
Abstract
Background
Glaucoma is the leading cause of irreversible blindness worldwide. Emerged evidence has shown that glaucoma is considered an immune system related disorder. The gut is the largest immune organ in the human body and the gut microbiota (GM) plays an irreversible role in maintaining immune homeostasis. But, how the GM influences glaucoma remains unrevealed. This study aimed at investigating the key molecules/pathways mediating the GM and the glaucoma to provide new biomarkers for future predictive, preventive, and personalized medicine.
Methods
Datasets from the primary open-angle glaucoma (POAG) patients (GSE138125) and datasets for target genes of GM/GM metabolites were downloaded from a public database. For GSE138125, the differentially expressed genes (DEGs) between healthy and POAG samples were identified. And the online Venn diagram tool was used to obtain the DEGs from POAG related to GM. After which GM-related DEGs were analyzed by correlation analysis, pathway enrichment analysis, and protein–protein interaction (PPI) network analysis. Human trabecular meshwork cells were used for validation, and the mRNA level of hub genes was verified by quantitative real-time polymerase chain reaction (RT-qPCR) in the in vitro glaucoma model.
Results
A total of 16 GM-related DEGs in POAG were identified from the above 2 datasets (9 upregulated genes and 7 downregulated genes). Pathway enrichment analysis indicated that these genes are mostly enriched in immune regulation especially macrophages-related pathways. Then 6 hub genes were identified by PPI network analysis and construction of key modules. Finally, RT-qPCR confirmed that the expression of the hub genes in the in vitro glaucoma model was consistent with the results of bioinformatics analysis of the mRNA chip.
Conclusion
This bioinformatic study elucidates NFKB1, IL18, KITLG, TLR9, FKBP2, and HDAC4 as hub genes for POAG and GM regulation. Immune response modulated by macrophages plays an important role in POAG and may be potential targets for future predictive, preventive, and personalized diagnosis and treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glaucoma is the leading cause of irreversible blindness worldwide [1]. It is predicted that there will be over 1.18 hundred million glaucoma patients in 2040 [1]. This disease is characterized as progressive and irreversible loss of retinal ganglion cells (RGCs) mainly due to the increased intraocular pressure (IOP) [2]. The cause of glaucoma is a complex of multiple reasons, such as age and genomic mutations [3,4,5,6]. Meanwhile, systemic diseases such as hypertension, thyroid disease, diabetes, and dyslipidemia have been reported to increase the incidence of glaucoma [7, 8]. At present, the clinical methods for early diagnosis and treatment of glaucoma are very limited. Due to the selective retinal ganglion cells (RGCs) damage and characteristic visual field defects, a considerable number of patients with late-stage glaucoma can still maintain a distance vision of more than 5.0, which leads to subjective neglect and missed diagnosis. Meanwhile, in terms of treatment, the intraocular pressure of glaucoma patients is mainly reduced or controlled through 3 methods: medicine, laser, and surgery, to achieve the purpose of protecting the patient’s visual function [9,10,11]. However, for some glaucoma patients, although the intraocular pressure has been controlled at normal level, visual field defects continue to develop, eventually leading to irreversible blindness. High intraocular pressure is the main factor of glaucoma, but it is not the only factor. The pathogenesis of glaucoma has not been thoroughly studied so far. Moreover, emerged evidence has suggested glaucoma could be considered an immune disorder [12,13,14]. Glaucoma can be divided into multiple subtypes including primary open-angle glaucoma (POAG), primary angle closure glaucoma (PACG), and secondary glaucoma [15]. Among these subtypes, POAG consist of 74% of glaucoma according to the data from the clinical field [1].In POAG, both the anterior and posterior segments of the eye are affected, and serious damage may be caused upon the trabecular meshwork (TM) and optic nerve (ON) [16]. The trabecular meshwork, located within the iridocorneal angle, is the main pathway for the drainage of the aqueous humor (AH) out of the eye, and its dysfunction leads to IOP elevation [17]. Thus, trabecular meshwork (TM) is the key structure during the process of glaucoma including POAG.
POAG and immune response are not traditionally perceived to be related, but recently, mounting evidence suggests the pathogenesis in POAG is related to immune system disorders [18, 19]. The gut is considered the largest immunological organ in the human body with an irreplaceable role in regulating immune homeostasis [20]. Researches have shown that there is over 1014 microbes in the human digestive system: from the stomach to the distal colon, and intestinal microbes are highly varied from individuals [21]. The gut microbiota (GM) and its metabolites have adverse health effects on the human body, and GM dysbiosis can result in a variety of chronic diseases, including metabolic diseases, gastrointestinal diseases, cardiovascular diseases (the gut-heart-axis), and neurodegenerative diseases (the gut-brain-axis) [22,23,24,25,26]. Among them, Flammer syndrome (FS) is a series of special clinical symptoms and signs caused by abnormal blood supply caused by primary vascular dysfunction. Studies have shown that the diseased eating of patients with FS reduces intestinal microflora diversity and negatively impacts the microbiome by evidence of an intestinal dysbiosis, while a dysfunctional GM can further aggravate the symptoms of FS [27].
Recently, the concept of gut-eye-axis (Fig. 1) have been raised [28], as in human body, biological signals released by the intestinal microbes could lead to immune response from the eye. Researchers have shown series of ocular diseases such as uveitis, macular degeneration [29], diabetic retinopathy [30], and corneal disease [31] relevant to GM dysbiosis. Studies also discovered the possible link between the gut microbiota and glaucoma, for example: irritable bowel disease patients have higher risk of glaucoma [32], and there is distinct difference in gut microbiota composition and serum metabolic phenotype between POAG patients and healthy individuals [33, 34]. Thus, the modification of the gut microbiota in metabolic syndrome and its related chronic diseases stands as a prominent objective within the realm of microbiome research, with the aim of enabling the clinical application of probiotics [35, 36]. The increasing scientific and clinical evidence highlight the impact of microbial influence on distal sites such as the skin, heart, and central nervous system [37]. Despite the above evidence, the knowledge of how the gut microbiota correlated and influenced the process of POAG targeting predictive, prevention, and personalization medicine (PPPM) is still very limited.

The conception atlas of the gut-eye-axis
Working hypothesis
Thus, understanding the specific mechanism and mode of action of immune response and immune regulation in POAG will help to obtain a novel approach for the early diagnosis of POAG, to expand effective neuroprotective strategies, and to develop personalized treatment methods, which are in-line with the principle of PPPM. Recently, several studies have revealed how non-ocular factors contribute to the diagnosis of ocular diseases, for example: red blood cell distribution width (RDW) could be a potential laboratory parameter for disease prediction of primary angle-closure glaucoma (PACG) [38]. Moreover, systemic inflammatory biomarkers and metabolomics have been proved to allow early identification and timely implementation of replacement therapies for ocular diseases [39, 40].
In this study, we analyzed RNA-seq data from primary open-angle glaucoma (POAG) patients and co-analyzed them with the genetic data from the gut microbiota database. We found several genes/pathways differentially expressed in POAG patients that are gut microbiota related, among which are critical genes/pathways who played an important role in regulating macrophages differentiation and polarization. Our findings could be regarded as biomarkers for the early identification of POAG and could be developed into a personalized treatment therapy from the prospect of gut microbiota regulation. Our research provided a new perspective to expand the understanding of the gut-eye-axis, the identification of hub genes and pathways could represent potential therapeutic strategies for glaucoma, and point towards future predictive, preventive, and personalized medicine (PPPM/3PM).
Methods and materials
Study design and data collection
Datasets of POAG patients were downloaded from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). The keyword “glaucoma” was used to search related gene expression datasets and non-human tested specimens were excluded. The GSE138125 [41] data set consisted of the gene expression profiles of 4 POAG patients and 4 normal controls. Datasets of the gut microbiota were obtained from gutMGene (http://bio-annotation.cn/gutmgene/home.dhtml), which is a database for target genes of gut microbes and microbial metabolites released by Harbin Medical University.
Differentially expressed gene analysis
For GSE138125, the differentially expressed genes (DEGs) between healthy and POAG samples were screened by the “limma” package in R software. The genes with P-value < 0.05 and logFC > 1 were to be DEGs. In addition, the online Venn diagram tool was used to obtain the gut microbiota-related DEGs (http://bioinformatics.psb.ugent.be/webtools/Venn/). Then, the “heatmap” and “ggplot2” packages in R software are used to draw a heatmap, a volcano plot, and a box plot, respectively, to visualize the gut microbiota-related DEGs [42]. The co-DEGs obtained from the two datasets were visualized using the R packages’ “complex heatmap” and “ggplot2” to generate the heatmaps and volcano maps, respectively. These co-DEGs were retained for subsequent analysis. After screening out the gut microbiota-related genes, the “complex heatmap” and the “ggplot2” R packages were used for visualization, generating heatmaps, and volcano maps, respectively.
Pathway enrichment analysis and functional annotation
The above differentially expressed gut microbiota-related genes in POAG were submitted to GO function enrichment analysis, which consisted of biological process (BP), cellular component (CC), and molecular function (MF), and the Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling pathway enrichment analysis using an R package “clusterProfiler”. The enriched GO terms and KEGG pathways with an adjusted P-value < 0.05 was selected.
PPI network construction and hub genes identification
The protein–protein interaction (PPI) network was analyzed using the Search Tool for the Retrieval of Interacting Genes (STRING; http://string-db.org). An interaction with a combined score > 0.4 was selected and used to construct a PPI network with Cytoscape software (version 3.9.0). Hub genes were identified for further analysis by using the cytoHubba plugin.
Cell culture and cell grouping
Human trabecular meshwork cell was purchased from the IMMOCELL company and cultured in DMEM/F12 medium containing 15% fetal bovine serum (FBS) (Gibco, USA) and 1% Antibiotic–Antimycotic (Gibco, USA) at 37 °C with 5% carbon dioxide. When the cell density reached 80%, it was washed with PBS (Gibco, USA) and treated with 0.05% trypsin (Gibco, USA) for passage at the proportion of 1:3.
The logarithmic growth phase cells with good growth condition were inserted into 6-well plates with 1.2 × 106 cells per well. The cells were divided into two groups: the H2O2-treated group to mimic glaucoma in vitro and the control. The H2O2-treated group was cultured in the medium containing 200 nM H2O2, and the normal group was cultured in the SG medium. For H2O2 treated group, 200 µM H2O2 was added to each well and cultured at 37 °C for 24 h to create the in vitro glaucoma model.
RNA extraction and RT-qPCR
RNA was extracted from human trabecular meshwork stem cells using Trizol reagent (Fisher Scientific, #15–596-018) and cDNA generated using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories). For each treatment group, 3 biological replicates were collected. RT-qPCR was performed on a CFX384 Touch Real-Time PCR Detection System (Bio-Rad Laboratories) using the 2X SYBR Green Pro TaqHS Premix (Accurate Biotechnology Co.). Primer sequences are provided in Table 1. Gene expression fold change was calculated using the comparative CT method (2 − ΔΔCT) and the expression in untreated human trabecular meshwork cells was normalized to 1 as the control [43]. GAPDH was used as the housekeeping gene in these experiments.
Statistics
RT-qPCR data were analyzed utilizing Prism 9 (version 9.5.0) software, the gene expression fold change was analyzed using a one-way ANOVA and the difference between groups were statistically significant when P-value < 0.05.
Results
In this study, we analyzed RNA-seq data of human trabecular meshwork tissue collected from 4 POAG patients and 4 healthy controls. We further co-analyzed 1606 differential expressed genes (DEGs) screened out from POAG dataset and co-analyzed those DEGs with the gut microbiota database gutMGene v1.0, which contains target genes of gut microbiota and gut microbiota metabolites. The results are presented as follows.
Identification of GM-related differentially expressed genes in POAG and correlation analysis
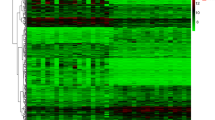
1606 DEGs were screened between POAG patients and healthy controls using the “limma” package. Compared with datasets obtained from gutMGene v1.0, a total of 16 differentially expressed GM-related genes in POAG patients were identified after taking the intersection of the Venn, including 9 common upregulated genes and 7 common downregulated genes. These genes were identified as GM-related DEGs in POAG (Fig. 2 and Table 2).

Differentially expressed gut microbiota-related genes in POAG patients and normal control. A The heatmap of 16 differentially expressed GM-related genes in POAG patients. Red represents upregulated genes and blue represents downregulated genes. GM, gut microbiota, B volcano plot of 16 differentially expressed GM-related genes in POAG patients. The red dots in the picture represent significantly upregulated genes, blue dots represent significantly downregulated genes, black dots represent genes that are not differentially expressed, and the five genes that are most significantly upregulated or downregulated are marked; C a total of 201 differentially expressed GM-related genes shown in Venn diagram; D the boxplot of 16 differentially expressed GM-related genes in POAG and normal control, including 9 upregulated genes and 7 downregulated genes
To explore the expression correlation of these 16 GM related DEGs in POAG, the correlation analysis has been performed by bioinformatics methods. The results showed that there was a high correlation between upregulated genes and downregulated genes, respectively (Fig. 3).

Correlation analysis of 16 differentially expressed GM-related genes in POAG patients and normal control. A and B correlation heatmap
Pathway enrichment analysis
To further explore the underlying biological processes of these GM related DEGs in POAG, gene ontology (GO) enrichment analysis, Kyoto Encyclopedia of Genes, and Genomes (KEGG) pathway enrichment analysis were performed. The results showed that in terms of GO analysis, the genes were mainly enriched in signaling pathways related to regulation of macrophage activity such as: positive regulation of granulocyte macrophage colony stimulating factor production, granulocyte macrophage colony stimulating factor production, positive regulation of macrophage-derived foam cell differentiation.
Meanwhile, KEGG analysis showed that DEGs related to GM were mainly enriched in PD-L1 expression and PD-L1 checkpoint pathway in cancer and a series of parasite diseases including Chagas disease and Amoebiasis (Fig. 4 and Table 3).

Pathway enrichment analysis of 16 differentially expressed GM-related genes in POAG, including BPs, CCs, and MFs. A Bar plot of the enriched GO terms, B bar plot of enriched KEGG terms, and C chordal graph of the enriched GO terms. It shows the correlation between the upregulated differentially expressed GM-related genes in POAG and the first 10 enriched GO pathways. D Eight diagrams of the enriched GO terms. GO, gene ontology; BPs, biological processes; CCs, cellular components; MFs, molecular functions
Relationship between enriched pathways
The relationship between the above enriched pathways was shown (Fig. 4). There were 7 common GM-related genes in POAG patients in the 10 most prominent pathways, which are CCL16, nuclear factor kappa B subunit 1(NFKB1), interleukin 18(IL18), KITLG, TLR9, MUC2, and HDAC4. The 10 most prominent pathways include positive regulation of granulocyte macrophage colony-stimulating factor production, maintenance of the gastrointestinal epithelium, regulation of macrophage-derived foam cell differentiation, response to interleukin 1, and positive regulation of leukocyte proliferation. Besides, we analyzed the expression of differential genes in the significantly enriched pathway and showed the results in the heatmap-like functional classification map (Fig. 5).

Correlations between the enriched pathways A relationships between the enriched GO pathways, B common genes in the top enriched GO pathways, C heatmap–like functional classification. Genes that are most significantly upregulated or downregulated are marked
PPI network construction of GM related DEGs and identification of hub genes
To discover the potential relationships between proteins encoded by GM-related DEGs in glaucoma and to identify hub genes, the PPI network of the DEGs was screened by Search Tool for the Retrieval of Interacting Genes (STRING), including 16 nodes and 6 edges, with a PPI enrichment P-value < 0.0153 (Fig. 6A). Then, the obtained results were imported into Cytoscape software for visual analysis. The interaction number of each gene was also shown. The PPI network was analyzed by Cytoscape plug-in CytoHubba to identify hub genes (Fig. 6B). The top 6 genes identified as potential hub genes are: nuclear factor kappa B subunit 1(NFKB1), interleukin 18(IL18), KITLG, TLR9, FKBP2, and HDAC4.

PPI network and identification of hub genes. A PPI network of the differentially expressed GM-related genes in POAG patients, constructed by STRING. B Significant gene module. STRING, Search Tool for the Retrieval of Interacting Genes
Validation of the hub gene expression in the in vitro glaucoma model
In this study, we incubated human trabecular meshwork cells with H2O2 (200 µM) to simulate a glaucoma model in vitro. We performed RT-qPCR to validate the expression change of the hub genes IL18, HDAC4, TLR9, and NFKB1. NFKB1, TLR9, and HDAC4 were predicted to be upregulated in glaucoma and their mRNA level increased according to the RT-qPCR data (Fig. 7), meanwhile, IL18 was predicted to be downregulated in glaucoma and its mRNA level was also increased according to RT-qPCR.

The mRNA level of 4 hub genes. The mRNA level of NFKB1, IL18, TLR9, and HDAC4 were measured in human TM cells by RT-qPCR. P-values were calculated using one way ANOVA. *P < 0.05; ***P < 0.001; ns, non-significant; RT-qPCR, quantitative real-time polymerase chain reaction
Discussion
Glaucoma has a huge impact on public health worldwide and primary open-angle glaucoma (POAG) is responsible for over 70% of glaucoma [8]. Current consensus on glaucoma is not solely based on intraocular pressure (IOP), especially in cases of normal-tension glaucoma (NTG). Although high IOP is a well-established risk factor for glaucoma, there is increasing evidence that other factors, such as vascular and neurodegenerative mechanisms [19, 20], also play a role in the development and progression of the disease. Lowering IOP is currently the only intervention available [21]. However, clinical evidence indicates that lowering IOP does not prevent progression in all POAG patients [22]. Thus, non-IOP factors are involved in POAG. Considering these findings, the treatment of glaucoma has shifted from a sole focus on lowering IOP to a more individualized approach that considers other factors that may be contributing to the disease. Previous studies have revealed inflammation and immune response mediate the process of POAG from several different aspects [9], yet much remains to be understood with regards to cellular processes regulating the maintenance of the TM and its relevance to IOP.
Recent advances have identified the significant associations between the gut microbiota and several disorders in multiple organs besides the digestive tract [44, 45]. Emerging evidence has suggested the gut microbiota could be the regulator of several central nervous system disorders, including the ocular system disorders [46]. For example, the gut microbiota has been recognized as an important contributor to anorexia nervosa as an extreme cases of the FS phenotype, furthermore, FS is strongly associated with glaucomatous optic neuropathy [27]. Although data from the clinical field are showing the possible link between the gut microbiota and the occurrence of POAG [33, 47,48,49], the mechanism underlying its connections and the key molecules remained unknown.
In this study, we aimed at discovering the key genes and pathways in POAG that are gut microbiota related. After analysis, NFKB1, IL18, TLR9, FKBP2, KITLG, and HDAC4 were recognized as hub genes in POAG patients and in the database of gut microbiota regulation. They showed the most remarkable correlation with regulation and stimulation of leukocytes such as macrophages. Thus, we conclude these 6 hub genes might play important roles via affecting the activity of macrophages during the progression of POAG and in regulating gut microbiota. We further verified the expression level of 4 hub genes (NFKB1, TLR9, HDAC4, and IL18) through RT-qPCR. The above 4 hub genes were shown to be regulating immune response from different aspects. Based on our RT-qPCR results, the mRNA level of all 4 genes was increased in H2O2-treated group compared to the control.
NFKB1 has been reported to be playing a critical role in suppressing inflammation, aging and cancer [50]. NFKB1−/− mice have reduced numbers of conventional and plasmacytoid dendritic cells in response to TLR9 stimulation [51]. Moreover, NFKB1−/− bone marrow-derived macrophages show an increased expression of interferon-inducible genes, including IFN-β, in response to TLR-4 and TLR-9 stimulation [52]. Toll-like receptor 9 (TLR9) is an intracellular TLR, expressed in different immunological and non-immunological cells [53]. The gut microbiota is a source of TLR ligands, including TLR9. Though its role in glaucoma via regulating gut microbiota remained mystical, previous research has indicated that TLR9 deficiency induces osteoclastic bone loss via gut microbiota-associated systemic chronic inflammation [54], thus, it is possible that the TLR9 mediate the occurrence of glaucoma through chronic inflammation process. Targeting immune responses mediated by TLR9 is a potential therapeutic strategy for preventing disease-associated inflammation and autoimmune diseases [55]. Several previous studies have also described a wide range of oxidative stress-related makers which are found in glaucomatous patients, including the activation of the NFKB pathway and the upregulation of pro-inflammatory cytokines [56].
Little is known of what the function of HDAC4 in glaucoma is, but emerging evidence has shown HDAC4 has its importance in regulating neurogenesis [57]. In vivo selective inhibition of HDAC4 via MS-275 (entinostat) or LMK-235 could prevent ongoing RGC degeneration [58]. Reduced HDAC4 expression is associated with blood–brain barrier (BBB) breakdown contributing to ischemia/reperfusion injury-induced infarct in ischemic stroke model rats, while increased HDAC4 expression ameliorates BBB injury, contributing to the reduced infarct volume [59].
IL18 is a pro-inflammatory cytokine which has varies function in different cell types [60]. As an important regulator for both innate and acquired immune response, IL18 plays an important role in inflammatory/autoimmunity diseases. Research has shown the intestinal epithelial cell secretion of IL18 is necessary for gut microbiota homeostasis (dysbiosis) [61]. Meanwhile, increased IL18 expression was observed in glaucomatous trabecular meshwork [62]. Our bioinformatic prediction showed downregulation of IL18 while the RT-qPCR result showed significantly increased mRNA level of IL18, and RT-qPCR result is consistent with the most previous research. In that case, we hypothesized that the reason for the downregulation of IL18 showed in bulk RNA-seq data is due to the small sample number.
Pathways enriched in both GO and KEGG database are mostly related to the activity of leukocytes especially macrophages. Substantial amount of research concerning the role of the immune system in glaucoma has been performed in the recent years. Researchers have provided evidence of macrophages participating in the process of glaucomatous pathogenesis [63], studies analyzing the trabecular meshwork of patients with POAG found macrophages in this tissue by scanning electron microscopy [64], and the presence of macrophage inhibitory factor were observed in healthy human doners’ TM. Meanwhile, it is shown that using macrophage-activator drug Zymosan could induce neurodegenerative effect in glaucoma model [65]. Moreover, a study has revealed enhanced expression of vascular regulation gene ABC1 in leukocytes as a potential marker for the diagnostics and ex vivo molecular monitoring of glaucoma [66]. Nevertheless, TM has been shown to display characters of several cell types including expressed behavior pattern of macrophages when it operates to maintain homeostasis of IOP [67], which further support the accuracy of our bioinformatic analysis. It seems like macrophages played the role of the “double sided sword” in glaucoma. Still, deeper discovery of the protective factors of the macrophages is needed to bring forward a therapeutic approach.
In this study, we utilized a hydrogen peroxide (H2O2) model to mimic glaucomatous ocular hypertension in vitro. H2O2 induces acute oxidative stress to TM which leads to the pathogenesis of POAG [68], this model was proved to be a useful platform which resulted in TM cellular dysfunction without the destruction of the TM structure [69], which made this model suitable for the hypothesis that gut microbiota dysbiosis regulates TM cellularity through innate immune response.
Gut microbiota and its metabolites contribute to a variety of diseases with inflammation components via the regulation of macrophages, for example, intracranial aneurysm [70], IBD (inflammatory bowel disease) [71], Alzheimer’s disease [72], and more. Gut microbiota-derived metabolite SCFAs (short-chain fatty acids) promotes anti-inflammatory functions of the macrophages [73], as another gut microbiota metabolite TMAO (trimethylamine-N-oxide) induces M1 macrophage polarization [74]. Specific strains such as L. delbrueckii subsp. bulgaricus IMV B-7281 and Lactobacillus spp have been shown to significantly stimulate macrophages to synthesize inflammatory factors such as IL-12, which plays a key role in the activation of cell-mediated immunity. Besides, the heterogeneity of the bacterial cell wall of these probiotics can be used as an ideal biomarker for predicting host-bacterial interaction, which has important clinical significance to facilitate the development of personalized probiotics and probiotics treatments [74]. Our research showed that the 6 most critical glaucoma pathogenic genes, such as the NFKB1, IL18, and TLR9, are closely relevant to strains such as Bifidobacterium adolescentis and Lactobacillus paracasei JS1, which may affect the progression of the disease through metabolites such as Equol, Butyrate, and Indole (Supplement Fig. 1). Currently, it is still unclear how gut microbiota effects glaucoma via the regulation of macrophages, it will be crucial to testify this mechanism in the real world. The gut-eye-axis is a relatively new concept that has gained attention in the scientific community in recent years.
Utilizing non-ocular laboratory examinations to help with early identification of ocular diseases has been proven to have a great potential [75, 76], for example, key molecules and pathways affected by glaucoma pathology can be developed into a predictive diagnosis strategy [77]. Meanwhile, microbiome phenotypes can be utilized as parameters of predictive medicine, aiding in the recognition of patients’ predispositions and assessment of treatment responses [35]. The incorporation of various phenotype markers enables effective monitoring of microbiome modulation. In our case, we identified hub genes and pathways linked POAG with gut microbiota, which could be further used as biomarkers indicating POAG at an early stage from common non-invasive examination including fecal examinations.
The modification of the gut microbiota in metabolic syndrome and associated chronic diseases is among the leading tasks of microbiome research and is needed for the clinical use of probiotics [36].
Recent evidence has shown that dietary interventions have the potential to counteract systemic immunological disorders and neuroinflammation diseases by modulating and restoring gut microbiota balance [27, 78]. However, there is a lack of evidence for the implications of microbiome modification in improving metabolic health, particularly when applied in a personalized manner. In the field of developing new strategy for glaucoma in the manner of 3P medicine, probiotics also have strong and well-documented preventive potential. Thus, the gut-eye-axis hypothesis is still an emerging field of research, and further studies are needed to fully understand the mechanisms behind this relationship and to establish a causal link towards future predictive and preventive medicine.
In the end, our study has its limitations. We showed gut microbiota could be influencing the occurrence of glaucoma via affecting macrophages, but there is lack of evidence from patients with glaucoma and gut microbiota dysbiosis at the same time. Moreover, the sample number of POAG patients is relatively small (n = 4). As a matter of fact, the human trabecular meshwork tissue is very rare, and incomparable with other animal or in vitro cell models. Considering our results are statistically significant, we believe that the four groups of samples can still reflect the situation of POAG. Meanwhile, the condition of systemic diseases was not reported for both the 4 POAG patients and the 4 controls. Recent studies have reported in POAG patients, a significant prevalence of systemic diseases is observed, including arterial hypertension, dyslipidemia, and diabetes [7, 8, 79, 80]. Given that all three mentioned systemic diseases are known to induce inflammatory process and macrophages activation, there is a possibility of overlapping effects in the context of findings related to POAG.
In a word, as a pilot study, our observations provide expanding insight of the gut-eye-axis and possible therapeutic target for future predictive, preventive, and personalized treatment of glaucoma.
Conclusion and expert recommendations
This study identified IL18, TLR9, NFKB1, HDAC4, FKBP2, and KITLG as hub genes in glaucoma that are gut microbiota-related as well as showed that the regulation of immune response by gut microbiota is associated with glaucoma. Those key molecules and pathway could contribute to the early prediction of POAG, thus, reduce the negative effect such as economic and clinical burden associated with unnecessary medical treatments. Our findings offered strong evidence to prove the existence of the gut-eye-axis and support the potential of systemic immune therapy for targeting preventive and personalized treatment of POAG, especially for those with gut microbiota dysbiosis [81], which could further provide expanding insight in-line with the paradigm shift from a reactive treatment to a 3PM.
Data availability
Datasets of POAG patients (GSE138125) were downloaded from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). Datasets of gut microbiota were obtained from gutMGene (http://bio-annotation.cn/gutmgene/home.dhtml).
Code availability
All software applications used are included in this article.
References
Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90(3):262–7.
Shen J, Wang Y, Yao K. Protection of retinal ganglion cells in glaucoma: current status and future. Exp Eye Res. 2021;205:108506.
Wiggs JL, Pasquale LR. Genetics of glaucoma. Hum Mol Genet. 2017;26(R1):R21–7.
Challa P, Schmidt S, Liu Y, et al. Analysis of LOXL1 polymorphisms in a United States population with pseudoexfoliation glaucoma. Mol Vis. 2008;14:146.
Rowan S, Jiang S, Korem T, et al. Involvement of a gut-retina axis in protection against dietary glycemia-induced age-related macular degeneration. Proc Natl Acad Sci U S A. 2017;114(22):E4472–81.
Li H, Ye Z, Li Z. Identification of the potential biological target molecules related to primary open-angle glaucoma. BMC Ophthalmol. 2022;22(1):188.
Erb C, Gast U, Schremmer D. German register for glaucoma patients with dry eye. I. Basic outcome with respect to dry eye. Graefes Arch Clin Exp Ophthalmol. 2008;246(11):1593–601.
Lin HC, Chien CW, Hu CC, et al. Comparison of comorbid conditions between open-angle glaucoma patients and a control cohort: a case-control study. Ophthalmology. 2010;117(11):2088–95.
Yadav KS, Rajpurohit R, Sharma S. Glaucoma: current treatment and impact of advanced drug delivery systems. Life Sci. 2019;221:362–76.
Ma A, Yu SWY, Wong JKW. Micropulse laser for the treatment of glaucoma: a literature review. Surv Ophthalmol. 2019;64(4):486–97.
Lim R. The surgical management of glaucoma: a review. Clin Exp Ophthalmol. 2022;50(2):213–31.
Tezel G. The immune response in glaucoma: a perspective on the roles of oxidative stress. Exp Eye Res. 2011;93(2):178–86.
Tezel G. Immune regulation toward immunomodulation for neuroprotection in glaucoma. Curr Opin Pharmacol. 2013;13(1):23–31.
Tezel G, Wax MB. The immune system and glaucoma. Curr Opin Ophthalmol. 2004;15(2):80–4.
Coleman AL. Glaucoma. Lancet. 1999;354(9192):1803–10.
Saccà SC, Gandolfi S, Bagnis A, et al. From DNA damage to functional changes of the trabecular meshwork in aging and glaucoma. Ageing Res Rev. 2016;29:26–41.
Buffault J, Labbé A, Hamard P, et al. The trabecular meshwork: structure, function and clinical implications. A review of the literature. J Fr Ophtalmol. 2020;43(7):e217–30.
Zhang T, Xie X, Lu F. Primary open-angle glaucoma: neuroendocrine-immune disorder? Med Hypotheses. 2014;83(4):514–5.
Williams PA, Marsh-Armstrong N, Howell GR. Neuroinflammation in glaucoma: a new opportunity. Exp Eye Res. 2017;157:20–7.
Takiishi T, Fenero CIM, Câmara NOS. Intestinal barrier and gut microbiota: shaping our immune responses throughout life. Tissue Barriers. 2017;5(4):e1373208.
Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124(4):837–48.
Chen Y, Zhou J, Wang L. Role and Mechanism of gut microbiota in human disease. Front Cell Infect Microbiol. 2021;11:625913.
Matsiras D, Bezati S, Ventoulis I et al. Gut failure: a review of the pathophysiology and therapeutic potentials in the gut-heart axis. J Clin Med. 2023;12(7):2567.
Peh A, O’donnell JA, Broughton BRS, et al. Gut microbiota and their metabolites in stroke: a double-edged sword. Stroke. 2022;53(5):1788–801.
Ahlawat S, Asha Sharma KK. Gut-organ axis: a microbial outreach and networking. Lett Appl Microbiol. 2021;72(6):636–68.
Trøseid M, Andersen G, Broch K, et al. The gut microbiome in coronary artery disease and heart failure: current knowledge and future directions. EBioMedicine. 2020;52:102649.
Bubnov R, Golubnitschaja O. Flammer syndrome, disordered eating and microbiome: interrelations, complexity of risks and individual outcomes. In: Golubnitschaja O, editor. Flammer syndrome: from phenotype to associated pathologies, prediction, prevention and personalisation. Cham: Springer International Publishing; 2019. p. 317–30.
Floyd JL, Grant MB. The gut-eye axis: lessons learned from murine models. Ophthalmol Ther. 2020;9(3):499–513.
Rowan S, Jiang S, Korem T, et al. Involvement of a gut–retina axis in protection against dietary glycemia-induced age-related macular degeneration. Proc Natl Acad Sci. 2017;114(22):E4472–81.
Yempén REA, Venzel R, Campos MCP, et al. Gut microbiota: a potential therapeutic target for management of diabetic retinopathy? Life Sci. 2021;286:120060.
Napolitano P, Filippelli M, Davinelli S, et al. Influence of gut microbiota on eye diseases: an overview. Ann Med. 2021;53(1):750–61.
Mcpherson ZE, Sorensen HT, Horvath-Puho E, et al. Irritable bowel syndrome and risk of glaucoma: an analysis of two independent population-based cohort studies. United Eur Gastroenterol J. 2021;9(9):1057–65.
Gong H, Zhang S, Li Q, et al. Gut microbiota compositional profile and serum metabolic phenotype in patients with primary open-angle glaucoma. Exp Eye Res. 2020;191:107921.
Gong H, Zeng R, Li Q, et al. The profile of gut microbiota and central carbon-related metabolites in primary angle-closure glaucoma patients. Int Ophthalmol. 2022;42(6):1927–38.
Golubnitschaja O, Bubnov R. Microbiome in Lean individuals: phenotype-specific risks and outcomes. In: Boyko N, Golubnitschaja O, editors. Microbiome in 3P medicine strategies: the first exploitation guide. Cham: Springer International Publishing; 2023. p. 87–99.
Bubnov R, Spivak M. Pathophysiology-based individualized use of probiotics and prebiotics for metabolic syndrome: implementing predictive, preventive, and personalized medical approach. In: Boyko N, Golubnitschaja O, editors. Microbiome in 3P medicine strategies: the first exploitation guide. Cham: Springer International Publishing; 2023. p. 133–96.
Reid G, Abrahamsson T, Bailey M, et al. How do probiotics and prebiotics function at distant sites? Benef Microbes. 2017;8(4):521–33.
Chen Q, Zhao B, Wang MY, et al. Associations between the red blood cell distribution width and primary angle-closure glaucoma: a potential for disease prediction. EPMA J. 2019;10(2):185–93.
Li S, Qiu Y, Yu J, et al. Association of systemic inflammation indices with visual field loss progression in patients with primary angle-closure glaucoma: potential biomarkers for 3P medical approaches. EPMA J. 2021;12(4):659–75.
Zhang Q, Wang N, Rui Y, et al. New insight of metabolomics in ocular diseases in the context of 3P medicine. EPMA J. 2023;14(1):53–71.
Zhang F, Zhao Y, Cao M, et al. The potential role of long noncoding RNAs in primary open-angle glaucoma. Graefe’s Arch Clin Exp Ophthalmol. 2021;259(12):3805–14.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):1–21.
Schmittgen TI, Ivak KJ. Analyzing real-time PC’R data by the comparative (2 (“ I’) method: J 1. Nat Protoc. 2008;3(6):1101–8.
Jangi S, Gandhi R, Cox LM, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun. 2016;7:12015.
Scher JU, Littman DR, Abramson SB. Microbiome in inflammatory arthritis and human rheumatic diseases. Arthritis Rheumatol. 2016;68(1):35–45.
Cryan JF, O’riordan KJ, Cowan CSM, et al. The microbiota-gut-brain axis. Physiol Rev. 2019;99(4):1877–2013.
Zhang Y, Zhou X, Lu Y. Gut microbiota and derived metabolomic profiling in glaucoma with progressive neurodegeneration. Front Cell Infect Microbiol. 2022;12:968992.
Skrzypecki J, Izdebska J, Kamińska A, et al. Glaucoma patients have an increased level of trimethylamine, a toxic product of gut bacteria, in the aqueous humor: a pilot study. Int Ophthalmol. 2021;41(1):341–7.
Mcpherson ZE, Sørensen HT, Horváth-Puhó E, et al. Irritable bowel syndrome and risk of glaucoma: an analysis of two independent population-based cohort studies. United Eur Gastroenterol J. 2021;9(9):1057–65.
Cartwright T, Perkins ND, L Wilson C. NFKB1: a suppressor of inflammation, ageing and cancer. FEBS J. 2016;283(10):1812–22.
O’keeffe M, Grumont RJ, Hochrein H, et al. Distinct roles for the NF-kappaB1 and c-Rel transcription factors in the differentiation and survival of plasmacytoid and conventional dendritic cells activated by TLR-9 signals. Blood. 2005;106(10):3457–64.
Cheng CS, Feldman KE, Lee J, et al. The specificity of innate immune responses is enforced by repression of interferon response elements by NF-κB p50. Sci Signal. 2011;4(161):ra11.
Saber MM, Monir N, Awad AS, et al. TLR9: A friend or a foe. Life Sci. 2022;307:120874.
Ding P, Tan Q, Wei Z, et al. Toll-like receptor 9 deficiency induces osteoclastic bone loss via gut microbiota-associated systemic chronic inflammation. Bone Res. 2022;10(1):42.
Sameer AS, Nissar S. Toll-like receptors (TLRs): structure, functions, signaling, and role of their polymorphisms in colorectal cancer susceptibility. Biomed Res Int. 2021;2021:1157023.
Vernazza S, Tirendi S, Bassi AM, et al. Neuroinflammation in primary open-angle glaucoma. J Clin Med. 2020;9(10):3172.
Kong Q, Hao Y, Li X, et al. HDAC4 in ischemic stroke: mechanisms and therapeutic potential. Clin Epigenetics. 2018;10(1):117.
Schlüter A, Aksan B, Fioravanti R, et al. Histone deacetylases contribute to excitotoxicity-triggered degeneration of retinal ganglion cells in vivo. Mol Neurobiol. 2019;56(12):8018–34.
Zhang Q-Y, Wang Z-J, Sun D-M, et al. Novel therapeutic effects of leonurine on ischemic stroke: new mechanisms of BBB integrity. Oxid Med Cell Longev. 2017;2017:7150376.
Yasuda K, Nakanishi K, Tsutsui H. Interleukin-18 in health and disease. Int J Mol Sci. 2019;20(3):649.
Macia L, Tan J, Vieira AT, et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun. 2015;6:6734.
Micera A, Quaranta L, Esposito G, et al. Differential protein expression profiles in glaucomatous trabecular meshwork: an evaluation study on a small primary open angle glaucoma population. Adv Ther. 2016;33(2):252–67.
Bell K, Und Hohenstein-Blaul NVT, Teister J, et al. Modulation of the immune system for the treatment of glaucoma. Curr Neuropharmacol. 2018;16(7):942–58.
Sihota R, Goyal A, Kaur J, et al. Scanning electron microscopy of the trabecular meshwork: understanding the pathogenesis of primary angle closure glaucoma. Indian J Ophthalmol. 2012;60(3):183–8.
Yin Y, Cui Q, Li Y, et al. Macrophage-derived factors stimulate optic nerve regeneration. J Neurosci. 2003;23(6):2284–93.
Yeghiazaryan K, Flammer J, Wunderlich K, et al. An enhanced expression of ABC 1 transporter in circulating leukocytes as a potential molecular marker for the diagnostics of glaucoma. Amino Acids. 2005;28(2):207–11.
Stamer WD, Clark AF. The many faces of the trabecular meshwork cell. Exp Eye Res. 2017;158:112–23.
Yu AL, Fuchshofer R, Kampik A, et al. Effects of oxidative stress in trabecular meshwork cells are reduced by prostaglandin analogues. Invest Ophthalmol Vis Sci. 2008;49(11):4872–80.
Snider EJ, Hardie BA, Li Y, et al. A porcine organ-culture glaucoma model mimicking trabecular meshwork damage using oxidative stress. Invest Ophthalmol Vis Sci. 2021;62(3):18.
Shikata F, Shimada K, Sato H, et al. Potential influences of gut microbiota on the formation of intracranial aneurysm. Hypertension. 2019;73(2):491–6.
Na YR, Stakenborg M, Seok SH, et al. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol. 2019;16(9):531–43.
Kim MS, Kim Y, Choi H, et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut. 2020;69(2):283–94.
Schulthess J, Pandey S, Capitani M, et al. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity. 2019;50(2):432-445.e7.
Wu K, Yuan Y, Yu H, et al. The gut microbial metabolite trimethylamine N-oxide aggravates GVHD by inducing M1 macrophage polarization in mice. Blood. 2020;136(4):501–15.
Torres Crigna A, Link B, Samec M, et al. Endothelin-1 axes in the framework of predictive, preventive and personalised (3P) medicine. EPMA J. 2021;12(3):265–305.
Baek SU, Lee WJ, Park KH, et al. Health screening program revealed risk factors associated with development and progression of papillomacular bundle defect. EPMA J. 2021;12(1):41–55.
Golubnitschaja O, Yeghiazaryan K, Flammer J. Key molecular pathways affected by glaucoma pathology: is predictive diagnosis possible? EPMA J. 2010;1(2):237–44.
Pivovarova-Ramich O, Zimmermann HG, Paul F. Multiple sclerosis and circadian rhythms: Can diet act as a treatment? Acta Physiol (Oxf). 2023;237(4):e13939.
Plotnikov D, Huang Y, Khawaja AP, et al. High blood pressure and intraocular pressure: a mendelian randomization study. Invest Ophthalmol Vis Sci. 2022;63(6):29.
Trott M, Smith L, Veronese N, et al. Eye disease and mortality, cognition, disease, and modifiable risk factors: an umbrella review of meta-analyses of observational studies. Eye (Lond). 2022;36(2):369–78.
Golubnitschaja O, Liskova A, Koklesova L, et al. Caution, “normal” BMI: health risks associated with potentially masked individual underweight-EPMA position paper 2021. EPMA J. 2021;12(3):243–64.
Funding
This research was funded by grants from the National Natural Science Foundation of China (no. 81974134 awarded to prof. Xiaobo Xia and no. 81974137 awarded to prof. Siqi Xiong) and the National Key Research and Development Program of China (no. 2020YFC2008205 awarded to prof. Xiaobo Xia).
Author information
Authors and Affiliations
Contributions
Conceptualization, Si Chen; funding acquisition, Siqi Xiong and Xiaobo Xia; investigation, Siqi Xiong and Xiaobo Xia; methodology, Si Chen; project administration, Xiaobo Xia; software, Nan Wang; supervision, Siqi Xiong and Xiaobo Xia; validation, Si Chen and Nan Wang; writing—original draft, Si Chen and Nan Wang; writing—review and editing, Siqi Xiong and Xiaobo Xia. All authors will be informed about each step of the manuscript processing including submission, revision, and revision reminder via emails from our system or assigned assistant editor. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, S., Wang, N., Xiong, S. et al. The correlation between primary open-angle glaucoma (POAG) and gut microbiota: a pilot study towards predictive, preventive, and personalized medicine. EPMA Journal 14, 539–552 (2023). https://doi.org/10.1007/s13167-023-00336-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13167-023-00336-2