
Abstract
Gas hydrate inhibition is very key and has become more sensitive as oil and gas exploration goes into deeper terrains especially deep offshore as a result of technological advancement. Use of chemicals has been the most efficient and cost effective in these areas. These chemicals add to the cost of doing oil and gas business and also cause harm to the environment; hence, research has been going on for more eco-friendly and cost-efficient inhibitors. This study takes a look at a locally sourced surfactant as one of such inhibitors. Varying weight percentages of the LSS were screened in a locally fabricated laboratory mini flow loop of 39.4 m with an internal diameter of 0.5 inch mounted on an external frame work. The various pressure plots (pressure vs. time, change in pressure vs. time, initial and final pressures vs. time) show that the LSS used in very small percentages performed better than the synthetic inhibitor methanol (MeOH) used in higher weight percentage than the LSS. The final pressures for MeOH for 1–5 wt% were 104, 111, 123, 120 and 123 psi while those of the LSS were 115, 128, 125, 127 and 131 psi, respectively, for 0.01–0.05 wt%, respectively. This means that the system with LSS had more stable pressure values than those of MeOH. Similarly, the change in pressure at the end of 120 min for MeOH was 46, 39, 27, 30 and 27 psi against 35, 22, 25, 23 and 19 psi for LSS. This was an indication that more gas was used up in the system with MeOH than in the system with LSS. The mitigation capacity of the LSS in percentage was calculated to be 69.30, 80.71, 78.07, 79.82 and 83.3% for 0.01–0.05 wt% while MeOH had values of 59.65, 65.79, 76.32, 73.68 and 76.32% for 1–5 wt%, respectively. This showed that the LSS inhibited hydrates better than MeOH in all the weight percentages considered. There is need to harness and develop the LSS for gas hydrate mitigation because it performed better than MeOH which is a known toxicant to man, terrestrial and aquatic habitat.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
When gas molecules referred to as guest molecules and water called the host molecule balance out via hydrogen bonding, gas hydrate is said to form (Koh 2002). Depending on the stability of gust molecules, temperature and pressure conditions (Fleischer and Janda 2013), different gas hydrate structures, namely SI, SII and SH, can be formed. Of all these structures, the prominent structures encountered in oil and gas operations are SI and SII. When water and gas molecules are present under high pressure and low temperatures, hydrate may likely form. Its formation undergoes processes such as hydrate nucleation, growth and agglomeration (Metaxas et al. 2019). Hydrate agglomeration could block subsea flowlines and pipelines which can cause safety, economic and environmental threat (Zerpa et al. 2011). Although gas hydrates can be challenging for oil and gas operators, which has led to heavy investments to prevent them (Zerpa et al. 2011), they can hold more hydrocarbon than can be consumed by man (Makogon 2010).
Plugging takes place when pressure drop is increased due to decrease in flow caused by blockage of pipelines by the hydrate particles. When particles that are entrained in the flowing fluid, form stable structures that restrict further flow or when particles are deposited on the wall of pipelines, grow and cause constriction of the internal diameter of the pipeline or flowline till there is complete blockage, plugging is said to occur as a result of jamming. Although, when it comes to failure of a pipeline or flowline, a combination of factors are responsible (Strivastav et al., 2017 and Allen et al. 2001). In order to prevent this, gas hydrate plugs are either prevented or commercially managed through the use of chemicals which are categorized as thermodynamic hydrate inhibitors (THIs) and low-dosage hydrate inhibitors (LDHIs).
-
(1)
Thermodynamic hydrate inhibitors (THIs) This class of inhibitors acts by moving the hydrate stability of thermodynamic boundary conditions to lower temperatures and higher pressures (Hu et al. 2018; Chaudhary et al., 2018). They do not allow hydrate conditions to occur. Thermodynamic inhibition involves keeping the pipeline thermodynamic conditions outside of the hydrate stability region. Tactical insulation or heating of the pipe can keep the temperature too high for hydrates to form. During planned shut ins and restarts, the well heads and jumpers can be treated before the insulated flow lines reaches the hydrate stability regions. However, the problems associated with their usage include; high dosing requirements, especially at high water cuts which can make them an expensive, and environmentally unfriendly choice. THIs are effective at high weight percentages (say 50%) or fractions in line with water content (Kelland 2006).
Examples of thermodynamic hydrate inhibitors are methanol (MeOH), ethylene glycols (MEG) and triethylene glycol (TEG).
-
(2)
Low dosage hydrate inhibitors (LDHIs) They are so named because they are effective in low dosages of about 0.5 to 3wt of water content although some LDHIs mode of action has not been fully understood and show low degradability (Kelland 2006). LDHIs are classified into kinetic hydrate inhibitors (KHIs) and anti-agglomerants (AAs). Kelland (2006) gave an insight into LDHIs and also early simulations by Carver et al. (1995).
-
(a)
Kinetic hydrate inhibitors (KHIs) These are water-soluble polymers which delay the onset and agglomeration of hydrates. They reduce the rate of gas hydrate formation significantly and allow for production/transportation of fluids (gas and/or oil) during a certain time frame or interval (Kelland, 2006). KHIs tend to lose their effectiveness at higher subcooling (over 120 °C) (Gao 2009).
-
(b)
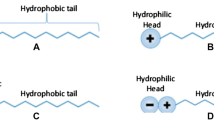
Anti-agglomerants (AAs) are surface active agents (Chua and Kelland 2013) that prevent agglomeration and if properly administered, produces a slurry of hydrates which is transportable. Anti-agglomerants (AAs) are mostly utilized for hydrate management where thermodynamic inhibition is no longer feasible (Anklam et al. 2008; Gao 2009). The use of AAs allows hydrates to form in the pipeline, but the chemical additives reduce the cohesion or clumping of the particles to one another and the adhesion of particles to the walls, such that hydrates flow as a slurry until the pipeline reaches higher temperature, lower pressure conditions, and the hydrates dissociate back into their constituent components. AAs have complex chemical compositions, but many contain surfactant-type chemical groups which are molecules with both hydrophilic and oleophilic functionalities, which allow for interaction with both the water and the oil in a pipeline (Chua and Kelland 2013; Zanota et al. 2005). An increase in salt concentrations increases their performance (Aman et al. 2015). AAs are known to be effective at high subcoolings and may possibly not slow the rate of growth but can prevent agglomeration to form large plugs. This action can prevent blockages in pipelines. Examples such as quaternary ammonium salts (QAs) are well-known AAs (Zanota et al. 2005). Most AAs are effective only at lower water cuts (< 30%).
LDHIs do not prevent the formation of hydrates completely because they do not cause shifts in hydrate equilibrium curves such that once hydrates form they cannot be removed by LDHIs (because the conditions of operation cannot be changed); therefore, a well using LDHIs is still at risk of forming hydrates and will still require THIs. The advantages of LDHIs is that they reduce OPEX and CAPEX. The disadvantage is that they their application field on large scale cause important problems due to the fact that they are poor biodegradable inhibitors (Jensen et al. 2010).
Typically, LDHIs are affected by oil composition, density, viscosity, emulsion forming tendency, amount of gas present in oil (gas–oil ratio, GOR) (Aman et al. 2017). The performance of LDHIs can be drastically affected by little changes in their molecular structure. It is common practice for systematic experiments to be carried which is aimed at identifying the optimum dosage and composition of LDHIs.
Another important way of managing gas hydrate is by preventing the fluid produced from entering the hydrate stability region during flow (through front end engineering design). This can be achieved via multiphase system modeling (Charlton et al. 2018). This is done by finding out the pipeline size, subsea system pumping requirement and the insulation thickness. This is imperative due to the changing nature of produced fluid as the well gets older, making it possible for the fluid to enter hydrate stability zone, causing blockages during flow as the field matures (Mehta et al. 2002). The internal surfaces of pipelines/ flowlines make for many nucleation sites and coldest point. Field trials and several experiments had suggested that deposition of hydrates often occurred on the wall/surfaces of the pipelines (Nicholas et al. 2009; Aman et al. 2014). Changes in rheology such as sloughing and jamming were indicators of hydrate plugging. To effectively prevent plug formation in offshore operations, knowledge of hydrate formation and flow behavior are key. Great efforts have been put in by researchers to get the knowledge of these key factors (Aman et al. 2010, 2011).
Farhadian et al (2020) carried out a study on a novel strategy which had potential. This was based on waterborne polymers (which were synthesized in the absence of a catalyst and according to Farhadian et al. 2019) for hydrate and corrosion risk management and incompatibility issues. They found out that the waterborne polymers (waterborne polyuria/ureathenes, WPUUs) performed well in delaying hydrate induction by 17 times and reduced growth rate by 7.7 times when compared to water. They reported that WPUUs are promising kinetic/anti-agglomerant methane hydrate and corrosion inhibitors, which will chart a new course in hydrate and corrosion risk management. Kumar et al., (2019) in their study used a simple apparatus and short experimental times to assess morphology, phase equilibria and apparent kinetics of methane hydrate in the presence of thermodynamic hydrate inhibitor ethane-1,2-diol (MEG) and sodium chloride (NaCl) and kinetic hydrate inhibitor polyvinyl-pyrrolidone (PVP). They found out that both NaCl and MEG acted as kinetic inhibitors at the studied concentrations and concluded that in all, the obtained morphology, kinetics and phase equilibria results were reproducible and consistent and that assessment of multiple subcooling in a single experiment was possible from gradient experiments. They opined that the 3-in-1 reactor technique should be used especially in assessment of potential inhibitors at critical turnaround times or when samples are limited in quantities.
In their study, Zhang et al. (2017) used a newly built up loop, pilot scale experiments to study typical hydrate plug phenomena exploring the reasons behind them. They used a tetrahydrofuran (THF) hydrate slurry@ 50 and 100 vol% with a typical anti-agglomerant KL-1. Based on morphologies, temperature variation as well as liquid loading and an anti-agglomerant on morphology and temperature, they found out that hydrate plugs were induced by hydrate deposition around the pipeline walls but not from the bed at the bottom of the pipeline. Also that AA compressed hydrate deposition and plug by dispersing hydrate particle agglomeration. They concluded that adhesive forces among hydrate particles can be reduced by AA and that hydrate agglomeration was loosened at lower subcooling but failed at higher subcooling. A study on the effect of soy lecithin as an anti-agglomerant using cetyltrimethylammoniumbromide (CTAB) on tetrahydrofuran (THF) hydrate was studied by Saikia and Mahto (2018) using an experimental setup built in house. It was found out that soy lecithin @ 0.1 wt% and 1°C in an experimental solution increased agglomeration time by 1440 min while @ 0.01 wt% it increased it by 1000 min. They concluded that soy lecithin a bio surfactant, can be effectively used as an anti-agglomerant additive to prevent hydrate plugging in oil/gas pipelines and drilling fluid flowlines.
An anti-agglomerant showing high effectiveness at low dosage in methane/natural gas hydrate over an entire water cut range was reported by Zhao et al. (2016). They used an improved formulation in various systems at constant high pressure (100 bar of natural gas) and high cooling rate (−8 °C/h) over water cut ranges of 30–80% in a closed rocking cell system with condensate liquid and crude oil as hydrocarbon liquid phase. They demonstrated the effectiveness of improved AA formulation in an extensive set of measurements. They found that increasing salinity can decrease the dosage of base chemical in formulation significantly.
Muhammad et al. (2016) presented a work on the various water-soluble polymers used for gas hydrate inhibition, including conventional and novel polymeric inhibitors along with their limitations. The work covered relevant properties of vinyl lactam, amide, dendremeric fluorinated and natural biodegradable polymers. A complete review of the modus operandi used to assess the performance of the polymeric inhibitor was given. It addressed recent developments, current and future challenges and field applications of a range of polymeric kinetic hydrate inhibitor.
Jiyeon et al. (2016) investigated the effect of 2-diamine piperazine (PZ) and hydrazine (HZ) on the formation of methane hydrate (CH4) with primary focus on thermodynamic, spectroscopic and computational analysis for their potential application in natural gas hydrate production and transportation. The methane hydrate equilibrium lines were shifted to low temperatures and high-pressure zones based on the concentrations of 2-diamines used. A slower hydrate growth and longer induction time as well as significant thermodynamic hydrate inhibition effect were shown by HZ than PZ.
The use of chemicals as a means of gas hydrate inhibition has been ongoing, and these are detrimental to the environment and humans. Local materials have been reported in enhanced oil recovery (Emuchay et al. 2013; Ojo et al. 2013) and also in the pharmaceutical industries use has been made of local extracts (Ezejiofor et al. (2013). Their use has been found to be effective in the area of flow assurance as local extracts have been used as hydrate inhibitors in lieu of the synthetic ones (Elechi et al. 2018, 2019, 2020 and Okon et al. 2018). This paper is a continuation of the use of such materials as gas hydrate inhibitors.
Materials and method
The materials used for this study are water, ice blocks (facilitated the attainment of hydrate formation temperature), compressed natural gas (CNG with mass of 14.5 g, 0.5 specific gravity), conventional hydrate inhibitor, methanol and locally sourced surfactant (LSS) for gas hydrate inhibition.
The LSS is of the plant family caricaceae. It was obtained from the botanical garden of University of Port Harcourt. The extracts were obtained by manual crushing of tender leaves, sieving with the aid of a cheese cloth and then collected in air tight containers and used immediately. Phytochemical screening showed the presence of some phytochemicals present in the LSS as shown in Table 1. They have characteristics that make them good inhibitors based on the polyphenolics and hydroxyl group they contain. It contains the –OH group just like methanol (MeOH). It contains oxygen bonded to hydrogen. It is pair of atom that is commonly found in organic compounds such as sugar (carboxylic acids) and alcohols. The anion [OH–] called hydroxide consists of a hydroxyl group. IUPAC (2015) rules say that the term hydroxyl refer to the radical OH only while the functional group-OH is called hydroxyl group. Its chemical formula is written as –OH where—refers to the carbon to which the hydroxyl group is bonded.
The extract is water soluble containing naturally occurring bioactive compounds like alkaloids, saponins, flavonoids and tannins. Flavonoids are polyphenolics that are anti-coagulants that prevent coagulation and act as powerful oxidants which protect against reactive oxygen. Tannins are a heterogenous group of high molecular weight polyphenolic compounds with capacity to form reversible and irreversible complexes. They are also found in fruits, legumes and grasses. Polyphenolics are a class of chemical compounds consisting of a hydroxyl group bonded directly to an aromatic hydrocarbon group. They are otherwise called poly hydroxyl phenols and are phytochemicals. They are a structural class of mainly natural products (chemical compounds or substance produced by a living organism that is found in nature (Quideau 2011). Condensed tannins are the most abundant poly phenols found in virtually all families of plants. Leaf tissues, the epidermis, bark layers, flowers and fruits have larger concentrations of poly phenols (Quideau et al. 2011).
Alkaloids are natural products that contain heterocyclic nitrogen atoms and are basic in character. They are naturally synthesized by a large number of organisms including animals, plants, bacteria and fungi. Saponins form a stable foam in aqueous solutions such as soap hence the name ‘saponin.’ Chemically, saponins as a group include compounds that are glycosylated steroids, hypernoids and steroid alkaloids. Saponins also prevent agglomeration and are surface acting agents with bubbles and foams acting as barriers that give good stability. Fourier transform infrared spectroscopy (FTIR) showed the presence of tannins, and alkaloids (with functional groups containing nitrogen, oxygen and carbon) with aromatic rings. The presence of flavonoids, tannins, saponins and alkaloids in plant extract contributes to its ability to inhibit hydrates just like conventional alcohols methanol and glycols.
Experimentals
The experimental setup used for this work is a 39.4-m-long, 0.5-inch I.D. locally fabricated laboratory mini flow loop housed in a PVC pipe (4 inches in diameter) used for the screening of the locally sourced and conventional inhibitors (Fig. 1).

Overview of experimental setup
The system is first flushed to remove debris before the actual experiments. For hydrate formation experiment, water is poured into the mixing vessel, at that point pump 3 is turned on and some quantity of water (about 435 ml) of water is let into the inner line via valve 4 (with pressure of about 25 psi), after which both pump and valve are closed. In order to build the pressure of the inner line to 150 psi, valve 1 and orifice are opened and then both are closed. Water enters into the cooling unit (in this case refrigerator) and is moved around the PVC by opening pump. Agitation and mixing of gas and water as well as circulation in the inner line is initiated by turning on pump 1. Pressures and temperatures readings are taken every 2 min for a total of 2 h. Pointers to formation of gas hydrates in this setup are: increase in loop temperature, a rapid decrease in system pressure, increase in pressure between inlet and outlet pressures (differential pressure), clogging of sample point valve and milky color of effluent). For hydrate mitigation experiments, water and inhibitor (depending on the concentration or weight percentage to be used) are poured into the mixing vessel and the experiment proceeds as earlier described.
Results and discussions
The performance of the local surfactant (LSS) was validated using pressure versus time and temperature versus time plots and superimposed with the pressure, temperature versus time plots for the uninhibited experiments. The performance is ascertained by the pressure value at the end of the experiment. A drastic temperature increase or pressure decline are pointers to hydrate formation for the uninhibited experiment while a regulated temperature or pressure profile is an indicator of the LSS’s ability to mitigate gas hydrate formation.
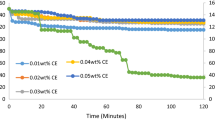
Pressure versus time for the various weight percentages of LSS and pressure versus time for uninhibited system is shown in Fig. 2. For 0.01 wt.% LSS, the pressure dropped from an initial pressure of 150 psi to 121 psi in 30 min, then to 118 psi in 60 min, 116 psi in 90 min and finally dropped to 115 psi at the end of 120 min. For 0.02 wt%, the temperature decreased to 135 psi in 30 min, 130 psi in 60 min, 129 psi in 90 min and finally to 128 psi at the end of the experiment. For 0.03 wt%, pressure dropped from 150 psi at the inception of the experiment to 133, 128, 126 and 125 psi in 30, 60, 90 and 120 min. For 0.04 wt%, the pressure decreased from 150 psi to 135, 131, 128 and 127 psi in the same time interval. Pressure decreases for 0.05 wt% in 30 min were 140 psi, then reduced to 131 in 60 min and was maintained till the end of 120 min.

Pressure versus time for 0.01–0.05 wt% locally sourced surfactant (LSS) and uninhibited experiment
For the uninhibited experiments, the pressure decreased from 150 psi to 113, 80, 40 and 36 psi for the same time frame. The various weight percentages of the LSS were able to regulate the pressure in the system, and the initial decreases were caused by the cold water flowing in the PVC pipe. Also, the gradual pressure decreases were occasioned by the formation of an impermeable layer between the fluids (gas and water) flowing in the inner line (Movareji et al. 2016). This was not the case for the uninhibited experiment where the pressure declined drastically from 150 to 36 psi at the end of 120 min. This drastic decrease was as a result of gas being used up to form hydrates due to large gas dissolution in water which is the initial gas hydrate formation phase. This was the resultant effect of release of latent heat due to exothermic nature of gas hydrate formation (Movareji et al. 2016).
Figure 3 is the temperature versus time for 0.01–0.05 wt% locally sourced surfactant and uninhibited experiments. The temperature decreases for 0.01 wt% was from 30 °C to 20.5 °C, 10 °C, in 30 and 60 min and then to 6.5 °C in 90 min and were maintained till the end of 120 min. 0.02 wt% had temperature decreasing from 30 to 14 °C, 7.5 °C, 7 °C and 6.5 °C in 30, 60, 90 and 120 min while for 0.03 wt%, the values were from 30 °C to 13, 9, 8 and 7.5 °C, respectively, for same time interval. For 0.04 wt% had temperature decreasing from 30 °C to 16.5, 11.5 and 7 °C in 30, 60,90 and 120 min The values reduced from 32 °C to 18, 9.5, 7.5 and 6.5 °C in 30, 60 90 and 120 min. In all the weight percentages, the temperature decrease was regulated and there was no drastic temperature increase which was noticed in the uninhibited experiment. After a steady decrease from 30 to 25 °C in 30 min, the temperature increased to 28, 29 and 29.5 °C in 60, 90 and 120 min. This increase led to large dissolution of methane gas in water which hastened agglomeration/ precipitation of hydrate as critical concentrations were reached (Zhang et al. 2017).

Temperature versus time for 0.01–0.05 wt% locally sourced surfactant and uninhibited experiment
In Fig. 3, the temperature increase was significant because from the plots, it is seen that for the inhibited experiment using 0.01–0.05 wt% of the surfactant, the temperature declined steadily and was well controlled. The sustained decrease in pressure was as a result of the inhibitors being able to regulate the temperature of the system which was observed in all the weight percentages but for the uninhibited, the temperature declined initially but after about 30 min, there was a spike which increased gradually and was sustained till the experiment ended. This sudden increase heralds the inception of gas hydrate agglomeration in the inner line owing to the exothermal nature (heat given off) of gas hydrate formation which may have been responsible for the temperature increase. As earlier stated in the discussion in Method section, one of the pointers to gas hydrate formation is a sudden spike in system’s temperature.
Gas hydrate mitigation comparison of LSS and MeOH
To assess the performance of the locally sourced surfactant (LSS) and the conventional inhibitor methanol (MeOH), various plots of pressure versus time, initial and final pressure versus time and change in pressure versus time were plotted. Furthermore, the mitigation capacity (MC) of the various weight percentages of both the local and conventional inhibitors was also calculated. It is worthy to note here that the LSS was used in low dosages of 0.01–0.05 wt% that is as low dosage hydrate inhibitor (LDHIs) while the conventional inhibitor was used as thermodynamic hydrate inhibitors (THIs) in high dosages of 1–5 wt%.
The conventional inhibitor, MeOH, was used in larger dosages of (1–5 wt%) while the LSS was used in smaller weight percentages (0.01–0.05 wt%) to see how they compare or contrast. The use of low dosages for the LSS was to ascertain the degree of their effectiveness or otherwise as a local surfactant for the study. Hence various weight percentages were compared.
The surfactant functions by adhering or adsorbing to the hydrate crystal structure via hydrogen bonding which forces the crystal to grow around and between the surfactant strands, thus preventing water from entering and stabilizing the hydrate cages. This adsorption occurs because of the interaction force between the surfactant molecules and the water molecule such that inhibition effect is attributed to the adsorption of the surfactants molecules through their functional groups on hydrate molecules. Water has very strong cohesive force which makes surface tension high, and as the surfactants adsorb (water molecules and surfactant molecules have low intermolecular forces hence decreases surface tension), they break these interactions.
The surfactant monomers disrupt the hydrogen bonding between water molecules at the surface, thus lowering the surface tension.
Pressure versus time plot
Figure 4 is the pressure versus time plot for 0.01, 0.02 and 0.05 wt% LSS and 1, 2 and 5 wt% MeOH. These plots are used as representatives of 0.03 and 0.4 wt% LSS and 3 and 4wt% MeOH. The pressure dropped from 150 psi to 121 in 30 min, 118 in 60 min and then increased to 116 psi in 90 min and finally to 115 psi at the end of 120 min for the LSS. For the MeOH inhibited system, the pressure decrease for the same time interval was from 150 psi to 114, 107,106 and 104 psi, respectively. The pressure in the LSS inhibited system was more regulated than that of the MeOH inhibited system. The pressure versus time plot for 0.02 wt% LSS and 2 wt% MeOH. Pressure decreases were 135, 130, 129 and 128 psi in 30, 60, 90 and 120 min for LSS while values for 2 wt% MeOH for same time interval were 120,117, 114 and 111 psi. The pressure versus time plots for 0.03, 0.04 and 0.05 wt% of LSS followed the same steady pressure decrease trend when compared to the 3, 4 and 5 wt% MeOH. In the weight percentages considered, the LSS inhibited system had better pressure control than the MeOH system. This shows that the LSS performed better than the conventional MeOH even when used in weight percentages that were 0.99, 0.98, 0.97, 0.96 and 0.95 less than those of MeOH. The uninhibited scenario had rapid pressure decrease from 150 psi to 113, 80, 40 and 36 psi in the same time interval. This was as a result of more of the gas dissolving in water such that the dissolving fluid could no longer take any more gas which led to the shortening of the time it takes for hydrate to form (Bishnoi and Natarajan 1996).

Pressure versus time plot for 0.01, 0.02 and 0.05 wt% LSS and 1,2 and 5 wt% MeOH and uninhibited experiment
Initial (Pi) and final (Pf) pressure plots
Another yard stick used in assessing the performance of the conventional and local inhibitor was the initial (Pi) and final (Pf) pressure. Basically, it is the final pressure because the initial pressure was the same for both the conventional and local inhibitors. The final pressure tells how much gas was used up or dissolved in the carrier fluid in order to form hydrates. The more the final pressure (Pf), the better the performance of the inhibitor.
The Pi and Pf versus time for 0.01–0.05 wt% LSS and 1–5 wt% MeOH and uninhibited experiment is shown in Fig. 5. At the end of the experiment (120 min), the pressure in the system using MeOH was 104 psi while that of the LSS system was 115 psi. For 2 wt% MeOH and 0.02 wt% LSS, the system with LSS had a Pf of 128 psi while that of MeOH was 111 psi. For 0.03 wt% LSS and 3 wt% MeOH, final pressure for the system using LSS was 125 psi while the system with MeOH had a value of 123 psi. For 0.04 wt% LSS and 4 wt% MeOH, the Pf for system using MeOH was 120 psi while that of the LSS was 127 psi. For 0.05 wt% LSS and 5 wt% MeOH, the values were 131 and 123 psi, respectively, at the end of the experiments. The LSS performed better than the conventional MeOH although both inhibitors inhibited gas hydrate formation better than in the uninhibited inhibited experiment where the final pressure at the end of the experiment was 36 psi meaning that almost all the gas in the system was used for gas hydrate formation. The cooling process contributed to the initial rapid pressure decrease due to large dissolution of gas in water (Movareji et al. 2016).

Pi and Pf versus time for 0.01–0.05 wt% surfactant and 1–5 wt% MeOH and uninhibited experiment
Change in pressure (∆P) plots
The change in pressure was also used to assess how the local and conventional inhibitors behaved in terms of suppression performance. The value of change in pressure tells how well the inhibitor was able to prevent gas hydrate formation. Figure 6 show the change in pressure versus time for 0.01, 0.02 and 0.05 wt% LSS and 1,2 and 5 wt% MeOH alongside the uninhibited experiment. The change in pressure for 0.01 wt% LSS at the end of 120 min was 35 psi while that of 1 wt% MeOH was 46 psi. Change in pressure for LSS was 11 psi less than that of MeOH. For 0.02 wt% LSS, the change in pressure was 22 psi while that of 2 wt% MeOH was 39 psi which is 17 psi more than that of LSS, while 0.05 wt% LSS and 5 wt% MeOH had ∆P values of 19 psi and 27 psi, respectively, at the end of 120 min. The other weight percentages (0.03/3, 0.04/4 wt%) of LSS and MeOH showed a similar trend as the those discussed above. Generally, both inhibitors had far lesser change in pressure values than the uninhibited experiment which had a change in pressure value of 114 psi which was an indication that almost all the gas was used up in gas hydrate formation (Jensen et al., 2005).

∆P versus time for 0.01, 0.02 and 0.05 wt% LSS and 1, 2 and 5 wt% MeOH and uninhibited experiment
It is worthy of note that even though the concentration of MeOH was higher than that of LSS in terms of weight percentage, it performed less than the LSS. This is very remarkable and speaks volume for the unconventional compound as gas hydrate inhibitor.
Mitigation capacity
The performance of the inhibitors was further ascertained by calculating the rate at which they mitigated gas hydrate formation.
Mitigation capacity, MC, is given as
Mitigation factor is given as
where.
ΔPinhibited is the difference between initial and final pressure of the inhibited experiment.
ΔPuninhibited is the difference between the initial and final pressures of the uninhibited experiment.
Pi is the initial pressure for both the inhibited and uninhibited systems.
Pinhibited is the final pressure for the inhibited systems.
Puninhibited is the final pressure for the uninhibited system.
The mitigation capacity (MC) versus weight % for the various weight percentages of LSS and MeOH is shown in Fig. 7. For 1/0.01 wt% MeOH and LSS, the MC values were 59.65 and 69.3%. At this weight percentage, over all the MC of LSS was 9.65% more than that of MeOH. The MC for 2 wt% MeOH was 65.79% while that of 0.02 wt% LSS was 80.70% and was able to mitigate gas hydrate formation 14.91% more than the mitigation ability of MeOH. The value of. 0.03 wt% LSS was 78.07% while that of 3 wt% MeOH was 76.32%, a value that was less than that of LSS by 1.75%. For 0.04/4 wt%, the values for LSS and MeOH were, respectively, 79.82 and 73.68%. LSS mitigated gas hydrates better than MeOH by 9.65%, while for 5 wt% MeOH, the MC value was 76.32% while 0.05 wt% LSS had a MC value of 83.3%, which is greater than the value of MeOH by 6.98%.

Mitigation capacity, MC, versus weight% for 0.01–0.05 wt% LSS and 1–5 wt% MeOH
From the analysis of the MC of the inhibitor, it is seen that the mitigation capacity for MeOH decreased after 3 wt% (which is considered the optimum weight % for MeOH). As concentration increased for MeOH, the mitigation capacity decreased at 4 wt% and then increased to the same weight percentage as 3 wt% at the end of the experiment as shown in Fig. 7. This is a case of over inhibition leading to decrease in effectiveness. For LSS, it was observed that after 0.02 wt%, the MC value decreased by 2.63% and 0.88%, respectively, after which it increased by 2.63% at 0.05 wt%. The decrease in MC could also be as a result of over inhibition, but it picked up at 0.05 wt%. 0.02 wt% is considered as optimum for LSS because above this concentration the MC dropped. Even though the MC at 0.05 wt% increased, the value was 2.63% more than the MC value at 0.02 wt% (this can be neglected). It is seen that LSS performed much better than the conventional MeOH in all the weight percentages considered.
The optimum weight fraction for LSS is considered to be 0.02 wt% because at this weight percentage, it had a better pressure drop control, had least change in pressure values and highest final pressure value when compared to the other weight percentages except for 0.05 wt%. The values gotten at 0.05 wt% are negligible such that inhibiting above that weight percentage (0.02wt%) is not economical. The MC for 0.02 wt% LSS is higher than that of all the wt.% (1–5 wt%) of MeOH.
Conclusions
The pressures in the system with LSS at the end of 120 min for the various weight percentages were more regulated with values of 115, 128, 125, 127 and 131 psi while those of MeOH inhibited system were 104, 111, 123, 120 and 123 psi, respectively, at the end of 120 min. For the uninhibited system, the pressure values were 113, 80, 40 and 36 psi in 30, 60, 90 and 120 min.
The change in pressure in the systems showed the LSS inhibited system to have values of 35, 22, 25, 23 and 19 psi for 0.01–0,05 wt%, respectively, at the end of 120 min while values for MeOH were 46, 39, 27, 30 and 27 psi, respectively, for the same time frame. The MC for the various weight percentages of the LSS was 69.3, 80.7, 78.07, 79.82 and 83.3%, respectively, while values for MeOH were 59.65, 65.79, 76.32, 73.68 and 76.32% for each weight percentage considered.
From all these, it is clear that the LSS performed far better than the conventional MeOH in all weight percentages considered even though they were used in much higher dosages than the LSS. The weight percentage that is to be considered optimal for inhibition for both inhibitors is 0.02/3 wt% for LSS and MeOH, respectively. At this weight, the mitigation capacity was highest after which it began to decrease and then increased at 5 wt% (for MeOH). For LSS after this weight percentage, the MC decreased and then increased to a value of 83.3% which was gotten at the end for 0.05 wt%. The difference between the MC at 0.02 wt% and 0.05 wt% is 2.63%. This is not so significant and inhibiting above 0.02 wt% for LSS would not be economical. The LSS should be considered and harnessed for gas hydrate inhibition given its performance moreover it is more eco-friendly than methanol which is a known toxicant.
Availability of data
The data used in this manuscript are not available to the public because of patent issues.
References
Allen E, Henshaw J, Smith P (2001) A review of particle agglomeration. Dorchester (UK): AEA Technology plc.49.http//hdl.handle.net/10068/615769
Aman ZM, Dieker LE, Aspenes G, Sum AK, Slaon ED, Koh CA (2010) Influence of model oil with surfactants and amphiphilic polymers on cyclopentane hydrate adhesion forces. Energy fuels 24:5441–5445
Aman ZM, Brown EP, Sloan ED, Sum AK, Koh CA (2011) Interfacial mechanisms governing cyclopentane clathrate hydrate adhesion/cohesion. Phys Chem Chem Phys 13:19796–19806
Aman ZM, Sloan ED, Sum AK, Koh CA (2014) Adhesion force interaction between cyclopentane hydrate and physically and chemically modified surfaces. Phys Chem Chem Phys 16(45):25121–25128
Aman ZM, Haber A, Ling NNA, Thornton A, Johns ML, May EF (2015) Effect of brine salinity on the stability of hydrate-in-oil dispersions and water-in-oil emulsions. Energy Fuel 29:7948–7955
Aman ZM, Syddall WGT, Haber A, Qin Y, Graham B, May EF, Johns ML, Pickering PF (2017) Characterization of crude oils that naturally resist hydrate plug formation. Energy Fuel 31:5806–5816
Anklam M, York J, Helmerich L (2008) Effects of anti-agglomerants on the interactions between hydrate particles. AIChE J 54(2):565–574
Carver TJ, Drew MGB, Rodger PM (1995) Inhibition of crystal-growth in methane hydrate. J Chem Soc Faraday T 91:3449–3460
Charlton TB, Di Lorenzo M, Zerpa LE, Koh CA, Johns ML, May EF, Aman ZM (2018) Simulating hydrate growth and transport behavior in gas-dominant flow. Energy Fuel 32:1012–1023
Chaudhari P, Zerpa LE, Sum AK (2018) A correlation to quantify hydrate plugging risk in oil and gas production pipelines based on hydrate transportability parameters. J Nat Gas Sci Eng 58:152–161
Chua PC, Kelland MA (2013) Study of the gas hydrate anti-agglomerant performance of a series of n-Alkyl-tri(n-butyl) ammonium bromides. Energy Fuel 27:1285–1292
Elechi VU, Ikiensikimama SS, Ajienka JA, Akaranta O, Onyekonwu MO, Odutola TO, Okon OE (2018) Gas hydrate inhibition in a simulated offshore environment using local inhibitor. SPE Paper 193439 presented at Nigeria annual international conference and exhibition, held in Lagos Nigeria
Elechi VU, Ikiensikimama SS, Ajienka JA, Akaranta O, Onyekonwu MO, Okon OE (2019) Comparative analysis of gas hydrate inhibitors: local and conventional, SPE 197429 presented at the Abu Dhabi international petroleum exhibition & conference held in Abu Dhabi, UAE, 11–14 November 20119
Elechi VU, Ikiensikimama SS, Ajienka JA, Akaranta O, Onyekonwu MO, Okon OE (2020) The use of a bench-scale high pressure apparatus to evaluate the effectiveness of a locally sourced material as gas hydrate. Sci Afr. https://doi.org/10.1016/j.sciaf.2020.e00300
Emuchay D, Onyekonwu MO, Ogolo NA, Ubani CE (2013) Breaking of emulsions using locally formulated demulsifiers. SPE Paper 167528 presented at the Nigeria annual international conference and exhibition held in Lagos, Nigeria
Ezejiofor AN (2013) Effect of Aqueous Leaves of costus afer Ker gawl on the Liver and Kidney of Male Albino Wistar Rat. Anc Sci Life J 33(1):4–9
Farhadian A, Varfolomeev MA, Abdelhay Z, Emelianov D, Delaunay A, Dalmazone D (2019) Accelerated methane hydrate formation by ethylene diamine tetraacetamide as an efficient promoter of methane storage with foam formation. Ind Eng Chem Res 58(19):7752–7760
Farhadian A, Varfolomeev MA, Kudbanov A, Rezaeisadat M, Nurgaliev DK (2020) Waterborne polymers as kinetic/anti-agglomerant methane hydrate and corrosion inhibitors: a new and promising strategy for flow assurance. J Nat Gas Sci Eng. https://doi.org/10.1016/j.jngse.2020.103235
Fleischer EB, Janda KC (2013) Prediction of clathrate structure type and guest position by molecular mechanics. J Phys Chem A 117:4001–4010
Gao S (2009) Hydrate risk management at high water-cuts with anti-agglomerant hydrate inhibitors. Energy Fuels 23:2118–2121
Hu Y, Sa JH, Lee BR, Sum AK (2018) Universal correlation for gas hydrates suppression temperature of inhibited systems: III. Salts and Organic Inhibitors. AIChE J 64:4097–4109
IUPAC (2015) World Chemistry Congress. https://doi.org/10.1515/ci-2016-0128
Jensen L, Ranlov H, Thonsen K, Von Holms N (2010) Inhibition of methane hydrate formation by ice structuring proteins. Ind Eng Chem Res 49:1486–1492
Jiyeon L, Eunae K, Yongwon S (2016) Dual inhibition effects of deamines on the formation of methane gas hydrate and their significance for natural gas production and transportation. Energy Convers Manage 124:578–586. https://doi.org/10.1016/j.enconman.2016.07-054
Kelland MA (2006) History of the development of low dosage hydrate inhibitors. Energy Fuel 20:825–847
Koh CA (2002) Towards a fundamental understanding of natural gas hydrates. Chem Soc Rev 31:157–167
Kumar A, Veluswamy HP, Linga P, Kumar R (2019) Molecular level investigations and stability analysis of mixed methane-tetrahydrofuran hydrates: implications to energy storage. Fuel 236:1505–1511
Makogon YF (2010) Natural gas hydrates - a promising source of energy. J Nat Gas Sci Eng 2:49–59
Mehta AP, Hebert PB, Cadena ER, Weatherman JP (2002) Fulfilling the promise of low dosage hydrate inhibitors: journey from academic curiosity to successful field implementation. OTC 14057, Offshore Technology Conference; Houston, TX, USA, 6–9 May 2002
Metaxas PJ, Lim VWS, Booth C, Zhen J, Stanwix PL, Johns ML, Aman ZM, Haandrikman G, Crosby D, May EF (2019) Gas hydrate formation probability distributions: induction times, rates of nucleation and growth. Fuel 252:448–457
Movareji MK, Charffarkhah A, Sadeghi A (2016) Effect of three representative surfactants on methane hydrate formation rate and induction time. Egypt J Pet. Vol 26. pp 231–339. www.elsevier.com/locate/egyjp
Muhammad SK, Ibnelwaleed AH, Abdullah S, Nicolas VS (2016) Application of various water-soluble polymers in gas hydrate inhibition. Renew Sustain Energy Rev 60:206–225. https://doi.org/10.1016/j.rser.2016.01-092
Nicholas JW, Koh CA, Sloan ED, Nuebling L, He H, Horn B (2009) Measuring hydrate/ice deposition in a flow loop from dissolved water in live liquid condensate. AIChE J 55:1882–1888
Ojo OV, Onyekonwu MO, Ogolo NA, Ubani CE (2013). Effect of viscosity of alkaline/surfactant/polymer in enhanced oil recovery in heterogeneous sands. SPE Paper 167550 presented at the Nigeria annual international conference and exhibition held in Lagos, Nigeria
Okon EO, Ajienka JA, Ikiensikimama SS, Elechi VU, Odutola TO (2018) Use of locally formulated inhibitor from agro wate for gas hydrate inhibition in a mini flow loop. Int J Sci Eng Inv (IJSEI) 7(83):104–112
Quideau S (2011) Why bother with poly phenols? Groupe Polyphenols
Quideau SP, Deffieut D, Douat-Casassus CL, Pougsegu L (2011) Plant polyphenols: chemical properties, biological activities and synthesis. Angew Chem Int Ed 50(3):586–621. https://doi.org/10.1002/aniee.201000044
Saikia T, Mahto V (2018) Evaluation of soy lecithin an eco-friendly biosurfactant clathrate hydrate antiagglomerant additives. J Surfactants Deterg 21(1):101–111. https://doi.org/10.1002/jsde/2018
Srivastava V. et al. (2017) CHR consortium meeting: flow loop data analysis & bedding modeling
Zanota ML, Dicharry C, Graciaa A (2005) Hydrate plug prevention by quaternary ammonium salts. Energy Fuels 19(2):584–590
Zerpa LE, Salager JL, Koh CA, Sloan ED, Sum AK (2011) Surface chemistry and gas hydrates in flow assurance. Ind Eng Chem Res 50:188–197
Zhang P, Wu Q, Mu C (2017) Influence of temperature on methane hydrate formation. Sci Rep. https://doi.org/10.1035/541598-017-08430-7
Zhao H, Sun M, Firoozabadi A (2016) Anti-aggloeration of natural gas hydrate in liquid condensate and crudeoil at constant pressure. Fuel 180:187–193. https://doi.org/10.1010/j.fuel-2016.03.029
Acknowledgement
The authors acknowledge the World Bank Africa Centre of Excellence for Oilfield Chemicals Research and the Institute of Petroleum Studies Laboratory, University of Port Harcourt, Nigeria, for their support in carrying out this research. We also acknowledge Dr. Obumneme O. Okwonna, for editing the final version of the manuscript.
Funding
This project was not funded by any external body.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Elechi, Virtue Urunwo and Okon, Efiong Okon contributed to material preparation and data collection. Elechi, Virtue Urunwo analyzed data and first drafted. Okon, Efiong Okon contributed to editing and reviewing of draft manuscript. Ikiensikimama, Sunday Sunday (also reviewd manuscript), Akaranta Onyewuchi and Ajienka Joseph Atubokiki contributed to supervision and comments on previous version of manuscripts.
Corresponding author
Ethics declarations
Conflict of interest
The authors whose names are listed above certify that they have no affiliation or involvement in any organization or entity with any financial interest or non-financial interest in the subject matter or material discussed in this manuscript.
Ethical approval
All authors approved final manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elechi, V.U., Ikiensikimama, S.S., Ajienka, J.A. et al. Mitigation capacity of an eco-friendly locally sourced surfactant for gas hydrate inhibition in an offshore environment. J Petrol Explor Prod Technol 11, 1797–1808 (2021). https://doi.org/10.1007/s13202-021-01127-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13202-021-01127-z