylene—associated hydroperoxides (APP) and isolated hydroperoxides (IPP). The concentration of APP was determined by reacting with KI in glacial acetic acid–organic solvent at 20 °C and IPP concentration was found by titration with KI in chloroacetic acids at 40 °C. The “active oxygen sites” concentration was expressed in mol O2/kg OPP [22].
Gel studies
PP might crosslink if a vinyl monomer is grafted onto it, especially grafting percentage was high. If the crosslinking happened, the structures and properties of polymer would be deteriorated to a great extent, which is not expected. It is reported that PP and grafted polymer would be dissolved in hot xylene completely within 30 min and the crosslinked polymer cannot be dissolved forever. If there is any gel present in xylene solution, crosslinking will be proved. So, gel content of grafted copolymers can be determined by Soxhlet extracting fractionation method. Accurately weighed grafting samples were boiled in xylene for 2 h. The undissolved part was washed with hot xylene at 80 °C and then cooled with acetone and dried till constant weight. Blank experiments were carried out using nascent PP powders.
Gel content = w e/w g × 100 wt%, where w e is weight of undissolved part after extraction and w g is weight of samples before extraction [9].
Characterization
FTIR spectra of selected samples of grafted copolymers were recorded on BrukerEquinox55 FTIR spectrophotometer. The PP samples were processed in 100 μm thick films by compression moulding of 0.1–0.2 g between Teflon-covered aluminum sheets under atmospheric pressure at 200 °C for 5 s.
Thermogravimetric analysis (TGA) of grafted copolymers was carried out on a Diamond TG/DTA thermal analyzer at a heating rate of 10 °C/min from room temperature to 500 °C.
Differential scanning calorimetric analysis (DSC) was studied with a Perkin-Elmer DSC7 thermal analyzer. The analysis was carried out at a constant heating or cooling rate of 10 °C/min in the temperature range of 50–250 °C under a nitrogen atmosphere. The apparent crystallinity (Xc) of virgin PP and grafted samples was calculated by the following equation:
where ΔH m is the melt enthalpy of the sample, ΔH 0 is the melt enthalpy of the hypothetical iPP with crystallinity of 100%, which was 209 J/g [4].
Scanning electron microscope (SEM) experiments were performed using a JSM-6360LA SEM. The samples were fractured in liquid nitrogen, and the cross-sectional surface was pre-coated with gold before the SEM analysis.
Results and discussion
Effects of hydroperoxidation temperature and time, oxidant dosage and average PP particle diameter on the content of hydroperoxides and G p, G p are illustrated in Figs. 1, 2, 3, 4, 5, and 6 and Table 1, respectively. Polypropylene is of porous structure; its internal surfaces of micropores are far larger than its external surfaces. Under normal conditions, it is difficult for monomers diffusing uniformly into microspores of HPP. With the aid of scCO2, monomers were impregnated uniformly into the HPP to form particle reactor. The effects of swell temperature, pressure, time on G p and G e are shown in Figs. 7, 8, and 9, respectively. To find out the optimal grafting operation conditions, effects of monomers concentrations and ratio, grafting reaction time and temperature were studied systematically, and the results are shown in Figs. 10, 11, 12, and 13, respectively.

Influence of hydroperoxidation temperature on hydroperoxides concentration. (hydroperoxidation time = 2 h, oxidant dosage = 0.5 g)

Influence of hydroperoxidation temperature on G p and G e (swell condition: 37 °C, 3 h and 8.5 MPa; grafting reaction condition: 80 °C and 8 h; w monomer:w PP = 30 wt %, x(PETA:St,mol:mol) = 1:1; other conditions were the same as those in Fig. 1)

Influence of hydroperoxidation time on hydroperoxides concentration (hydroperoxidation temperature = 80 °C, oxidant dosage = 0.5 g)

Influence of hydroperoxidation time on G p and G e (swell condition: 37 °C, 3 h and 8.5 MPa; grafting reaction condition: 80 °C and 8 h; w monomer:w PP = 30 wt%, x(PETA:St,mol:mol) = 1:1; other conditions were the same as those in Fig. 3)

Influence of oxidant dosage on hydroperoxides concentration (hydroperoxidation temperature = 80 °C, hydroperoxidation time = 2 h)

Influence of oxidant dosage on G p and G e (swell condition: 37 °C, 3 h and 8.5 MPa; grafting reaction condition: 80 °C and 8 h; w monomer:w PP = 30 wt%, x(PETA:St,mol:mol) = 1:1; other conditions were the same as those in Fig. 5)

Influence of swell temperature on G p and G e (swell condition: 3 h and 8.5 MPa; grafting reaction condition: 80 °C and 8 h; w monomer:w PP = 30 wt %, x(PETA:St,mol:mol) = 1:1; other conditions were the same as those in Fig. 2)

Influence of swell pressure on G p and G e (swell condition: 37 °C and 3 h; grafting reaction condition: 80 °C and 8 h; w monomer:w PP = 30 wt%, x(PETA:St,mol:mol) = 1:1; other conditions were the same as those in Fig. 2)

Influence of swell time on G p and G e (swell condition: 37 °C and 8.5 MPa; grafting reaction condition: 80 °C and 8 h; w monomer:w PP = 30 wt %, x(PETA:St,mol:mol) = 1:1; other conditions were the same as those in Fig. 2)

Influence of grafting temperature on G p and G e (swell condition: 37 °C, 3 h and 8.5 MPa; grafting reaction time: 8 h; w monomer:w PP = 30 wt%, x(PETA:St,mol:mol) = 1:1; other conditions were the same as those in Fig. 2)

Influence of grafting time on G p and G e (swell condition: 37 °C, 3 h and 8.5 MPa; grafting reaction temperature: 80 °C; w monomer:w PP = 30 wt%, x(PETA:St,mol:mol) = 1:1; other conditions were the same as those in Fig. 2)

Influence of monomers concentration on G p and G e (swell condition: 37 °C, 3 h and 8.5 MPa; grafting reaction condition: 80 °C and 8 h; x(PETA:St,mol:mol) = 1:1; other conditions were the same as those in Fig. 2)

Influence of monomers molar ratio on G p and G e. (swell condition: 37 °C, 3 h and 8.5 MPa; grafting reaction condition: 80 °C and 8 h; w monomer:w PP = 30 wt%; other conditions were the same as those in Fig. 2)
Figures 1 and 2 show influence of hydroperoxidation temperature on hydroperoxides concentration and G p, G p. As can be seen from Fig. 1, the hydroperoxides concentration increases and passes through a maximum at 80 °C as the hydroperoxidation temperature increases. The number of IPP (P–O–O–H) is higher than APP (P–O–O–P′) in procedure of oxidizing PP, which is the same as in literature [22]. Oxidizing PP molecules are mainly initiated by free radicals from decomposition of oxidant BPO. As is known, the half-life of BPO varies intensely with temperature. The decomposition rate of BPO speeds up with temperature increases. Initial increase in hydroperoxides concentration is caused by the increase of free radicals. At high hydroperoxidation temperature, the activated sites of PP will be saturated when free radicals reach a certain concentration, then excess free radicals will become deactivation. At the same time, the hydroperoxidation groups produced decompose simultaneously. So the hydroperoxides concentration is reduced. Consequently, G p and G e (shown in Fig. 2) increase initially with hydroperoxidation temperature, and then decrease.
Figures 3 and 4 illustrate the dependence of hydroperoxides concentration and G p, G p on hydroperoxidation time in the range of 1.0–3 h. As can be seen from the figures, hydroperoxides concentration increases with time but slowly downwards after 2 h. The half-life of BPO at 80 °C is 460 min. Hydroperoxides concentration enhances with the time, but hydroperoxide groups decompose at the same time. It can be observed in Fig. 4 that G p and G e decrease after a period of 2 h although they rapidly increase initially.
It was observed in Fig. 5 that with increasing oxidant dosage of BPO, the hydroperoxides concentration increases constantly. This means that the amount of free radicals from decomposing of BPO increases. However, Fig. 6 shows that G p and G e increase firstly, then level off after 0.5 g. When the oxidant dosage of BPO was low, the hydroperoxides site on PP was also low. There are no sufficient active sites produced on the back chains of PP. After 0.5 g, the active sites on PP were sufficient for grafting polymerization. So, G p and G e become independent of oxidant dosage. Furthermore, an increasing of oxidant dosage not only induces degradation of PP, but also increases auto-polymerization of monomers which causes G p and G e to slightly decrease.
Table 1 illustrates the effect of average PP particle diameter on hydroperoxides concentration, G p and G e. As can be seen from Table 1, all of the experimental variables-hydroperoxides concentration, G p and G e, increase as the average PP particle diameter diminishing except 0.1–0.56 mm. Oxidation reaction of PP was carried out mostly on its surfaces and micropores. It is known that surfaces and micropores of solid increase with decreasing diameter. Large surfaces and micropores mean the contact with oxidant in hydroperoxidation is easier, so the hydroperoxides concentration increases. However, the small particles accumulate easily, which is not favorable for oxidation. PP particles are capable of absorbing a larger amount of monomer than the films. And the smaller the diameter of PP pellets, the larger the specific surface areas and the more of crystalline deficiency areas and micropores. Therefore, it facilitates the monomers to diffuse into the inner and the surfaces of PP. The smaller of grain diameter, the easier of monomer penetrating; therefore, G p and G e increase. But small pellets are easy to adhere to the reactor. In this work, the mix of above five diameters of PP was used. It must be stressed that the size of the PP powder has little effect on the grafting percentage in the melt and solution grafting process.
Figure 7 illustrates the dependence of G p and G e on swell temperature in the range of 32–53 °C. The other conditions were the same as those in Fig. 2. It is clear in Fig. 7 that G p and G e reach a maximum at 37 °C, then decrease. This phenomenon can be explained as following: The critical temperature of the supercritical dioxide is 31.1 °C. Swell of PP and penetrating of monomers into PP are easy as raising temperature. But it is well known that the density of CO2 decreases with temperature at a constant volume and pressure. Therefore, the solvent ability of CO2 is weakened as the temperature is raised; the amount of monomers was diminished in the PP phase. Consequently, the samples prepared have a relatively lower grafting level when swell temperature was higher than 37 °C.
Figure 8 exhibits that G p and G e reach a maximum at scCO2 swell pressure of 8.6 MPa, which can be explained as following. On one hand, increasing scCO2 pressure induces an increase in swell content of PP substrate, which makes the diffusion of the monomer into HPP easier and is advantageous for subsequent grafting reaction. On the other hand, the solvent ability of scCO2 is continuously changed with density. An increase in pressure at a constant temperature and volume results in increased solubility of CO2, which is not favorable to the sorption of the monomers in the PP. The resulting maximum at 8.6 MPa illustrates a balance of the two opposite factors.
Figure 9 shows G p and G e plotted as the function of swell time. It can be seen that G p and G e increase rapidly in the initial stage and reach a constant value after a period of 3 h. This result indicates that the adsorption of monomers in HPP substrate reached equilibrium in 3 h.
Effects of grafting reaction temperature and time, monomers concentration and ratio on G p and G e are shown in Figs. 10, 11, 12, and 13, respectively. As can be known from Fig. 10, with reaction temperature varying from 50 to 90 °C, G p and G e increase rapidly in the beginning and level off above 80 °C. Grafting reaction is initiated by free radicals and its reaction rate is depended mainly on the free radicals concentration of thermal decomposition of HPP. Below 50 °C, peroxide linkages on HPP backbone are inert and thermal decomposition of HPP may not take place. As a result, lower G p and G e were observed. As HPP decompose rate speeds up with grafting temperature, the decomposition rate is compatible with the grafting polymerization rate; so monomers can be grafted onto PP efficiently. The increased grafting level can also be attributed to the increasing diffuse rate of monomers on HPP chain at high temperature. G p and G e were 27.9 and 91.2%, respectively, at 80 °C, which illustrates this method is very efficient in obtaining a high G p and G e.
To study the effect of reaction time on G p and G e, grafting time is varied from 6 to 10 h, and the results are presented in Fig. 11. It is observed that the grafting level initially increases and then G p and G e level off after 8 h. This is easy to understand that the amounts of monomers and active sites from HPP run out with grafting time collapses.
Figure 12 shows effect of monomers concentration on G p and G e. It can be seen that G p increases constantly with the concentration of the monomers but G e decreases straightly. As the concentration of monomers goes high, more monomers will be impregnated into the HPP matrix. As we all know, scCO2 is a swelling agent for PP; however, it mainly swells the amorphous regions of PP matrix. Because of the existence of crystalline domains in PP matrix, it can only swell the polymer to some extent. When the swelling degree is fixed, the increase of monomer concentration cannot increase the grafting percentage once the absorbance balance is obtained. When the monomers conceration increases constantly, redundant monomers would self-polymerize instead of grafting onto PP backbone. The results are in agreement with the findings obtained by Zhimin Liu [17].
To increase G p and G e of the PETA, St was used as a co-monomer. Figure 13 shows the influence of the St/PETA molar ratio on the grafting products. In the presence of St, G p and G e of PETA were always higher than those in the absence of St. St improved the grafting reactivity of PETA. G p and G e reached a maximum when the molar ratio of PETA and St was approximately 1:1 and then decreased when the concentration of St was higher than that of PETA, because St has the ability of providing the electron to capture radicals on the HPP backbone as rapidly as possible [24]. Therefore, PETA could be significantly improved in the presence of St. However, when St was higher than PETA, a part of the St monomer reacted with PETA to form the poly(styrene-co-pentaerythritol triacrylate) copolymer, while the remaining St monomers might preferentially react with HPP macroradicals to form relatively stable styryl macroradicals. As a result, G p and G e decreased.
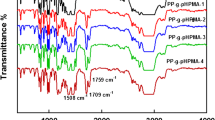
Fourier transform infrared (FTIR) spectroscopy is frequently adopted to characterize modified polymers. Figure 14 demonstrates the IR spectra of virgin PP and the grafted PP samples with different G p. It can be seen that there is an absorption peak around 1730 cm−1 in the spectra of PP-g-PETA; this peak is assigned to the stretch of the carbonyl group (–C=O). There are two absorption peaks around 694 and 742 cm−1 in the spectra of PP-g-St, and these are out-of-plane bending vibration of the C–H bonds in the aromatic, which cannot be observed from the virgin PP sample, and above-mentioned absorption peaks can be observed simultaneously in the spectra of PP-g-PETA/St. It was proved that monomers were grafted onto the PP. However, the peak of C=C can not be observed in the wavenumbers ranging from 1600 to 1800 cm−1, which indicates that cross-linking reaction may be occurred in grafting polymerization reaction.

FT-IR spectrum of samples
To investigate the thermal stability of PP-g-PETA/St, the TGA thermographs of pure PP, PP-g-PETA/St samples with different grafting percentages are shown in Fig. 15. The initial, 50% and end of decomposition temperature of the virgin and grafted PP samples are listed in Table 2. It can be seen that the modified samples show a higher onset thermal degradation temperature and a lower weight loss at a particular temperature. Besides, the thermal stability of the grafted copolymers was observed to increase with enlarge of grafting percentage. When PETA/St was grafted onto PP, grafting polymerization reaction is to substitute PETA/St branch chain for hydrogen atom from tertiary carbon atom of PP. The higher of grafting percentage, the side chains become more dense or longer. Furthermore, cross-linking reaction would occur easily at the higher G p. Due to multi-functional groups of PETA, an interpenetrating polymer network structure may be formed. These special structures will lead to gel present, which cannot dissolve in xylene solution. According to Table 3, the gel content of grafted sample increases with G p. This indicates the higher of G p, the greater probability of crosslinking presence.

TGA thermographs of virgin PP and PP-g- PETA/St with different G p
Figure 16 shows SEM micrographs of surfaces of pure PP, as well as different grafting percentages of PP-g-PETA/St samples. As can be seen from Fig. 16 b, some micropores with various sizes are present in PP, which permits monomers to enter interior of particles and form particle reactors. The morphologies of the grafted sample (Fig. 16c and d) were significantly different from those of the original sample. This was an indication that the monomers were grafted onto the PP, and also indicates that these grafted molecules can be uniformly distributed in the PP matrix under the assistant of scCO2.

SEM photographs of the inner and surface of samples a outer surface of a PP particle; b cut section of a PP particle; c outer surface of a PP-g-PETA/St particle, G p = 9.2%; d cut section of a PP-g-PETA/St particle, G p = 18.3%
To investigate their crystallization and melting behavior, DSC of different grafting percentages of PP-g-PETA/St samples were performed. The crystallization behaviors of the samples are shown in Figs. 17 and 18. The Tm, Tc, ΔH m, ΔH c and X c obtained from the DSC curves are listed in Table 4.

The cooling curves of pure PP and different PP-g-PETA/St samples

The melting curves of pure PP and different PP-g-PETA/St samples
Figure 17 shows DSC cooling scans at 10 °C/min for pure iPP and grafted product. It can be found from the figure that the iPP crystallization temperature (Tc) is 105.2 °C, specific enthalpy of crystallization (Hc) is 90.1 J/g, the crystallization temperatures of the grafted polymers (the G p of 9.2 and 18.3%) are 107.8 °C and 112.2 °C and their heats of crystallization are 93.3 and 95.6 J/g, respectively. As shown in Table 4, both Tc and T m of sample 1 and sample 2, compared with pure PP, increased obviously and their Xc were also larger than that of PP. These features suggested that the existence of the grafted branches segments promoted the crystallization of PP by a nucleation effect. The grafted branches would disrupt the regularity of PP chain structure and increase the spaces between chains, which would bring about the heterogeneous nucleation in PP. So the temperature of crystallization increased as the G p is increasing. The increased density of effective nuclei results in the completion of the crystallization process in a short time at a higher temperature.
Figure 18 shows DSC melting curves for pure iPP and grafted product. It can be found that the melting temperature (T m) and specific fusion enthalpy of samples increase with more monomers grafted onto PP, which also indicates more percentage of crystallization in the grafted polymers.
Conclusion
In this study, a novel strategy was designed for the preparation of PP-g-PETA/St grafting copolymer by impregnating PETA and St into HPP with scCO2 as a solvent and swelling agent, followed by free radical polymerization within the swollen PP matrix after releasing CO2. The grafting level can be controlled flexibly by changing condition such as hydroperoxidation, scCO2 swell condition and grafting reaction parameters. Optimum results could be obtained at the hydroperoxidation condition of 80 °C, 2 h, 0.5 g oxidant dosage, the scCO2 swell condition of 37 °C, 8.5 MPa, 3 h and the grafting condition of 80 °C, 8 h, m monomer:m PP = 30 wt%, x(PETA:St,mol:mol) = 1:1. It was found that the scCO2-assisted grafting process of PETA and St onto PP did have some scientifically interesting facts: the grafting efficiency was greater than 90% in most of the cases, and no serious cross-linking reaction occurred during the grafting process. FT-IR and SEM analysis proved that PETA and St were grafted simultaneously onto PP chains. TGA results showed that the grafted samples had a better thermal performance than pure PP. DSC results suggested that the grafted samples exhibited higher crystallization temperature, higher melting temperature, and higher crystallinity compared with the pure PP.
References
Wang K, Wang S, Fei W, P Y, Liu W, Zhai W, Zheng W (2016) A new strategy for preparation of longchain branched polypropylene via reactive extrusion with supercritical CO2 designed for an improved foaming approach. J Mater Sci 51:2705
Wang LL, Wu JJ, Zhang ZB, Zhou J, He XC, Yu HY, Gu JS (2016) Methoxypolyethylene glycol grafting on polypropylene membrane for enhanced antifouling characteristics-effect of pendant length and grafting density. Sep Sci Technol 164:81
Zhou S, Zhao S, Xin Z (2015) Preparation and foamability of high melt strength polypropylene based on grafting vinyl polydimethylsiloxane and styrene. Polym Eng Sci 55:251
Kunita MH, Rinaldi AW, Girotto EM (2005) Grafting of glycidyl methacrylate onto polypropylene using supercritical carbon dioxide. Eur Polym J. 41:2176
Prabhu KN, Macko T, Brüll R, Remerie K, Tacx J, Garg P, Ginzburg A (2016) Separation of maleic anhydride grafted polypropylene using multidimensional high-temperature liquid chromatography. J Chromatogr A 1441:96
Wang H, Chen W (2006) Radiation-induced grafting of binary monomers containing HALS onto polypropylene. Phys Chem 75:138
Hassan MS, Ibrahim HM (2016) Characterization and antimicrobial properties of metal complexes of polypropylene fibers grafted with acrylic acid using gamma irradiation. Polym Adv Technol 27:532
Boyer C, Boutevin B, Robin JJ (2005) Study of the synthesis of graft copolymers by a reactive process. Influence of the copolymer structure on the adhesion of polypropylene onto poly(vinylidene fluoride) polymer. Polym Degrad Stab 90:326
Patel AC, Brahmbhatt RB, Rao PV, Rao KV, Devi S (2000) Solid phase grafting of various monomers on hydroperoxidized Polypropylene. Eur Polym J 36:2477
Yanga C, Zhe X, Zhang M, Wang M, Wu G (2017) Radiation effects on the foaming of atactic polypropylene with supercritical carbon dioxide. Radiat Phys Chem 131:35
Liu Z, Dong Z, Han B (2002) Composites prepared by the polymerization of styrene within supercritical CO2-swollen polypropylene. Chem Mater 14:4619
Tai H, Wang W, Howdle SM (2005) Dispersion polymerization of vinylidene fluoride in supercritical carbon dioxide using a fluorinated graft maleic anhydride copolymer stabilizer. Macromolecules 38:1542
Cao K, Shen ZC, Yao Z, Qu BW, Pang XB, Lu ZQ, Li Y, Chen ZH (2010) New insight into the action of supercritical carbon dioxide for grafting of maleic anhydride onto isotactic polypropylene by reactive extrusion. Chem Eng Sci 65:1621
Galia A, De Gregorio R, Spadaro G (2004) Grafting of maleic anhydride onto isotactic polypropylene in the presence of supercritical carbon dioxide as a solvent and swelling fluid. Macromolecules 37:4580
Wang K, Pang Y, Liu W, Wu F, Zheng W (2016) A new approach designed for improving in situ compatibilization of polypropylene/polystyrene blends via reactive extrusion with supercritical CO2 as the processing medium. J Supercrit Fluid 118:203
Wang Y, Liu Z, Han B, Chen G (2004) pH Sensitive polypropylene porous membrane prepared by grafting acrylic acid in supercritical carbon dioxide. Polymer 45:855
Liu Z, Song L, Dai X, Yang G, Han B, Jian X (2002) Grafting of methyl methylacrylate onto isotactic polypropylene film using supercritical CO2 as a swelling agent. Polymer 43:1183
Liu T, Hu GH, Tong GS, Zhao L, Cao GP, Yuan WK (2005) Supercritical carbon dioxide assisted solidstate grafting process of maleic anhydride onto polypropylene. Ind Eng Chem Res 44:4292
Bao JB, Junior AN, Weng GS, Wang J, Fang YW, Hu GH (2016) Tensile and impact properties of microcellular isotactic polypropylene (PP) foams obtained by supercritical carbon dioxide. J Supercrit Fluid 111:63
Zhang A, Zhang Z, Gong Y (2004) Grafting of MAH and polypropylene in supercritical CO2 medium. Acta Polym Sin 2:292
Gong Y, Zhang Z, Zhang A (2004) Dual Grafting of MAH/styrene onto polypropylene using supercritical CO2 fluid. Chin J Appl Chem 21:914
Yiming W, Yu P, Yilong W, Guangpeng W, Yanjuan W, Nikitin LN, Xiaobing L (2008) Bulk graft modification of polyolefin membranes by combining pre-irradiation-induced graft and supercritical CO2-swelling polymerization. J Supercrit Fluid. 44:62
Citovický P, Chrástová V (1996) The copolymerization of styrene and maleic anhydride initiated by peroxides of isotactic polypropylene. Eur Polym 32:153
Yilu Z, Zhifang G, Liming Z, Lisha P, Zheng T, Sujuan P, Pang X, Qiang L, Nai X (2015) Mechanochemistry: a novel approach to graft polypropylene with dual monomers (PP-g-(MAH-co-St)). Polym Bull 72:1949
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, D., Wang, J. Grafting dual polar monomers onto hydroperoxidized polypropylene with the assistant of supercritical carbon dioxide. Appl Petrochem Res 7, 169–179 (2017). https://doi.org/10.1007/s13203-017-0190-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-017-0190-5