
Abstract
Boron- and barium-doped vanadium–magnesium oxide catalysts (BVMgO and BaVMgO) were synthesized by the wet impregnation method and were used for the oxidative dehydrogenation of n-octane at different n-octane/O2 molar ratios. The catalysts were characterized by ICP-OES, in situ XRD, 51V MAS NMR, SEM, EDX and TGA-DSC. The catalytic tests were carried out in a continuous flow fixed bed reactor. The incorporation of boron and barium as dopants into the VMgO system resulted in catalysts with some differences in properties, such as the details of phase transformations, porosity and degree of hydration. The catalytic performance was affected by the strength of the oxidative environment. Moreover, incorporation of barium enhanced the catalytic performance of VMgO, while the addition of boron adversely affected the performance of VMgO over all n-octane/O2 molar ratios with regard to both activity and oxidative dehydrogenation selectivity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The gas-to-liquid processes (GTL) that are becoming more and more widely used for fuel production, produce long-chain alkanes, including n-octane, as by-products. Due to the increasing demand for alkenes and aromatics in the polymer industry, there is a growing tendency in the petrochemical industry to the usage of these readily available alkanes to produce alkenes and aromatics. Although the non-oxidative dehydrogenation of alkanes has already been commercialized [1, 2], the oxidative dehydrogenation (ODH) represents a potential alternative route for the production of olefins and aromatics, as no thermodynamic equilibrium limitations or coke formation are expected. Moreover, possibilities for cracking product formation may be minimized by employing ODH, as operating at relatively low temperatures would be possible. The great challenge in the ODH, however, is to stop the reaction at intermediate stages (formation of olefins and aromatics), and not allow it to proceed further to form the thermodynamically stable carbon oxides (undesirable combustion products) [3]; especially when a strong oxidant such as oxygen (air) is used. The catalyst’s properties, as well as the operational conditions, are crucial in determining which of the above products will dominate.
Research on the catalytic activation of n-octane was dominated by the non-oxidative activation [4–13], and when oxygen was used for such activation (usually from air) the focus was on the production of hydrogen, syngas, as well as the short-chain olefins [14–20]. A common feature in these studies was that the linear octenes and styrene were either not formed or formed in small amounts. This is consistent with the fact that long-chain alkanes are prone towards cracking. In this context, the catalyst choice seems to be crucial in securing a successful pathway from n-octane to linear octenes and aromatics, especially in a reaction environment that contains oxygen. The ODH of alkanes in general is believed to proceed via a redox mechanism [21]. The participation of lattice oxygen in this mechanism was inferred from pulse experiments and isotopic studies [3, 22, 23]. Selectivity to the dehydrogenation product is deemed to be affected by the acid–base character of the catalyst, the mobility of the lattice oxygen, the atomic arrangements around the active sites, and the reducibility of the cation involved in the active centres [21, 24]. It is clear from this, and from the mechanism of alkane activation, that in the oxidative activation of alkanes, the catalyst choice is very important in determining the selectivity to a certain class of products, e.g. dehydrogenation, oxygenates, or cracking products. The VMgO system was not known for the formation of either oxygenates or cracked products during the dehydrogenation of short-chain alkanes [21, 25–30]. Thus, VMgO catalysts were employed in our laboratories for the ODH of n-octane to produce octenes and C8 aromatics [31–33]. In these studies, reasonable selectivities for the dehydrogenation products were obtained.
The modification of properties of a given catalyst, and hence its catalytic performance, may be attempted by employment of dopants (promoters). The dopants can influence the acid–base character of the catalysts, the metal–oxygen bond, electronic structure around the active sites and the isolation of these sites [34, 35]. The properties of the VMgO catalysts may be influenced by the addition of a suitable promoter, and hence its catalytic properties may also be modified. Different dopants have been studied with the VMgO catalysts, and different effects were observed [28, 36–42]. Boron- and barium-doped catalysts were employed in the ODH of n-octane at n-octane/oxygen molar ratio of 0.8 [43]. Boron and barium are different in their acid–base properties, electronegativity, as well as other properties like atomic size and charge/radius ratio. Therefore, it is likely that they will modify the VMgO system in different ways.
Besides the intrinsic properties of the VMgO system that favour the dehydrogenation route over oxygenates and cracking products formation, factors like the type of oxidant used as well as the alkane to oxygen ratio are also critical in determining the selectivity pattern to different products. The MgO support itself has been shown to make a minimal contribution to n-octane activation [44]. Since long-chain alkanes are prone to cracking, the dehydrogenation of such alkanes like n-octane to produce octenes and C8 aromatics must be conducted with extra caution. In our previous studies in n-octane activation, catalyst’ activity and selectivities were found to be influenced by the n-octane to oxygen ratios [32, 45]. Thus, the investigation of a factor like n-octane/oxygen ratios during the n-octane dehydrogenation over these doped VMgO catalysts would be informative.
Experimental
Catalyst synthesis and characterization
Magnesium oxide was prepared from magnesium oxalate by a procedure similar to that outlined in [31]. For the synthesis of the barium doped catalyst (BaVMgO), 700 ml of a hot aqueous solution that contained 1.030 g of ammonium metavanadate was added to 4.6427 g of MgO. The resultant solution was magnetically stirred and heated to 80 °C. After 30 min, 200 ml of a hot aqueous solution that contained 0.0626 g of barium carbonate was added to the hot solution that contained magnesium and vanadium. Heating and magnetic stirring was continued till a paste was formed. The paste was placed overnight in an oven at 110 °C. The catalyst so-obtained was ground, thoroughly mixed and was then calcined at 550 °C for 5 h (the temperature ramping during the calcination was 1.5o/min). The calcined catalyst was pelletized, crushed, and sieved to a 600–1000 μm mesh size. The same procedure was performed for the synthesis of the boron-doped catalyst (BVMgO); boric acid was used as a source for boron. The synthesis of the undoped catalyst (VMgO) is discussed elsewhere [31].
In situ XRD was performed in a flow of air (20 ml/min) using a Bruker AXS diffractometer equipped with a TCU 750 temperature control unit and operating at 40 kV and 40 mA. The source of radiation was Cu Kα. The 51V magic angle spinning nuclear magnetic resonance (MAS NMR) spectra were recorded at 600 MHz on a Bruker Avance III 600 NMR Spectrometer fitted with a Bruker Solid State Probe. The catalyst sample was packed in a cylindrical zirconia rotor that was rotated at a speed of 12 kHz during the data acquisition. SEM images were obtained using a LEO 1450 Scanning Electron Microscope. EDX was carried out using a Jeol JSM 6100 Scanning Electron Microscope equipped with a Bruker EDX detector. Samples for SEM images were coated with gold using a Polaron SC Sputter Coater, while those for the EDX were coated with carbon in a Jeol JEE-4C Vacuum Evaporator. Thermal analysis (TGA and DSC) was performed on an SDT Q600 TGA-DSC instrument in static air atmosphere. The sample (ca. 5 mg) was contained in an alumina crucible. The temperature was raised from ambient to 1000 °C at a heating rate of 10o/min.
Catalytic testing
The catalytic testing was carried out in a fixed bed continuous flow reactor that operated in a down flow mode. The reactor tube was stainless steel; the tube was 220 mm in length and 10 mm in internal diameter. Air was used as an oxidant and nitrogen as a make-up gas. The flow rates of nitrogen and gaseous n-octane were varied to give the required total flow (134 ml/min) and the n-octane/O2 molar ratios (0.1, 0.4, 0.8, 1.2 and 1.6). Before conducting any catalytic experiment, the air and nitrogen flow rates were set to the desired values using air and nitrogen rotameters, respectively, and the flow of each gas was further rechecked using a Perkin Elmer PE 1000 electronic flowmeter. An HPLC pump was used to deliver the n-octane to the reactor. The volume of the gas phase during the catalytic run was determined by a Ritter Drum-Type Gas Metre. The products of the catalytic testing were analyzed off-line using a Perkin-Elmer Clarus500 Gas Chromatograph equipped with both an FID and a TCD; the FID for the organic products and the TCD for the carbon oxides. For the capillary column attached to the FID (PONA), the carrier gas was hydrogen that flowed at 2.0 ml/min. For the capillary column attached to the TCD (Carboxen 1006 PLOT), the carrier gas was helium that flowed at 6.0 ml/min. An 870 KF Titrino plus (Metrohm) Karl Fischer Titrator was used to determine the weight percentage of water in both the organic and aqueous layers that formed by the catalytic tests. The catalyst volume was 1 ml (ca. 0.45 g) and the catalyst pellets sizes were between 600 and 1000 μm. The catalytic tests were carried out at 450 °C and at a gas hourly space velocity (GHSV) of 8000 h−1. The VMgO catalysts (doped and undoped) were tested at different n-octane to oxygen molar ratios, as it has been shown in our previous studies of n-octane activation that this parameter (n-octane/oxygen ratio) has a great influence on the catalytic activity and selectivity [32, 45]. The flow rates of gaseous n-octane/air/nitrogen in these experiments were 1.5/72/61, 6/72/56, 12/72/50, 18/72/44 and 24/72/38, which correspond to n-octane/O2 molar ratios of 0.1, 0.4, 0.8, 1.2 and 1.6, respectively. Conversion and selectivity were calculated on a carbon basis (carbon balances were in the range of 97–101%), and all experiments were carried out in duplicate.
Results and discussion
Characterization
The synthesized catalysts were characterized by ICP-OES, in situ XRD, MAS NMR spectroscopy, SEM, EDX and TGA-DSC. As determined by the ICP-OES, the concentrations of boron and barium (by weight) in the doped VMgO catalysts were 1.3 and 1.2%, respectively.
In situ XRD
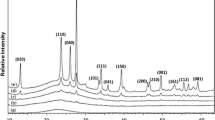
In situ XRD was carried out on the uncalcined catalysts in a bid to investigate phase transformations. These XRD experiments were conducted under a flow of air, i.e. under conditions similar to those under which the calcination of the catalysts was performed. The importance of phase transformations is that they are believed to affect the chemical and textural properties of the resultant catalyst after calcination [46, 47]. The in situ XRD diffractograms of BVMgO and BaVMgO catalysts are given in Fig. 1a, b, respectively. The lines assignments of these diffractograms were made according to [25, 46, 48]. At 50 °C, the XRD diffractograms of the doped catalysts (BVMgO and BaVMgO) showed lines characteristic of Mg(OH)2 at 2θ equal to 18.5°, 38°, 50.5° and 59° (d spacings of 4.78, 2.37, 1.8 and 1.57 Å, respectively). At 100 and 150 °C, the diffractograms of these doped catalysts were still characterized by the existence of Mg(OH)2 as a main crystalline phase. However, at these temperatures, the diffractograms of each of the doped catalysts also showed a weak line that is assignable to the magnesium orthovanadate; at 2θ equal to 35.5° (d spacing of 2.52 Å) for BVMgO and at 2θ equal to 23° (d spacing of 3.7 Å) for BaVMgO (Fig. 1a, b, respectively). A noticeable difference between BVMgO and BaVMgO is that the diffractogram of the BVMgO catalyst at 200 °C (Fig. 1a) showed a line attributable to MgO at 2θ equal to 43o (d spacing of 2.1 Å); however, Mg(OH)2 is still the dominant crystalline phase of both catalysts. This phase composition continues up to 300 °C. At 350 °C, the existence of MgO as the main crystalline phase can be inferred from the lines at 2θ equal to 43° and 62° (d spacings of 2.11 and 1.5 Å, respectively); the line assignable to Mg(OH)2 (d spacing of 2.37 Å) was still present for both doped catalysts. Worth mentioning here is that Oganowski et al. [49] attributed this line to the existence of a hydrated form of magnesium vanadate [Mg3(OH)2–V2O7(H2O)2]. A noticeable difference between the doped catalyst on one hand and the undoped catalysts on the other hand was that MgO was detected in the undoped catalyst (VMgO) for the first time at 350 °C and became the dominant crystalline phase at 400 °C and above [32]; i.e. both dopants caused the crystalline MgO to form from Mg(OH)2 at lower temperatures. For the doped catalysts (BVMgO and BaVMgO), the only crystalline phases detected at 400, 450, 500 and 550 °C were MgO by the lines at 2θ equal to 43 and 62° (d spacings of 2.11 and 1.50 Å, respectively) and magnesium orthovanadate by the line at 2θ equal to 44° (d spacing of 2.05 Å). Worth noting is that the existence of the orthovanadate in the undoped catalyst at these temperatures, viz. 400, 450, 500 and 550 °C, was indicated by the lines at the d spacings of 3.04 and 2.5 Å [32]. This indicates that magnesium orthovanadate in the doped catalysts (BVMgO and BaVMgO) exhibited different crystal planes than those shown by the undoped catalysts (VMgO).

a In situ XRD of the BVMgO catalyst [filled circle for Mg(OH)2, filled triangle for MgO, and filled square for Mg3(VO4)2]. b In situ XRD of the BaVMgO catalyst [filled circle for Mg(OH)2, filled triangle for MgO, and filled square for Mg3(VO4)2]
51V magic angle spinning nuclear magnetic resonance (MAS NMR)
In our previous study [43], the MAS NMR spectroscopy on the calcined catalysts (doped and undoped), indicated that vanadium exists as tetrahedrally coordinated V5+ species in coordination environments similar to that of orthovanadate. To get more insights, the MAS NMR spectroscopy was performed on the uncalcined catalysts. The 51V MAS NMR spectroscopy of the undoped catalyst (Fig. 2a) shows a band at –660 ppm. This, according to [50, 51], suggests that vanadium in the undoped catalyst (VMgO) exists as dimeric VO4 tetrahedra in a coordination environment similar to that of V2O7 4−, i.e. with the corner-sharing of two VO4 tetrahedra through a bridging oxygen atom. In a different observation, the spectra of the doped catalysts (BVMgO and BaVMgO) showed a band at –545 ppm (Fig. 2b, c, respectively). According to the assignments in [50, 51], this indicates that vanadium in the doped catalysts exists as isolated VO4 tetrahedra in a coordination environment similar to that of VO4 3−. This suggests that the incorporation of boron and barium into the VMgO system hinders the corner sharing of the VO4 tetrahedra. This is consistent with the observation from the in situ XRD results that the dopants cause the MgO to form from Mg(OH)2 at lower temperatures compared to that of the undoped catalyst; it was reported that the formation of the orthovanadate phase is somewhat associated with the transformation of Mg(OH)2 into MgO during the calcination of the VMgO catalyst [49].

51V MAS NMR spectra of the uncalcined catalysts:( a) VMgO, (b) BVMgO, and (c) BaVMgO
Scanning electron microscopy (SEM)
As Fig. 3 shows, the SEM micrographs of the three catalysts display fluffy, rough surfaces with a platelet-like structure. The surfaces of VMgO and BVMgO (Fig. 3a, b) show more cavities than that of BaVMgO (Fig. 3c), which indicates that these two catalysts are more porous than BaVMgO; this is in an agreement with the values obtained from the pore volume analysis [43], viz. 0.75, 0.75 and 0.64 cm3/g for VMgO, BVMgO and BaVMgO, respectively.

SEM micrographs (20,000 times magnification) of a VMgO, b BVMgO, and c BaVMgO
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC)
As suggested by the TGA and DSC results (Table 1), the three catalysts generally exhibit similar thermal behaviour. In each of the three catalysts, the thermal analysis shows an endothermic effect at around 130 °C, which is likely to be due to the removal of weakly bonded water molecules. Considering the weight loss for this effect (Table 1), the higher value in the case of BVMgO indicates that this catalyst is more hydrated than the other two. This is consistent with the acidic character of boron which is likely to enhance interaction with the lone pairs of electrons in the water molecules. Another weight loss was recorded at 350–390 °C. Considering the assignments in [49, 52], this weight loss may be attributed to the removal of strongly bonded water molecules. Interestingly, this weight loss was not accompanied by observable heat change. This suggests that the removal of these strongly bonded water molecules was accompanied by a form of crystallization, and that the endothermic effect induced by the removal of water was equated by the exothermic effect of the postulated crystallization.
Catalytic activity
It is clear from Fig. 4 that the catalytic activity is sensitive to the change in the n-octane to oxygen molar ratios. Enriching the reactant mixture with oxygen causes a significant increase in the conversion of n-octane. The oxygen-rich reactant mixture represents a strong oxidative environment that enhances the formation of the thermodynamically stable carbon oxides, which results in pushing the equilibrium far to the right, and consequently leads to high conversion. As Fig. 4 displays, the boron-doped catalyst shows the lowest conversion at all the ratios. The lower conversion over BVMgO is in agreement with the TPR data [43], as this catalyst shows the highest values for both Tmax and onset temperature when compared to BaVMgO and VMgO. This lower conversion over the BVMgO catalyst may be attributed to the decrease in the mobility of the lattice oxygen that is plausibly induced by the acidic and highly electronegative boron metal [43]. As Fig. 4 shows, the difference in conversion between the undoped (VMgO) and the barium-doped (BaVMgO) catalysts is widened at the n-octane to oxygen ratios of 0.4 and 0.1 (which corresponds to 4.5 and 1.1% (v/v) of gaseous n-octane in the reactant mixture); in these n-octane-lean environments, competitive adsorption between the components of the reactant mixture is likely to contribute much to the conversion.

n-Octane conversion and selectivity to carbon oxides at different n-octane/O2 ratios
Selectivity to 1-octene and styrene
Generally, high demand exists for linear alpha alkenes. 1-Octene and styrene are valuable products that are used in the petrochemicals and polymers industry; in fact, polystyrene is one of the most important polymers. As Fig. 5 displays, selectivity to 1-octene goes parallel to the enrichment of the reactant mixture with n-octane (mild oxidative environments), which in turn is accompanied by low conversions. This is consistent with the following conceivable sequential reaction [33]:
In this consecutive reaction, a strong oxidative environment will shift the reaction far to the right, and thereby lowers the selectivity to 1-octene; the opposite is true for the weak oxidative environment (n-octane-rich mixture). In the above reaction, the stable aromatic nucleus in styrene, and in C8 aromatics in general, represents a driving force that pushes the reaction far to the right; in support of this was that only small amounts of octadienes and octatrienes were formed.

Selectivity to 1-octene at different n-octane/O2 ratios
Opposite to the trend shown by 1-octene, enriching the reactant mixture with n-octane causes a noticeable decrease in the selectivity to styrene (Fig. 6). Consistent with the consecutive reaction postulated, enriching the reactant mixture with oxygen (strong oxidative environment), which is accompanied by an increase in the conversion, causes a noticeable increase in styrene selectivity, presumably by pushing the reaction far to the right. The exception to this, however, was at the n-octane/oxygen ratio of 0.1, as a noticeable decrease in the styrene selectivity was observed. This decrease in the styrene selectivity at the ratio of 0.1 may be attributed to the extremely oxygen-rich environment where the formed styrene may be degraded, via secondary combustion, to form the thermodynamically stable carbon oxides. In support of this was that a steep increase in selectivity to COx was observed when moving from the ratio 0.4 to 0.1 (Fig. 4). The BaVMgO catalyst exhibited the highest selectivity to styrene at the ratios 0.4, 0.8, and 1.2, while BVMgO catalyst showed the lowest selectivity to styrene at all the ratios. Worth mentioning is that the above-discussed observations, that related to the 1-octene and styrene selectivity, are applicable to the selectivity to octenes and C8 aromatics in general. To support these observations and to provide more data related to the main products of the catalytic testing, selectivity for octenes, C8 aromatics (ethylbenzene, styrene and o-xylene) and COx are given in Table 2.

Selectivity to styrene at different n-octane/O2 ratios
Dehydrocyclization ability
For the catalytic tests over the doped and the undoped catalysts, octenes and C8 aromatics represent the major ODH products. As indicated by the postulated reaction scheme, as well as by our previous studies [31, 33], octenes are precursors to C8 aromatics and that further dehydrogenation and cyclization of the formed octenes will eventually lead to the formation of C8 aromatics. Therefore, the dehydrocyclization ability of these catalysts may be indicated by comparing the amounts of the formed octenes to the combined amounts of octenes and C8 aromatics (Fig. 7). As can be inferred from Fig. 7, the cyclization power increases (more aromatics and less octenes) as the n-octane/O2 ratio decreases, i.e. as the reactant mixture is enriched with oxygen. Also, over all the n-octane/O2 ratios, the dehydrocyclization power of BaVMgO is greater than that of BVMgO, which indicates the better oxidative dehydrogenation performance of the former. In this respect, the barium-doped catalyst also exceeds the undoped one at the ratios of 0.4, 0.8 and 1.2. The variation in the dehydrocyclization power at different n-octane to oxygen ratios can also be seen when inspecting the selectivity of ethylbenzene and styrene. As shown in Fig. 8, the ethylbenzene selectivity gets closer to the styrene selectivity at the relatively higher n-octane/O2 ratios (1.2 and 1.6); while in the oxygen-rich environments (ratios of 0.1 and 0.4), the selectivity to styrene is obviously greater than that to ethylbenzene. As styrene results from deeper dehydrogenation of n-octane when compared to ethylbenzene, this again suggested that the dehydrogenation performance is enhanced by enriching the reactant mixture with oxygen.

Dehydrocyclization trends over the three catalysts at different n-octane/O2 ratios

Selectivity of ethylbenzene and styrene
As elucidated above, the catalytic results of the ODH of n-octane over the two doped catalysts (BVMgO and BaVMgO) at different n-octane/O2 ratios indicated that the performance of the barium-doped catalyst was better than that over the boron-doped catalyst in both activity and selectivity to ODH products (octenes and C8 aromatics).
Characterization of the used catalysts
To investigate any possible chemical or textural changes, characterization was carried out on the used catalysts; namely, powder XRD, EDX and ICP-OES. Worth mentioning is that the time-on-stream during the catalytic testing was 120 h. In a similar observation to those reported in [32, 53] for the used VMgO, XRD on the used BVMgO and BaVMgO catalysts indicates no change in the crystalline phases, as only magnesium oxide and magnesium orthovanadate were detected. Regarding the textural properties (Table 3), the catalytic testing induced an increase in the average crystallite sizes, as calculated from the XRD using the Scherrer equation. In the same trend as that observed for the undoped catalysts [32], the increase in the crystallite size goes parallel to the catalytic activity, which again suggests that this change is probably due to the redox cycle and the accompanying order/disorder reconstruction. Based on the EDX results (Table 3), it may be said that there was no observable change in vanadium concentration at the surface of BaVMgO catalyst, as well as for VMgO. For the BVMgO catalyst, however, a slight change (a decrease) in the vanadium surface concentration may have been induced by the catalytic testing; however, worth mentioning here is that the ICP-OES test on the used catalysts did not show elemental change.
Conclusions
Investigation of the phase transformations during the catalysts calcination indicated that in the boron-doped catalyst the MgO started to form at lower temperature, namely 200 °C. Also, for both doped catalysts, MgO was the dominant crystalline phase at 350 °C; for the undoped catalyst, MgO became the main crystalline phase at 400 °C and above. The magnesium orthovanadate phase existed in the doped catalysts; however, it exhibited crystal planes different from those shown by the undoped catalyst. Some differences between the BaVMgO and BVMgO were also observed, e.g. the former is less porous, while the latter is more hydrated. Moreover, no changes in the crystalline phases were detected in the used catalysts; however, some changes that related to the textural properties were observed. The catalytic testing showed that the doped catalysts maintained the main features (merits) of the undoped VMgO catalysts with regard to the low propensity to form oxygenates and cracking products under all n-octane/O2 molar ratios. At all ratios, the BVMgO showed the lowest activity and ODH selectivity. Enriching the reactant mixture with oxygen (strong oxidative environment) increased the conversion, but at the same time enhanced the formation of the combustion products (carbon oxides). In this context, selectivity to 1-octene increased as the ratio of n-octane to oxygen increased; the opposite was true for styrene (except for the n-octane to oxygen ratio of 0.1). The highest selectivity to styrene was observed over the BaVMgO catalyst at n-octane/O2 molar ratios of 0.4, 0.8 and 1.2. In fact, the dehydrocyclization ability in general was greater over this catalyst (BaVMgO) at these ratios. Generally, the barium-doped catalyst (BaVMgO) showed a better performance than the boron-doped catalyst (BVMgO) with regard to both the catalytic activity and the ODH selectivity.
References
Hornaday GF, Ferrell FM, Mills GA (1961) Manufacture of mono- and diolefins from paraffins by catalytic dehydrogenation. In: McKetta JJ Jr (ed) Advances in petroleum chemistry and refining, vol 4. Interscience, Pans
Bhasin MM, McCain JH, Vora BV, Imai T, Pujado PR (2001) Dehydrogenation and oxydehydrogenation of paraffins to olefins. Appl Catal A Gen 221:397–419
Hodnett BK (2000) Heterogeneous catalysis oxidation: fundamental and technological aspects of the selective and total oxidation of organic compounds. Wiley, New York, pp 66–101
Denayer JF, Baron GV, Vanbutsele G, Jacobs PA, Martens JA (2000) Evidence for alkylcarbenium ion reaction intermediates from intrinsic reaction kinetics of C6–C9 n-alkane hydroisomerization and hydrocracking on Pt/H-Y and Pt/USY zeolites. J Catal 190:469–473
Akhmedov VM, Al-Khowaiter SH, Al-Refai JK (2003) Hydroconversion of C5–C8 alkanes over Zr-containing supported catalysts prepared by metal vapour method. Appl Catal A Gen 252:353–361
Jongpatiwut S, Sackamduang P, Rirksomboon T, Osuwan S, Resasco DE (2003) n-Octane aromatization on a Pt/KL catalyst prepared by vapour-phase impregnation. J Catal 218:1–11
Kuznetsov PN (2003) Study of n-octane hydrocracking and hydroisomerization over Pt/HY zeolites using the reactors of different configurations. J Catal 218:12–23
Grau JM, Yori JC, Vera CR, Lovey FC, Condo AM, Parera JM (2004) Crystal phase dependent metal-supported interactions in Pt/SO42–ZrO2 catalysts for hydroconversion of n-alkanes. Appl Catal A Gen 265:141–152
de Lucas A, Valverde JL, Sanchez P, Dorado F, Ramos MJ (2005) Hydroisomerization of n-octane over platinum catalysts with or without binder. Appl Catal A Gen 282:15–24
de Lucas A, Ramos MJ, Dorado F, Sanchez P, Valverde JL (2005) Influence of the Si/Al ratio in the hydroisomerization of n-octane over platinum and palladium beta zeolite-based catalysts with or without binder. Appl Catal A Gen 289:205–213
de Lucas A, Sanchez P, Dorado F, Ramos MJ, Valverde JL (2005) Effect of the metal loading in the hydroisomerization of n-octane over beta agglomerated zeolite based catalysts. Appl Catal A Gen 294:215–225
Altwasser S, Welker C, Traa Y, Weitkamp J (2005) Catalytic cracking of n-octane on small-pore zeolites. Microporous Mesoporous Mater 83:345–356
Szechenyi A, Solymosi F (2006) n-Octane aromatization on Mo2C-containing catalysts. Appl Catal A Gen 306:149–158
Yanhui W, Diyong W (2001) The experimental research for production of hydrogen from n-octane through partially oxidizing and steam reforming method. Int J Hydrogen Energy 26:795–800
Zhang J, Wang Y, Ma R, Wu D (2003) Characterization of alumina-supported Ni and Ni–Pd catalysts for partial oxidation and steam reforming of hydrocarbons. Appl Catal A Gen 243:251–259
Subramanian R, Panuccio GJ, Krummenacher JJ, Lee IC, Schmidt LD (2004) Catalytic partial oxidation of higher hydrocarbons: reactivities and selectivities of mixtures. Chem Eng Sci 59:5501–5507
Qi A, Wang S, Fu G, Wu D (2005) Autothermal reforming of n-octane on Ru-based catalysts. Appl Catal A Gen 293:71–82
Williams KA, Schmidt LD (2006) Catalytic autoignition of higher alkane partial oxidation on Rh-coated foams. Appl Catal A Gen 299:30–45
Panuccio GJ, Schmidt LD (2006) Increasing olefins by H2 and CH4 addition to the catalytic partial oxidation of n-octane. Appl Catal A Gen 313:63–73
Panuccio GJ, Schmidt LD (2007) Species and temperature profiles in a differential sphere bed reactor for the catalytic partial oxidation of n-octane. Appl Catal A Gen 332:171–182
Mamedov EA, Cortes Corberan V (1995) Oxidative dehydrogenation of lower alkanes on vanadium oxide-based catalysts. The present state of the art and outlooks. Appl Catal A Gen 127:1–40
Burch R, Swarnakar R (1991) Oxidative dehydrogenation of ethane on vanadium–molybdenum oxide and vanadium-niobium-molybdenum oxide catalysts. Appl Catal 70:129–148
Andersen PJ, Kung HH (1993) The effect of oxygen binding energy on the selective oxidation of butane over V/γ-Al2O3. Stud Surf Sci Catal 75:205–217
Kung HH, Michalakos PM (1993) Catalytic selective oxidation, ACS symposium series. Hattingh L, Washington DC, pp 389–408
Chaar MA, Patel D, Kung MC, Kung HH (1987) Selective oxidative dehydrogenation of butane over V-Mg-O catalysts. J Catal 105:483–498
Chaar M, Patel D, Kung HH (1988) Selective oxidative dehydrogenation of propane over V-Mg-O catalysts. J Catal 109:463–467
Blasco T, Lopez Nieto JM, Dejoz A, Vazquez MI (1995) Influence of the acid-base character of supported vanadium catalysts on their catalytic properties for the oxidative dehydrogenation of n-butane. J Catal 157:271–282
Kung HH, Kung MC (1997) Oxidative dehydrogenation of alkanes over vanadium–magnesium-oxides. Appl Catal A Gen 157:105–116
Blasco T, Lopez Nieto JM (1997) Oxidative dehydrogenation of short chain alkanes on supported vanadium oxide catalysts. Appl Catal A Gen 157:117–142
Dejoz A, Lopez Nieto JM, Marquez F, Vazquez MI (1999) The role of molybdenum in Mo-doped V–Mg-O catalysts during the oxidative dehydrogenation of n-butane. Appl Catal A Gen 180:83–94
Elkhalifa EA, Friedrich HB (2010) Oxidative dehydrogenation of n-octane using vanadium–magnesium oxide catalysts with different vanadium loadings. Appl Catal A Gen 373:122–131
Elkhalifa EA, Friedrich HB (2011) On the effect of hydrocarbon/oxygen ratios during the dehydrogenation of n-octane over a VMgO catalyst. Catal Lett 141:554–564
Elkhalifa EA, Friedrich HB (2014) Oxidative dehydrogenation and aromatization of n-octane over VMgO catalysts obtained by using different MgO precursors and different precursor treatments. J Mol Catal A: Chem 392:22–30
Thomas JM, Thomas WJ (1997) Principles and practice of heterogeneous catalysis. VCH, Weinheim
Bowker M (1998) The basis and applications of heterogeneous catalysis. Oxford University Press, Oxford, UK
Patel D, Andersen PJ, Kung HH (1990) Oxidative dehydrogenation of butane over orthovanadates. J Catal 125:132–142
Valenzuela RX, Mamedov EA, Cortes Corberan V (1995) Effect of different additives on the performance of V-Mg-O catalysts in the oxidative dehydrogenation of propane. React Kinet Catal Lett 55:213–220
Stern DL, Michaels JN, DeCaul L, Grasselli RK (1997) Oxydehydrogenation of n-butane over promoted Mg-V-oxide based catalysts. Appl Catal A Gen 153:21–30
Sugiyama S, Hashimoto T, Morishita Y, Shigemoto N, Hayashi H (2004) Effects of calcium cations incorporated into magnesium vanadates on the redox behaviours and the catalytic activities for the oxidative dehydrogenation of propane. Appl Catal A Gen 270:253–260
Sugiyama S, Hashimoto T, Tanabe Y, Shigemoto N, Hayashi H (2005) Effects of the enhancement of the abstraction of lattice oxygen from magnesium vanadates incorporated with copper(II) cations on the oxidative dehydrogenation of propane. J Mol Catal A: Chem 227:255–261
Klisinska A, Loridant S, Grzybowska B, Stoch J, Gressel I (2006) Effect of additives on properties of V2O5/SiO2 and V2O5/MgO catalysts II. Structure and physicochemical properties of the catalysts and their correlations with oxidative dehydrogenation of propane and ethane. Appl Catal A Gen 309:17–27
Jin M, Lu P, Yu GX, Cheng ZM, Chen LF, Wang JA (2013) Effect of additives doping on catalytic properties of Mg3(VO4)2 catalysts in oxidative dehydrogenation of cyclohexane. Catal Today 212:142–148
Elkhalifa EA, Friedrich HB (2015) Effects of boron and barium dopants on VMgO catalysts employed in the oxidative dehydrogenation of n-octane. Kinet Catal 56:212–221
Elkhalifa EA, Friedrich HB (2014) Magnesium oxide as a catalyst for the dehydrogenation of n-octane. Arab J Chem. doi:10.1016/j.arabjc.2014.10.002
Dasireddy VDBC, Singh S, Friedrich HB (2013) Activation of n-octane using vanadium oxide supported on alkaline earth hydroxyapatites. Appl Catal A Gen 456:105–117
Corma A, Lopez Nieto JM, Paredes N (1993) Preparation of V-Mg-O catalysts: nature of active species precursors. Appl Catal A Gen 104:161–174
Corma A, Lopez Nieto JM, Paredes N (1993) Influence of the preparation methods of V-Mg-O catalysts on their catalytic properties for the oxidative dehydrogenation of propane. J Catal 144:425–438
Burch R, Crabb EM (1993) Homogeneous and heterogeneous contributions to the oxidative dehydrogenation of propane on oxide catalysts. Appl Catal A Gen 100:111–130
Oganowski W, Hanuza J, Kepinski L, Mista W, Maczka M, Wyrostek A, Bukowski Z (1998) New intermediate phases in formation at V–Mg–O catalyst. J Mol Catal A: Chem 136:91–104
Hayakawa S, Yoko T, Sakka S (1993) 51V NMR studies of crystalline divalent metal vanadates and divanadates. Bull Chem Soc Jpn 66:3393–3400
Blasco T, Lopez Nieto JM (1996) Nuclear magnetic resonance studies on supported vanadium oxide catalysts. Colloids Surf A Physicochem Eng Asp 115:187–193
Holgado MJ, San Roman S, Malet P, Rives V (2005) Effect of the preparation method on the physicochemical properties of mixed magnesium–vanadium oxides. Mat Chem Physic 89:49–55
Michalakos PM, Kung MC, Jahan I, Kung HH (1993) Selectivity patterns in alkane oxidation over Mg3(VO4)2–MgO, Mg2V2O7, and (VO)2P2O7. J Catal 140:226–242
Acknowledgements
The authors acknowledge the financial support from the National Research Foundation (NRF) and the Technology and Human Resources for Industry Programme (THRIP), (South Africa). Thanks also go to the University of KwaZulu-Natal (South Africa) and University of Khartoum (Sudan).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Elkhalifa, E.A., Friedrich, H.B. Dehydrocyclization of n-octane over boron- and barium-doped V-Mg-O catalysts: influence of n-octane/oxygen ratios. Appl Petrochem Res 7, 23–32 (2017). https://doi.org/10.1007/s13203-016-0174-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-016-0174-x