
Abstract
Arbuscular mycorrhizal fungi (AMF) are obligate symbionts of c. 80% of land plants, having enormous ecological and economic impact, as they often improve crop plant nutrition and yield. DNA-based identification with molecular markers is used to analyze AM fungal communities in the field, but reaching species level taxonomic resolution remains challenging. Thus, currently there is no consensus on how to analyze high-throughput sequences and assign them into species. Here, a new sequencing strategy combined with taxonomic affiliations implemented with an evolutionary placement algorithm (EPA) was established. It is based on sequencing a c. 450 bp region of the large subunit (LSU) ribosomal rRNA gene with the MiSeq-Illumina platform. The method is suitable for the discrimination of closely related AMF species and was used to study host-AMF preferences in roots of Pequin pepper, soybean and orange at one location in the arid northeast of Mexico. Twenty AM fungal species from 13 genera were detected. Phylogenetic affiliation of reads to species revealed crop preferential associations. In Pequin pepper roots, several Rhizophagus species represented most of the community, Rhizophagus clarus being the most abundant. The soybean AM fungal community was dominated by Rhizophagus irregularis and Funneliformis mosseae and that of orange by several species of Dominikia, some of them only found in this crop. Unraveling the AMF-plant preferences of important crops by an affordable and robust sequencing method, combined with phylotaxonomic AMF species resolution, is an important tool to obtain taxonomic units that are meaningful in both biological and ecological studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Arbuscular mycorrhizal fungi (AMF, abbreviation also used for ‘arbuscular mycorrhizal fungal’) have received great attention because they form symbiotic relations with the most relevant food crops for human consumption (FAO 2018). AMF are obligate symbionts of c. 80% of land plants, providing them with a wide range of benefits such as improved nutrient uptake, particularly phosphorus (P), water supply, soil structure, protection against pathogens and carbon recycling (Rillig et al. 2002; Tawaraya et al. 2012; Wehner et al. 2011; Zhao et al. 2015).
Historically, AMF communities have been characterized by spore morphology studies but this method may be misleading as presence of spores, which are resting stages, does not necessarily represent the active community (Renker et al. 2005; Hempel et al. 2007) and, e.g., spatiotemporal variations may be mistaken by sporulation dynamics (Pringle and Bever, 2002). For intradical AMF, the identification by morphological characteristics is not possible and therefore sequence based characterization has become standard.
When using molecular methods to delimit AMF species, an extended DNA barcoding region amplified by AMF specific primers (Krüger et al. 2009) with species level phylogenetic resolution (Stockinger et al. 2010) has proven to be successful for species characterization and for molecular ecological field studies (Senés-Guerrero et al. 2014). This 1.5 kb barcode comprises a part of the SSU rRNA gene, the complete ITS region (ITS1, 5.8S, ITS2 rRNA gene) and c. 800 bp of the LSU rRNA gene.
Because of its length, high-throughput sequencing of the 1.5 kb AMF barcode fragment has only been attempted with Pacific BioSciences (PacBio) Single-Molecule Real-Time (SMRT) sequencing (Schlaeppi et al. 2016). However, SMRT sequencing is not the most commonly used high-throughput sequencing technology due to its high error rate and limited throughput (Ardui et al. 2018).
In several molecular ecological studies on AMF a c. 350–400 bp fragment of the SSU rRNA gene was sequenced using the Illumina MiSeq sequencing platform. After sequencing, consecutive bioinformatics analyses consist of using similarity thresholds (usually 97%) to cluster reads into operational taxonomic units (OTUs) and BLAST them against public or curated databases (Drumbell et al., 2011). However, taxonomic classification to species is often impossible or unreliable due to the short read length and because the SSU rDNA region can only be reliably used to characterize genus level or above (Stockinger et al. 2009; Egan et al. 2018). Moreover, BLAST based approaches require reference sequences to make accurate taxonomic annotations but there are many unknown AMF species in unexplored areas (Öpik et al. 2013) which are not represented in databases and therefore their correct affiliation is restricted to described species (Berger et al. 2011). Another DNA-based strategy to achieve species level phylogenetic resolution of AMF was proposed by Senés-Guerrero and Schüßler (2016a), targeting a c. 760 bp of the LSU rRNA gene and using 454 GS-FLX+ pyrosequencing combined with an evolutionary placement algorithm (EPA) (Berger and Stamatakis 2011). This approach relied on an aligned reference sequence database (Krüger et al. 2012) together with a maximum-likelihood phylogenetic reference tree that served as a backbone to affiliate the high-throughput short reads via EPA. This approach requires more bioinformatic steps than a BLAST based method but proved to have the sufficient phylogenetic resolution to characterize AMF species. However, 454 pyrosequencing was phased out by mid-2016 and replaced by other next generation sequencing technologies, mainly by Illumina. Therefore, in this study we adapted the approach of Senés-Guerrero and Schüßler (2016b) to a new strategy using the MiSeq-Illumina platform to sequence a c. 450 bp fragment of the LSU-D2 rDNA region.
Understanding plant-AMF host or environmental specificity or preferences is of high interest, as it may help to improve current agricultural practices as well as the development of tailored-made biofertilizers and ensure their establishment after inoculation. Here, we developed a new MiSeq-based strategy to characterize AMF sequences to species and analyzed the AMF species communities of three important crops of the arid northeast of Mexico, Pequin pepper, soybean and orange, at one location. We focused on host-AMF species preferences and the effect of the developmental stage of the crops on the AMF community composition.
2 Materials and methods
2.1 Field site and sampling
Pequin pepper (Capsicum annuum var. glabriusculum), soybean (Glycine max) and orange trees (Citrus sinensis) were sampled from May until September 2017 (Table 1) from the Tecnologico de Monterrey experimental field site in Hualahuises, Nuevo Leon, Mexico (24°52.50’ N, 99°37.26’ W). The sites planted with soybean and pepper were in close proximity (10 m apart), whereas the orange trees were planted approx. 50 m from these crops (Fig. 1). Soils from beside the pepper and soybean site (soil sample 1) and within the orange trees orchard (soil sample 2) were analyzed from composite samples, each, of a 0–30 cm depth by the ISO certificated company Fertilab (Guanajuato, Mexico). Overall, no differences were found among the sites, except for iron, manganese and sulfur concentrations. The soils are of a medium clay-loam texture with pH 8.0 to 8.5, low organic matter (1.96–2.8%), moderately low water conductivity (2.6 cm/h), moderately high carbonate content (19.5–24.8%), low salinity (0.44–0.65 ds/m) and, among nutrients, show deficiency in phosphorus (5.7–9.0 ppm), zinc (0.43–0.54 ppm) and boron (0.22–0.58 ppm). For soil sample 1, manganese, iron and sulfur ranged from high to moderately high whereas for soil sample 2, they ranged from moderately low to very low (Electronic Supplementary Material 1). In the state Nuevo Leon, Mexico, around 612 mm of precipitation are expected annually and the climate at the field site is classified as semi-warm arid with an average annual temperature for 2016 of 23 °C (minimum 16, maximum 29) (SAGARPA, 2017). During the sampling period, the experimental field had 153 mm of rain (Electronic Supplementary Material 2).

Location of the experimental field site in Nuevo Leon, Mexico
Root samples from the three studied crops were collected in May, July and September 2017 with approximately two months between sampling dates. For soybean and Pequin pepper, each, four independent replicates were collected from three plant stages (young, mature and senescent), which corresponded to each sampling time (Table 1). Therefore, for pepper and soybean, plant stage cannot be separated from sampling time. The young stage was defined as flowering plants, mature stage when the plants had green fruits and senescent when fruits were ripe or already harvested. The orange trees, being mature 12-year-old plants, were sampled in triplicate each sampling time and the results obtained may rather indicate seasonal changes than plant stages. Secondary roots were collected in sterile Whirl-Pak bags from a soil depth of 0–10 cm using a sterile wooden stick and avoiding roots of invasive plants. To select the plant sample each field was divided in sections and a plant replicate was taken randomly from each section. The samples were maintained at 4 °C during transportation to the laboratory, where they were processed within two days after arrival.
2.2 Sample preparation and DNA extraction
All 33 root samples stored at 4 °C were washed with tap water and preserved in 96% ethanol in 50 ml tubes at −20 °C. Lateral roots were then cut into twenty 1-cm segments under sterile conditions and stored in 96% ethanol in 2 ml tubes at −20 °C. For DNA extraction, ethanol was removed with a pipette and samples were dried at 60 °C in a sterile environment to evaporate the remaining ethanol. Then samples were rehydrated with 200 μl of DNase-free molecular grade water and DNA was extracted with the FastDNA Spin Kit for soil (MP Biomedicals, Santa Ana, CA, USA) following the manufacturer’s instructions with the lysing matrix type A, but with one extra ¼ inch ceramic bead to assure complete rupture of the roots (Senés-Guerrero et al. 2014). DNA was stored at −20 °C until sequencing library preparation.
2.3 MiSeq sequencing
AMF DNA amplification was conducted with a nested PCR approach. Each PCR was done in triplicate. For the first PCR, the primer mixtures SSUmAf and LSUmAr described by Krüger et al. (2009) were used. These AMF-specific primers amplify an approximately 1.8 kb fragment covering a part of the SSU rRNA gene, the ITS region (ITS1–5.8S-ITS2) and a part of the LSU rRNA gene. DNA was amplified in a 15 μl PCR mixture containing 0.5 μl of DNA extract, 0.75 pmol of each primer and 7.5 μl of the 2x Phusion High-Fidelity PCR mastermix (New England Biolabs, Ipswich, MA, USA). Cycling conditions included initial denaturation at 99 °C for 5 min, followed by 35 cycles of denaturation at 99 °C for 10 s, annealing at 60 °C for 30 s and extension at 72 °C for 1 min; a final extension was conducted at 72 °C for 10 min (Krüger et al. 2009). Each PCR product was used as a template for a nested PCR to amplify a c. 450 bp fragment (LSU-D2 rDNA region). The forward primer LSUD2Af (5’-GTGAAATTGTTRAWARGGAAACG-3′) is modified from a primer designed for and tested in a previous 454FLX sequencing study (Krüger 2013; Schüßler A. and Krüger C., unpublished) and the reverse primer mix (LSUmBr) is as previously published (Krüger et al. 2009). Forward and reverse primers, respectively, were modified with the corresponding MiSeq adapters. Nested PCR was performed in a volume of 20 μl using 10 μl of the 2x Phusion High-Fidelity PCR mastermix, 1 pmol of each primer and 0.5 μl of the first PCR amplicon. The cycling conditions were 99 °C for 2 min, followed by 30 cycles of 99 °C for 10 s, 63 °C for 30 s, 72 °C for 20 s, a final extension step was conducted at 72 °C for 5 min. After nested PCR, amplicons were visualized in a 2% agarose gel. The nested PCR reaction triplicates for each sample were then pooled, purified using AMPure XP beads (Qiagen, Hilden, Germany) and visualized in a 2% agarose gel. Then, Nextera v2 indexes (Illumina, San Diego, CA, USA) were attached by an eight cycle PCR to tag each amplicon, using the same conditions as the nested PCR. For this, PCR cycling conditions were identical to the nested PCR reaction but using 8 cycles of amplification. PCR clean-up was performed using AMPure XP beads and indexed PCR products were observed in a 2% agarose gel; the expected amplicon size was 630 bp.
Library quantification was done with the Qubit® dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) in a Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA). PhiX DNA was used to spike the pooled library at a concentration of 30%. Paired-end sequencing (2 × 300 bp) was performed with a MiSeq Reagent Kit v3 (Illumina, San Diego, CA, USA) at a final loading concentration of 6.3 pM in a MiSeq System (Illumina, San Diego, CA, USA).
The sequencing run has been stored at the Sequence Read Archive at the NCBI with accession number PRJNA472054.
2.4 Bioinformatic data analysis
Raw sequence reads were analyzed using QIIME v1.9.1. (Caporaso et al. 2010) and the fungi Illumina pipeline from Bálint et al. (2014). First, paired ends were joined with a minimum 10 bp overlap and allowing an error rate of 25%. In the next step, sequences with a quality score lower than 25 and less than 70% of consecutive high-quality base calls were discarded. The resulting sequences were clustered into representative sequences (RS) using UCLUST (Edgar, 2010), in the following the term representative sequence (RS) is used for the sequences analyzed, each RS representing a cluster, with a similarity threshold of 98%. An open-reference approach was performed, using published sequences (Krüger et al. 2012) and additional corresponding sequences from recently described AMF species available in public repositories, as reference database to reduce the time and resources needed for clustering. After clustering, singletons were removed. The remaining RS were analyzed with USEARCH v10.0.240 (Edgar, 2010) to detect chimeric sequences. Afterwards, non-AMF sequences were removed from the remaining RS by BLAST against the NCBI Nucleotide database using the CLC Genomics Workbench (Qiagen, Hilden, Germany). The remaining RS, after quality filtering, chimera and non-AMF sequence removal, were used for downstream analyses.
2.5 Species delimitation
For AMF species delimitation, a reference phylogenetic tree was computed using published 1.5 kb reference sequences (Krüger et al. 2012) and additional corresponding sequences from recently described AMF species available in public repositories. This served as a backbone for the placement of the approximately 450 bp LSU-D2 RS. Sequences for the reference phylogenetic tree were aligned with MAFFT v7.311 (Katoh et al. 2002) and manually revised in CLC Genomics Workbench (Qiagen, Hilden, Germany). A maximum-likelihood phylogenetic tree was calculated with RAxML-HPC v.8 on XSEDE at the CIPRES Science Gateway (Miller et al., 2010) using the GTRGAMMA model and 1000 bootstraps (Stamatakis 2015).
As a second step, every RS was aligned to the previous 1.5 kb sequence alignment with MAFFT. Hence, the resulting alignment contains the c. 1.5 kb sequences which comprises the phylogenetic backbone and the c. 450 bp LSU-D2 representative sequences. This alignment file is used as an input at the CIPRES Science Gateway using the RAxML-HPC v.8 evolutionary placement algorithm (EPA) with the GTRGAMMA model (Berger and Stamatakis 2011; Berger et al. 2011). For each RS, EPA assigns a place in the phylogenetic tree that is used as a backbone. EPA was shown to be useful for the placement of short AMF reads with high species-resolving accuracy (Senés-Guerrero and Schüßler 2016a). It has been shown that maximum placement likelihood weights (PLW) of >0.5 usually are artifacts (Berger et al., 2011; Zhang et al., 2013). Therefore, such RS (0.75% of the total RS) were discarded. The resulting phylogenetic tree with the placement of the RS was visualized in Archaeopteryx Treeviewer v0.9920 (Han and Zmasek 2009). RS were manually annotated to a taxonomic level corresponding to the branch where they were affiliated in the phylogenetic backbone tree.
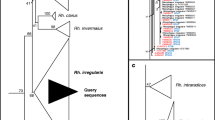
To validate EPA affiliation of the c. 450 bp LSU-D2 sequences, 138 sequence variants of eight Rhizophagus species, all sequences corresponding to the c. 1.5 kb extended AMF DNA-barcode (Krüger et al. 2012) were used to generate a maximum-likelihood reference phylogenetic tree, rooted with two sequences of Claroideoglomus claroideum. This tree was used as a backbone for testing short sequence affiliation. After trimming to the 450 bp LSU-D2 rDNA region corresponding to the amplicon sequenced by MiSeq, 21 highly variable sequence variants of the closely related, well characterized species Rhizophagus intraradices (5 sequences) and R. irregularis (16 sequences) were used as query sequences in EPA, to test for their correct affiliation.
2.6 Statistical analysis
All statistical analyses were performed in R 3.4.4 (R Core Team, 2018) using the vegan v2.4–6 (Oksanen et al. 2013) and BiodiversityR v2.9–2 (Kindt 2016) packages, unless stated otherwise. Rarefaction curves for each crop and sampling time were calculated using the rarefaction function of the vegan package. The read count matrix was square root normalized and analyzed by canonical analysis of principal coordinates (CAP) (Anderson and Willis, 2003) using the BiodiversityR function CAPdiscrim, based on Bray-Curtis dissimilarities and 999 permutations. Sample clustering shown in CAP was done with the ordiellipse function, which highlights groups with a 95% confidence. Beta-diversity among samples was calculated using the Bray-Curtis dissimilarity with the vegdist function of vegan. Beta-diversity changes among samples were tested by permutational MANOVA (PerMANOVA) using the adonis function of vegan and 999 permutations. To validate the obtained P values, the statistical power of the PerMANOVA test was corroborated using G*Power v3.1.9.2 (Faul et al. 2007) with an effect size of 0.6666. The effect size was calculated by taking into account the 33 samples, 9 groups (Pequin pepper, soybean, orange at three sampling times), 2 predictors (crop and sampling) and 1 response variable (species abundance). Regarding species presence/absence, a Fisher’s exact test was applied to determine significant differences in their presence in each crop. Whenever significant differences were found, post-hoc analysis was conducted with pairwise Fisher’s exact tests. This statistical analysis was performed using the rcompanion package of the R software.
3 Results
3.1 Validation of species-level affiliation
Using the artificially constructed R. intraradices and R. irregularis dataset, the EPA approach resulted in the correct affiliation of all 21 sequence variants with a maximum placement likelihood value <0.012; none of the sequences misplaced into incorrect branches (Electronic Supplementary Materials 3 and 4). The EPA approach therefore can correctly discriminate between closely related species.
3.2 Library generation and sequencing data analysis
A total of 33 samples were processed for library generation, which includes crops (n = 3), sampling times (n = 3) and replicates (n = 3–4). MiSeq sequencing, paired-end reads joining, quality filtering, clustering, and singletons removal resulted in 55,337 clusters. Of these, 5,769 putative chimera and 3,575 non-AMF clusters were removed, leaving 45,993 clusters from AMF (representing 1,315,642 reads with lengths between 357 and 592 bp) that were affiliated into the phylogenetic backbone tree. We use the term representative sequence (RS) for the sequences analyzed, each RS representing a cluster. Using RAxML-EPA, RS from the root samples of Pequin pepper, soybean and orange from one location (Fig. 1) were annotated to 20 AMF species from 13 genera. All samples were represented by at least 7,000 AMF reads. Rarefaction curves of the samples showed that the AMF species detected were close to, or reached, saturation, suggesting coverage of AMF species diversity in all samples (Electronic Supplementary Material 5).
3.3 AMF species diversity associated with the different crop plants
The AMF community of each crop was evaluated by determining its relative read abundance at the species (taking only into account reads assigned to species) and genus level (Fig. 2). At the genus level, the highest relative read abundance in pepper consisted of Rhizophagus (46%), followed by Septoglomus (21%), Sclerocystis (14%) and Funneliformis (13%). Pepper AMF species community presented a high abundance of Sclerocystis sinuosa (22%), Rhizophagus clarus (22%), Rhizophagus arabicus (8%) and Rhizophagus fasciculatus (8%). From first to third sampling of the pepper plants, Septoglomus and Sclerocystis abundances strongly increased over the three sampling times, from 2 to 33% and < 1 to 26%, whereas Dominikia and Funneliformis showed high relative abundances in the first (6% and 18%) and second (10% and 31%) samplings but a clear decline in the third (1%, both).

Relative read abundance of AMF species (a, b) and genera (c, d) for crop plants and sampling times. “Other species” or “other genera” contain species or genera that did not exceed 1% relative abundance in the samples. Dominikia spp. represent D. distichi, D. iranica, D. achra and unknown species of this genus
For soybean, the genera Rhizophagus (47%), followed by Funneliformis (29%), Kamienskia + Microkamienskia (5%) and Claroideoglomus (4%) dominated. The soybean AMF community was unique in terms of the presence of Microkamienskia perpusilla (6% relative abundance) and of Claroideoglomus hanlinii (5%). From first to third sampling, Rhizophagus relative abundances were high and doubled over sampling time (31%, 56% and 67%), while Funneliformis showed a opposite succession with highest abundance in the first sampling (62%), which then decreased (15% and 16%).
In terms of species richness, orange trees showed a more complex AMF community, with 18 species identified, when compared to Pequin pepper (15 species) and soybean (14 species). Orange was mainly colonized by Dominikia (44%) and Rhizophagus (22%) species. Dominikia species dominated regardless of sampling time (82%, 24%, 56%), whereas known species such as Rhizophagus irregularis increased in abundance after the first sampling (<1%, 25%, 12%) or showed irregular patterns e.g., Funneliformis mosseae (< 1%, 35%, 3%) and Rhizophagus intraradices (16%, < 1%, < 1%).
Generalist species such as F. mosseae and R. irregularis were observed in all crops, with highest combined relative read abundances in soybean (71%) followed by pepper (31%) and orange (28%). Most of the identified species occurred in both pepper and soybean but in different proportions. In contrast, the orange AMF community was dominated by Dominikia spp. (52%), which were also found in both pepper (7%) and soybean (3%), but in low abundance.
The presence or absence of AMF species among all replicates within each crop was evaluated (Fig. 3) to determine whether certain species were specifically or preferentially associating with a crop. One rationale for this is, that the read abundances are relative values and semi-quantitative, which in some cases may under- or overestimate the role of certain AMF. In all replicates, regardless of crop, the generalists F. mosseae and R. irregularis were present. In addition to the wide occurrence of generalist AMF, orange presented a unique set of five AMF: three Dominikia species (D. distichi, D. iranica and D. achra), Kamienskia bistrata and Dentiscutata savannicola (Fig. 3c). Rhizophagus prolifer was only observed in pepper (Fig. 3a). However, Fisher’s test indicated that the only significant differences were observed in pepper and soybean regarding Acaulospora scrobiculata and Rhizophagus arabicus (Electronic Supplementary Material 6).

Presence or absence data for AMF species in all study replicates for Pequin pepper, soybean and orange. Asterisks show significant differences per AMF species. Letters indicate species differences per crop. For Pequin pepper and soybean, samples 1–4, 5–8 and 9–12 represent the first, second and third sampling, respectively. For orange, samples 1–3, 4–6 and 7–9 represent the first, second and third sampling, respectively
3.4 Influence of crop species, sampling time and plant developmental stage on AMF species community composition
For pepper and soybean, the influence of plant developmental stages (which cannot be separated from sampling time) was analyzed for each crop. Differences in the global AMF species community composition at different sampling times was analyzed by merging all three crops by sampling month.
CAP analysis of the effect of crop species on AMF community composition resulted in a two-dimensional solution with 72.7% of correct allocations, which strongly separated three clusters corresponding to crop species (Fig. 4a). Furthermore, PerMANOVA analysis (Table 2) indicated a significant effect of the crop species on AMF community composition (P = 0.001). On the contrary, the effects of sampling time (analyzing all three crops together), plant developmental stage of pepper, and of soybean showed no significant influence of these variables on the AMF species community composition (Fig. 4b-d), which is supported by the two-way PerMANOVA (Table 2).

Canonical analysis of principal coordinates (CAP) of the AMF community among crop type (a), sampling time (b), and developmental stage of pepper (c) and soybean (d). Weight-calculated ellipses highlights groups with a 95% confidence level
4 Discussion
In this study, we aimed to implement a new Miseq-based high-throughput AMF species characterization strategy. The approach was used to analyze AMF communities associated with three economically important crops grown at one location, in an arid ecosystem in northeast Mexico.
4.1 AMF species delimitation method
By using a strategy based on a nested PCR followed by MiSeq-Illumina sequencing and a phylogenetic pipeline that used a reference phylogenetic tree and an evolutionary placement algorithm (EPA; Berger and Stamatakis 2011; Berger et al. 2011), we were able to annotate the studied AMF communities with high-throughput, at the species level. Egan et al. (2018) demonstrated that MiSeq-Illumina sequencing can recapture an AMF community from a mock community, even in terms of relative abundances, suggesting a robust technique. However, they found a problem in taxonomy assignment that may be due to the gene region used which was a 500 bp fragment of the SSU rDNA region that does not provide species level resolution for many AMF (Stockinger et al. 2010). Also, the lack of type sequence representation in databases was mentioned by Egan et al. (2018). Clearly, the selection of the DNA region is crucial to obtain sufficient phylogenetic resolution when characterizing sequences to the species level. A good candidate for species identification from field samples is the LSU-D2 rDNA region (Krüger et al. 2009; Stockinger et al. 2009), which is covered by the extended DNA barcode for AMF and thus characterized for most AMF species newly described. Therefore, we focused on this region to conduct in silico tests to define a primer set that amplifies DNA of all species for which LSU-D2 rDNA sequences are known (Krüger et al. 2012; additional sequence data from public databases). The amplified fragment had to be restricted to a maximum length of c. 550 bp, including primers and adapters, as recommended by Illumina for 2 × 300 bp paired-end sequencing.
As there was no reported bioinformatic pipeline for AMF species-level identification using Illumina MiSeq sequencing data, we adapted methods developed for 454-FLX sequencing (Bálint et al. 2014; Caporaso et al. 2012; Senés-Guerrero and Schüßler 2016a,b) to process and annotate the generated sequences. After raw sequence assembly and quality filtering, it was necessary to set a similarity threshold for sequence clustering. For this threshold, the length and variability of the sequenced region must be taken into account. Many studies have used 97% similarity to cluster AMF sequences (Bell et al. 2014; Lekberg et al. 2012), but this threshold depends on the taxonomic level of interpretation desired and on the variability of the DNA region studied. Based on the short length of the reads and the region used, which possesses an intraspecific rDNA sequence variation of up to 15.7% (Stockinger et al. 2010), we decided to use a similarity threshold of 98%. Distance matrices generated from sequence data of closely related species sets showed that no inter-species clustering occurred when using 98%. In comparison to a 97% threshold, this leads to a higher number of representative sequences (RS, corresponding to clusters), but avoids that clusters contain sequences from different species. Because the RS are later affiliated by a phylogenetic method, a split into more RS by using a 98% similarity level does not inflate the number of finally annotated AMF species by EPA, but it ensures that RS of different species are not merged during clustering.
EPA has been shown to be an accurate method to affiliate short sequences into a phylogenetic tree (Berger and Stamatakis 2011). Here, a maximum-likelihood phylogenetic tree based on published AMF sequences was used to affiliate the ~450 bp LSU-D2 rDNA RS derived from clustering the MiSeq sequencing reads. By this method, each query RS is placed at a tree node and this placement is accompanied by its likelihood. To exemplify that the method is suitable to distinguish closely related species, we used a set of ~450 bp sequences of R. irregularis and R. intraradices. EPA placed all sequences correctly, demonstrating that the selected c. 450 bp region can robustly be used for the molecular characterization of communities of AMF species, as it was previously shown for c. 800 bp 454-FLX-titanium sequences (Senés-Guerrero and Schüßler 2016b).
4.2 AMF communities associated with Pequin pepper, soybean and orange
Specificity of AMF to certain plant species or cultivars has been reported, however most studies have been conducted under laboratory or greenhouse conditions (Vandenkoornhuyse et al. 2003; Torrecillas et al. 2012; Van Geel et al. 2016) and may not reflect natural environments. Pequin pepper, soybean and orange are economically relevant crops in Mexico. Here, we show differences in AMF species communities colonizing these crops within the same experimental field site. Generalist species such R. irregularis (Peyret-Guzzon et al. 2016) and F. mosseae (Lekberg et al. 2012; Sýkorová et al. 2007) are fast, early-stage colonizers and adapted to disturbed systems like agricultural fields, and therefore were observed representing the majority of the AMF community at all sampling times. Nevertheless, crop-AMF associations differed in terms of abundance and presence of non-generalist AMF.
Pequin pepper and soybean crops were grown close to each other in the experimental field sharing 14 out of 15 AMF species found within its roots. Nevertheless, some differences due to the presence of A. scrobiculata and R. arabicus were significant. Moreover, in Pequin pepper, Rhizophagus arabicus, R. clarus, R. fasciculatus and S. sinuosa were found in higher abundances than in soybean, or also orange. Other reported AMF spore-based studies of pepper-associated AMF are congruent with our results. Carballar-Hernández et al. (2017) found R. fasciculatus, F. mosseae and S. sinuosa in relative high proportions in Poblano peppers at different agro-ecosystems in the central area of Mexico (Puebla state); they also found A. scrobiculata under moderate and high input of agrochemicals. Sánchez-Sánchez et al. (2018) reported F. mosseae and most likely R. irregularis (or R. fasciculatus; as R. intraradices) as highly abundant species in Pequin peppers sampled in the north and central area of Mexico (Coahuila and Zacatecas states); they also detected S. coremioides and R. clarus at some of the four sites studied. In this context it should be noted, that it is impossible to distinguish R. intraradices from R. irregularis morphologically in field samples (see discussion in Stockinger et al. 2009) and that, in general, the vast majority of spore morphology based reports of “R. intraradices” are of R. irregularis.
In agreement with our results, a high AMF diversity was reported for soybean (García de León et al. 2018). Moreover, a very high presence of R. irregularis, which has been shown to increase soybean yield (Niwa et al. 2018) was also observed. Besides, C. hanlinii and M. perpusilla were particularly abundant in soybean. These AMF species were recently described and there are no sufficient data to discuss their plant associations or distribution.
Citrus species are highly-mycorrhizal dependent crop plants that can be colonized by a wide diversity of AMF species (Wu et al. 2013). Here, the orange AMF community was very distinct as it was dominated by different species of the genus Dominikia that were not present in pepper and soybean. However, it cannot be ruled out that this preference may be due to a high presence of Dominikia in the soil closer to oranges or that changes in AMF community composition could be driven by contrasting concentrations of iron, manganese and sulfur. While it has been shown that AMF has an effect on plant nutritional status depending on micronutrient concentration (Rahman et al. 2020), information regarding the effect of micronutrients on AMF community dynamics is scarce.
A particular assembly of AMF species only found in a few replicates of orange was composed of D. achra, D. iranica, D. distichi, K. bistrata and D. savannicola. The Dominikia species have been described from maritime dunes, and although they have been suggested as important microorganisms with high colonization power for establishing, shaping and functioning of plant communities (Błaszkowski et al. 2018), their specific roles have not been elucidated. Other AMF species abundant in orange were F. mosseae, R. irregularis, R. intraradices and O. diaphana, the latter two species being very low (<1%) in relative abundance in pepper and soybean. In particular, F. mosseae has been found to confer drought tolerance when inoculated in citrus plants under greenhouse conditions (Wu et al. 2019). Additionally, the AMF community found in this study is similar to the spore-based AMF characterization of Citrus reticulata by Youpensuk et al. (2008) in Thailand and to the AMF species found in Citrus spp. surveyed in eastern Spain (Camprubí and Calvet 1996), which also showed high abundances of R. intraradices and R. irregularis (indistinguishable in spore based studies), R. fasciculatus and F. mosseae.
4.3 Influence of crop plant, sampling time and plant developmental stage on AMF community composition
In this study, we sampled three crops grown within a small experimental field site, thus under very similar environmental and soil conditions. The major variables that could have an effect on AMF community composition were host preference, sampling time and plant developmental stage. Only the variable crop plant (host) showed a significant effect on the AMF community composition. This was supported by PerMANOVA analysis using Bray-Curtis dissimilarities. Similar results had also been observed in other studies reporting a significant impact of host plant species on the AMF community (Martínez-García and Pugnaire, 2011; Pagano et al. 2011; Torrecillas et al. 2012).
Some studies showed seasonal effects (Liu et al. 2009), a difference in AMF communities between winter and summer (Drumbell et al. 2011) or that the AMF community composition was maintained throughout the warmer months (Rosendahl and Stukenbrock 2004; Santos-Gonzalez et al. 2007). However, in our study neither sampling time nor plant developmental stage were significant factors shaping AMF species communities. This disagrees with some studies that suggest that early plants are colonized mainly by AMF generalists, like R. irregularis and F. mosseae, followed by progressive changes in AMF species composition through plant development (Werner and Kiers 2014; Sýkorová et al. 2007). In our study, a significant impact of the plant stage could be lacking because of the relatively short developmental cycles of pepper and soybean. However, for perennial vine, with longer cycles, Schreiner and Mihara (2009) also showed that AMF phylotypes did not change during the growing season, but the phylotype richness declined as vineyards age increased beyond 20 years. Similarly, for coffee it was indicated that AMF spore counts decreased under coffee plants and their shade trees when their age exceeded 20 years (Muleta et al. 2008). We attribute the significantly different AMF community we found in the 12 y-old orange trees, when compared to pepper and soybean, as to be driven by the plant host, but we cannot exclude that it could also be attributed to some kind of aging effect, as young orange plants could not be studied at the same site.
5 Conclusions
The development of a new, affordable high-throughput monitoring strategy that allows the taxonomic affiliation of AMF communities at the species level helps to obtain ecologically meaningful information and to get insight on community dynamics. Combining the MiSeq sequencing of c. 450 bp LSU-D2 rDNA region with EPA phylogenetic annotation allowed us to characterize sequences to both described and unknown AMF species. With this strategy we found AMF-host plant preferences in the AMF species composition of three economically important crops growing in the same field site, under identical climatic and soil biochemical conditions.
Availability of data and material
The sequencing run has been stored at the Sequence Read Archive at the NCBI with accession number PRJNA472054.
References
Anderson M, Willis T (2003) Canonical analysis of principal coordinates: a useful method of constrained ordination for ecology. Ecology 84:511–525
Ardui S, Ameur A, Vermeesch JR, Hestand MS (2018) Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res 46:2159–2168
Bálint M, Schmidt PA, Sharma R, Thines M, Schmitt I (2014) An Illumina metabarcoding pipeline for fungi. Ecol Evol 4:2642–2653
Bell T, Hassan S, Lauron-Moreau A, Al-Otaibi F, Hijri M, Yergeau E, St-Arnaud M (2014) Linkage between bacterial and fungal rhizosphere communities in hydrocarbon-contaminated soils is related to plant phylogeny. ISME J 8:331–343
Berger SA, Krompass D, Stamatakis A (2011) Performance, accuracy, and web server for evolutionary placement of short sequence reads under maximum-likelihood. Syst Biol 60:291–302
Berger SA, Stamatakis A (2011) Aligning short reads to reference alignments and trees. Bioinformatics 27:2068–2075
Błaszkowski J, Ryszka P, Kozłowska A (2018) Dominikia litorea, a new species in the Glomeromycotina, and biogeographic distribution of Dominikia. Phytotaxa 338:241–254
Camprubí A, Calvet C (1996) Isolation and screening of mycorrhizal fungi from citrus nurseries and orchards and inoculation studies. HortScience 31:366–369
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R (2012) Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624
Carballar-Hernández S, Hernández-Cuevas L, Montaño N, Larsen J, Ferrera-Cerrato R, Taboada-Gaytán O, Montiel-González A, Alarcón A (2017) Native communities of arbuscular mycorrhizal fungi associated with Capsicum annuum L. respond to soil properties and agronomic management under field conditions. Agric Ecosyst Env 245:43–51
Core Team R (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna http://www.R-project.org/
Drumbell A, Ashton P, Aziz N, Feng G, Nelson M, Dytham C, Fitter A, Helgason T (2011) Distinct seasonal assemblages of arbuscular mycorrhizal fungi revealed by massively parallel pyrosequencing. New Phytol 190:794–804
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461
Egan CP, Rummel A, Kokkoris V, Klironomos J, Lekberg Y, Hart M (2018) Using mock communities of arbuscular mycorrhizal fungi to evaluate fidelity associated with Illumina sequencing. Fungal Ecol 33:52–64
FAO (2018) Food and agriculture Organization of the United Nations, Land Resources. FAOSTATS-Crops. http://www.fao.org/faostat/en/#data/QC. Accessed 16 March 2020
Faul F, Erdfelder E, Lang AG, Buchner A (2007) G*power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39:175–191
García de León D, Cantero J, Moora M, Öpik M, Davison J, Vasar M, Jairus T, Zobel M (2018) Soybean cultivation supports a diverse arbuscular mycorrhizal fungal community in Central Argentina. Appl Soil Ecol 124:289–297
Han M, Zmasek C (2009) phyloXML: XML for evolutionary biology and comparative genomics. BMC bioinformatics 10:356
Hempel S, Renker C, Buscot F (2007) Differences in the species composition of arbuscular mycorrhizal fungi in spore, root and soil communities in a grassland ecosystem. EnvironMicrobiol 9:1930–1938
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucl Acids Res 30:3059–3066
Kindt R (2016) BiodiversityR: package for community ecology and suitability analysis. R package version 1:8–8. http://CRAN.R-project.org/package=BiodiversityR
Krüger C (2013) Arbuscular mycorrhizal fungi for reforestation of native tropical trees in the Andes of South Ecuador. Dissertation. Ludwig-Maximilians-Universität München
Krüger M, Stockinger H, Krüger C, Schüßler A (2009) DNA-based species level detection of Glomeromycota: one PCR primer set for all arbuscular mycorrhizal fungi. New Phytol 183:212–223
Krüger M, Krüger C, Walker C, Stockinger H, Schüßler A (2012) Phylogenetic reference data for systematics and phylotaxonomy of arbuscular mycorrhizal fungi from phylum to species level. New Phytologist 193 (4):970–984
Lekberg Y, Schnoor T, Kjøller R, Gibbons SM, Hansen LH, Al-Soud WA, Sørensen SJ, Rosendahl S (2012) 454-sequencing reveals stochastic local reassembly and high disturbance tolerance within arbuscular mycorrhizal fungal communities. J Ecol 100:151–160
Liu Y, He L, An L, Helgason T, Feng H (2009) Arbuscular mycorrhizal dynamics in a chronosequence of Caragana korshinskii plantations. FEMS Microbiol Ecol 67:81–92
Martínez-García LB, Pugnaire FI (2011) Arbuscular mycorrhizal fungi host preference and site effects in two plant species in a semiarid environment. Appl Soil Ecol 48:313–317
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE), 14 Nov. 2010, New Orleans, LA pp 1–8
Muleta D, Assefa F, Nemomissa N, Granhall U (2008) Distribution of arbuscular mycorrhizal fungi spores in soils of smallholder agroforestry and monocultural coffee systems in southwestern Ethiopia. Biol Fertil Soils 44:653–659
Niwa R, Koyama T, Sato T, Adachi K, Tawaraya K, Sato S, Hirakawa H, Yoshida S, Ezawa T (2018) Dissection of niche competition between introduced and indigenous arbuscular mycorrhizal fungi with respect to soybean yield responses. Sci Rep 8:7419
Oksanen J, Guillaume-Blanchet F, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Henry M, Stevens H, Wagner H (2013) Vegan: community ecology package. R package version 2:0–10. http://CRAN.R-project.org/package=vegan
Öpik M, Zobel M, Cantero JJ, Davison J, Facelli JM, Hiiesalu I, Jairus T, Kalwij JM, Koorem K, Leal ME, Liira J, Metsis M, Neshataeva V, Paal J, Phosri C, Põlme S, Reier Ü, Saks Ü, Schimann H, Thiéry O, Vasar M, Moora M (2013) Global sampling of plant roots expands the described molecular diversity of arbuscular mycorrhizal fungi. Mycorrhiza 23:411–430
Pagano MC, Utida MK, Gomes EA, Marriel IE, Cabello MN, Scotti MR (2011) Plant-type dependent changes in arbuscular mycorrhizal communities as soil quality indicator in semi-arid Brazil. Ecol Indic 11:643–650
Peyret-Guzzon M, Stockinger H, Bouffaud ML, Farcy P, Wipf D, Redecker D (2016) Arbuscular mycorrhizal fungal communities and Rhizophagus irregularis populations shift in response to short-term ploughing and fertilisation in a buffer strip. Mycorrhiza 26:33–46
Pringle A, Bever JD (2002) Divergent phenologies may facilitate the coexistence of arbuscular mycorrhizal fungi in a North Carolina grassland. Am J Bot 89:1439–1446
Rahman MA, Parvin M, Das U, Ela EJ, Lee SH, Lee KW, Kabir AH (2020) Arbuscular mycorrhizal symbiosis mitigates Iron (Fe)-deficiency retardation in alfalfa (Medicago sativa L.) through the enhancement of Fe accumulation and sulfur-assisted antioxidant defense. Int J Mol Sci 21(6):2219. https://doi.org/10.3390/ijms21062219
Renker C, Blanke V, Buscot F (2005) Diversity of arbuscular mycorrhizal fungi in grassland spontaneously developed on area polluted by a fertilizer plant. Environ Pollut 135:255–266
Rillig M, Wright S, Eviner V (2002) The role of arbuscular mycorrhizal fungi and glomalin in soil aggregation: comparing effects of five plant species. Pant Soil 238:325–333
Rosendahl S, Stukenbrock E (2004) Community structure of arbuscular mycorrhizal fungi in undisturbed vegetation revealed by analyses of LSU rDNA sequences. Mol Ecol 13:3179–3186
Sánchez-Sánchez A, Salcedo-Martínez S, Mendoza-Villarreal R, Pinedo-Espinoza J, Moreno-Limón S (2018) Aislamiento e identificación de micorrizas arbúsculares (MA) asociadas a la rizósfera del chile piquín (Capsicum annuum var. aviculare L.). Investigación y Desarrollo en Ciencia y Tecnología de Alimentos 3:86–91
Santos-Gonzalez J, Finlay R, Tehler A (2007) Seasonal dynamics of arbuscular mycorrhizal fungal communities in roots in a seminatural grassland. Applied Environmental Microbiology 73:5613–5623
Schlaeppi K, Bender F, Mascher F, Russo G, Patrignani A, Camenzind T, Hempel S, Rillig M, van der Heijden M (2016) High-resolution community profiling of arbuscular mycorrhizal fungi. New Phytol 212:780–791
Schreiner P, Mihara K (2009) The diversity of arbuscular mycorrhizal fungi amplified from grapevine roots (Vitis vinifera L.) in Oregon vineyards is seasonally stable and influenced by soil and vine age. Mycologia 101:599–611
Senés-Guerrero C, Schüßler A (2016a) DNA-based characterization and identification of arbuscular mycorrhizal fungi species. In: Martin F and Uroz S. (eds) microbial environmental genomics (MEG). Methods in molecular biology, vol 1399. Humana press, New York, NY
Senés-Guerrero C, Schüßler A (2016b) A conserved arbuscular mycorrhizal fungal core-species community colonizes potato roots in the Andes. Fungal Divers 77:317–333
Senés-Guerrero C, Torres-Cortés G, Pfeiffer S, Rojas M, Schüßler A (2014) Potato-associated arbuscular mycorrhizal fungal communities in the Peruvian Andes. Mycorrhiza 24:405–417
SAGARPA (2017) Servicio de Información Agroalimentaria y Pesquera, Nuevo León: Infografía Agroalimentaria 2017. Mexico City
Stamatakis A (2015) Using RAxML to infer phylogenies. Curr Protoc Bioinformatics 51:6.14.1–6.14.14
Stockinger H, Walker C, Schüßler A (2009) ‘Glomus intraradices DAOM197198’, a model fungus in arbuscular mycorrhiza research, is not Glomus intraradices. New Phytol 183:1176–1187
Stockinger H, Krüger M, Schüßler A (2010) DNA barcoding of arbuscular mycorrhizal fungi. New Phytol 187:461–474
Sýkorová Z, Ineichen K, Wiemken A, Redecker D (2007) The cultivation bias: different communities of arbuscular mycorrhizal fungi detected in roots from the field, from bait plants transplanted to the field, and from a greenhouse trap experiment. Mycorrhiza 18:1–14
Tawaraya K, Hirose R, Wagatsuma T (2012) Inoculation of arbuscular mycorrhizal fungi can substantially reduce phosphate fertilizer application to Allium fistulosum L. and achieve marketable yield under field condition. Biol Fertil Soils 48:839–843
Torrecillas E, Alguacil MM, Roldán A (2012) Host preferences of arbuscular mycorrhizal fungi colonizing annual herbaceous plant species in semiarid mediterranean prairies. Appl Env Microbiol 78:6180–6186
Van Geel M, De Beenhouwer M, Lievens B, Honnay O (2016) Crop-specific and single-species mycorrhizal inoculation is the best approach to improve crop growth in controlled environments. Agron Sustain Dev 36:37
Vandenkoornhuyse P, Ridgway KP, Watson IJ, Fitter AH, Young JPW (2003) Co-existing grass species have distinctive arbuscular mycorrhizal communities. Mol Ecol 12:3085–3095
Wehner J, Antunes PM, Powell JR, Caruso T, Rillig MC (2011) Indigenous arbuscular mycorrhizal fungal assemblages protect grassland host plants from pathogens. PLoS One 6(11):e27381
Werner G, Kiers T (2014) Order of arrival structures arbuscular mycorrhizal colonization of plants. New Phytol 205:1515–1524
Wu QS, He JD, Srivastava AK, Zou YN and Kuča K (2019) Mycorrhiza enhances drought tolerance of citrus by altering root fatty acid compositions and their saturation levels. Tree Physiol: tpz039
Wu QS, Srivastava AK, Zou YN (2013) AMF-induced tolerance to drought stress in citrus: a review. Sci Hortic 164:77–87
Youpensuk S, Lordkaew S, Rerkasem B (2008) Arbuscular mycorrhizal fungi associated with tangerine (Citrus reticulata) in Chiang Mai province, northern Thailand, and their effects on the host plant. ScienceAsia 34:259–264
Zhao R, Guo W, Bi N, Guo J, Wang L, Zhao L, Zhang J (2015) Arbuscular mycorrhizal fungi affect the growth, nutrient uptake and water status of maize (Zea mays L.) grown in two types of coal mine spoils under drought stress. Appl Soil Ecol 88:41–49
Zhang J, Kapli P, Pavlidis P, Stamatakis A (2013) A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29:2869–2876
Acknowledgements
We are grateful to Cristina Chuck and Juan Valiente for giving us access to the plants in the field site, to Diego Díaz-Vázquez for his collaboration with geographic localization and to Maribel Díaz-Garza for help with the figures.
Funding
This study was funded as a Mexican-German bilateral scientific and technological cooperation project, by CONACYT-National Council of Science and Technology of Mexico (grant no. 267782) and BMBF Germany (grant no. 01DN17031). CSG postdoctoral fellowship no. 253929 was provided by CONACYT and Tecnologico de Monterrey Emerging Technologies Research Chair (GIEE EICIM01).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Electronic Supplementary Material 1.
Soil properties from the experimental field site (Hualahuises, Nuevo Leon, Mexico). S1 and S2 refer to Sample 1 and Sample 2, respectively. (PDF 150 kb)
Electronic Supplementary Material 2.
Precipitation in the experimental field site (Hualahuises, Nuevo Leon, Mexico) during the study. Sampling dates during 2017 are indicated. (PDF 34 kb)
Electronic Supplementary Material 3.
Tree branch affiliation of published, trimmed Rhizophagus sequences used to validate the method of taxonomic affiliation of closely related AMF species. Accession numbers correspond to the NCBI database; Tree branch corresponds to the affiliated branch number in the reference tree (Electronic Supplementary Material 4) (PDF 68 kb)
Electronic Supplementary Material 4.
Maximum-likelihood tree calculated with sequences of eight Rhizophagus species to validate the evolutionary placement algorithm with AMF sequences from the LSU-D2 region. The branches affiliated in Electronic Supplementary Material 3 are shown in red, together with their bootstrap support values (PDF 365 kb)
Electronic Supplementary Material 5.
Rarefaction curves of the number of AMF species detected per crop and sampling time (PDF 43 kb)
Electronic Supplementary Material 6.
Fisher’s exact test P-values for AMF species presence or absence data in all study replicates for Pequin pepper, soybean and orange (PDF 9 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Senés-Guerrero, C., Giménez, S., Pacheco, A. et al. New MiSeq based strategy exposed plant-preferential arbuscular mycorrhizal fungal communities in arid soils of Mexico. Symbiosis 81, 235–246 (2020). https://doi.org/10.1007/s13199-020-00698-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13199-020-00698-5