
Abstract
Polymastiidae Gray, 1867 is a worldwide distributed sponge family, which has a great significance for understanding of the demosponge deep phylogeny since the former order Hadromerida Topsent, 1894 has been recently split based on the molecular evidence and a new separate order has been established for the polymastiids. However, molecular data obtained from Polymastiidae so far are scarce, while the phylogenetic reconstruction based on morphology has faced a deficit of characters along with the vagueness of their states. The present study is a phylogenetic reconstruction of Polymastiidae based on novel data on two molecular markers, cytochrome oxidase subunit I and large subunit ribosomal DNA, obtained from a broad set of species. Monophyly of the family and nonmonophyly of four polymastiid genera are revealed, suggesting a high level of homoplasy of morphological characters, which are therefore not an appropriate base for the natural classification of Polymastiidae. Although the presented phylogenies cannot yet provide an alternative classification scheme, several strongly supported clades, which may be used as reference points in future classification, are recovered and three taxonomic actions are proposed: transfer of one species from Radiella to Polymastia Bowerbank, 1862; transfer of three species from Radiella Schmidt, 1870 to Spinularia Gray, 1867; and the consequent abandonment of Radiella.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Polymastiidae Gray (1867), with its 122 species from 15 genera and a worldwide distribution (Van Soest et al. 2015), is one of the key families in Demospongiae Sollas, 1885, the most diverse class of sponges. At the same time, Polymastiidae is one of the problematic taxa with a controversial classification (Plotkin et al. 2012). Classification of the demosponges has traditionally been based on the shape and arrangement of their skeletal elements, i.e., mineral spicules and organic fibers (Hooper and Van Soest 2002). The polymastiids possess a relatively simple spicule assortment providing a rather scant set of taxonomic characters (Plotkin et al. 2012). Polymastiidae comprises sponges of various body shapes, often bearing papillae and possessing a skeleton mainly composed of smooth monactines (Boury-Esnault 2002; see terminology of the sponge morphology in Boury-Esnault and Rützler 1997). Based on the latter feature, this family was until recently affiliated with the demosponge order Hadromerida Topsent, 1894. For the moment, only one morphological feature delimiting Polymastiidae from other demosponges is usually defined, the presence of a superficial cortical palisade made of spicules differing from those composing the choanosomal tracts in size and/or in shape (Boury-Esnault 2002; Plotkin and Janussen 2008). However, this feature is in fact also displayed by some taxa from other families, e.g., by Aaptos Gray, 1867 belonging to the Suberitidae Schmidt, 1870 (Plotkin et al. 2012).
Discrimination between polymastiid genera is based on the body shape (e.g., radial body in Radiella Schmidt, 1870 and columnar body in Tentorium Vosmaer, 1887), the architecture of the choanosomal skeleton (diffuse skeleton in Quasillina Norman, 1869 and Ridleia Dendy, 1888, reticulate skeleton in Weberella Vosmaer, 1885 and radial skeleton in the remaining 12 genera) and the presence of spicules other than the ordinary smooth monactines in the choanosome (in four genera), or in the cortex (in five genera) (Boury-Esnault 2002). However, in some cases, these characters are inconsistent. For example, Polymastia Bowerbank, 1862 is usually defined as sponges with a radial choanosomal skeleton and smooth monactines constituting both the choanosomal and cortical skeleton (Boury-Esnault 2002) even though several species traditionally affiliated with Polymastia display a reticulate skeleton (Plotkin et al. 2012) or extraordinary spicules in the choanosome or in the cortex (Kelly-Borges and Bergquist 1997). Other characters used in the taxonomy of polymastiids include the number of size categories of the ordinary monactines and the minute differences in their shape, the presence and architecture of additional cortical layers, and the anatomy of the papillae (Boury-Esnault 1987, 2002; Kelly-Borges and Bergquist 1997; Morrow and Boury-Esnault 2000; Plotkin and Janussen 2008). These characters are often unstable and provide poor taxonomic information (Plotkin et al. 2012). Particularly, they fail to discriminate between some morphologically similar polymastiids, which inhabit the polar and temperate waters of the northern and southern hemispheres, but do not occur in the tropics, and consequently, a bipolar distribution is presumed for these species (e.g., for Tentorium semisuberites (Schmidt 1870) and Radiella sarsi (Ridley & Dendy, 1886)).
Phylogenetic reconstruction of Polymastiidae based on 25 binary morphological characters (Plotkin et al. 2012) questioned the monophyly of the family, with Pseudotrachya hystrix (Topsent, 1890) not grouping with any other polymastiid and one of the outgroup species, Aaptos papillata (Keller, 1880), joining the main polymastiid clade, and demonstrated that Polymastia is polyphyletic. At the same time, molecular phylogenies of Polymastiidae have been never properly reconstructed. Until now, common phylogenetic markers as the barcoding regions of cytochrome oxidase subunit I (CO1) and the partial RNA from the large and small ribosomal subunits (28S and 18S) have only been obtained for a small number of polymastiid species, aiming to resolve a deep phylogeny of the class Demospongiae instead of addressing the relationships within Polymastiidae (Nichols 2005; Morrow et al. 2012, 2013; Redmond et al. 2013; Vargas et al. 2015). In all phylogenies resulting from these studies, Polymastia was nonmonophyletic, while the family Polymastiidae was monophyletic excluding two species from Nichols (2005). Furthermore, in all molecular phylogenies of Demospongiae, Polymastiidae and other hadromerid families appeared in remote clades that seriously contradicted the traditional classification based on their morphological similarities. Very recently, based on the molecular data, Morrow and Cárdenas (2015) proposed abandoning the order Hadromerida and establishing five new orders for the former hadromerids, with the order Polymastiida including only one family, the Polymastiidae. This proposal highlights the importance of the polymastiids in the context of the deep phylogeny of demosponges.
The purpose of the present study was to reconstruct the phylogeny of the family Polymastiidae based on two broadly used molecular markers, the 5′-end barcoding region of CO1 (Folmer et al. 1994) and a large region of 28S rRNA (helix B10 to helix E19, numeration of the helices according to De Rijk et al. (1999, 2000) and Wuyts et al. (2001)), employing a much larger set of polymastiid species than ever studied before. We also tested the monophyly of the family as well as the monophyly of its genera and traced the evolution of morphological characters along the branches of the consensus molecular tree.
Material and methods
Sampling and taxonomic identification
Eighty-seven polymastiid individuals were collected for our study and deposited in the natural history collections of four museums (see Table 1 for details). Both the individuals in toto assigned for morphological examination and the choanosomal pieces of about 1 cm3 for DNA extraction were fixed in 95–100 % ethanol. Sponge anatomy was examined under a light microscope on 500–700-μm-thick sections prepared using a precise saw with a diamond wafering blade after embedding of tissue pieces in epoxy resin. Isolated spicules were examined under a light microscope and SEM. Preparations and subsequent taxonomic identification followed well-known routines for polymastiids (Boury-Esnault 1987; Boury-Esnault and Bézac 2007; Plotkin and Janussen 2007, 2008).
Taxonomic scope
In our study, we included genetic data on 24 unambiguously identified species and ten operational taxonomic units (OTUs), of which four were identified to species level with some uncertainty and six could not be referred to any known species and were therefore only identified to genus level (Table 1). These species and OTUs belonged to seven polymastiid genera. Each genus was represented at least by the type species except for Radiella Schmidt, 1870, the type species of which, Radiella sol Schmidt, 1870, was unavailable and had an ambiguous status (see “Discussion”). Sequences from 19 species and nine OTUs were novel. Data on two species and one OTU were taken from GenBank and sequences from three species were both obtained by us and taken from GenBank. Two species were chosen as outgroups, the suberitid Suberites ficus (Johnston, 1842) and the tethyid Tethya citrina Sarà & Melone, 1965. Data on both species were taken from GenBank. This selection was based on the substantial morphological affinities between Suberitidae, Tethyidae Gray, 1848, and Polymastiidae and on the former affiliation of these three families with the order Hadromerida.
DNA extraction, amplification, and sequencing
DNA extractions were made with Qiagen DNeasy Blood and Tissue Minikits or DNeasy Plant Minikits following the manufacturer’s protocols (the latter was found to yield DNA of higher quantity and purity).
CO1 barcoding regions were amplified with the Ex Taq polymerase (TaKaRa) and the amplification program from Vargas et al. (2012). For most species and OTUs, we used the primers dgLCO1490/dgHCO2198 (Meyer 2003), which are slight modifications of the universal primers LCO1490/HCO2198 (Folmer et al. 1994). For one species, Polymastia corticata Rildey & Dendy, 1886, CO1 sequences of satisfactory quality could be obtained only with the primers jgLCO1490/jgHCO2198 (Geller et al. 2013).
Amplification of the partial 28S ribosomal DNA (rDNA) was performed with three pairs of primers designed by Morrow et al. (2012): Por28S-15F/Por28S-878R for sequencing of the region coding RNA from helix B10 to helix C1, Por28S-830F/Por28S-1520R for the region coding from D1 to D19, and Por28S-1490F/Por28S-2170R for the region coding from D20 to E18–E19. For most species, the amplification of this DNA piece succeeded with the Ex Taq polymerase and a “touchdown” program reported by Morrow et al. (2012), which was optimized by the doubling of the sequence extension time. In the cases of Polymastia thielei Koltun, 1964 and Polymastia uberrima (Schmidt, 1870), the amplification succeeded only with the LA Taq (TaKaRa) and the following protocol: 94 °C for 3 min (94 °C for 30 s, 50 °C for 45 s, 72 °C for 1 min) × 35 cycles, 72 °C for 7 min. Quality and quantity of the PCR products were estimated by agarose gel electrophoresis. The PCR products were purified with exonuclease 1 and shrimp alkaline phosphatase as described by Eilertsen and Malaquias (2013) and used for sequencing reactions with BigDye terminator 3.1 (Applied Biosystems, Waltham, MA, USA) following the protocol of the producer. Subsequent sequence reads were performed with an automated ABI 3730XL DNA Analyser (Applied Biosystems) in the University of Bergen.
Contigs were assembled with the application SeqMan of DNASTAR Lasergene 8.0 and manually checked for sequencing errors. The consensus sequences of contigs were trimmed to remove primer residuals and checked by nucleotide BLAST search (Altschul et al. 1990) against GenBank sequences to verify their polymastiid origin. Where BLAST searches revealed epi- or endofaunal contaminations, the PCR products were cloned using StrataClone PCR cloning kit (Agilent Technologies, StrataGene Products Division, Santa Clara, CA, USA) according to the manufacturer’s instructions, and 10–20 clones per product were sequenced by LGC Genomics GmbH (Berlin, Germany). If the direct sequence reads displayed double signals, extractions from the respective samples were repeated once again and the PCRs were repeated two or more times in order to exclude eventual cross-contamination and PCR errors. If these repetitive procedures confirmed the double signals in the reads, PCR products were cloned following the same protocol as used for the separation of the native and contaminating DNA fragments. The resulting clones were checked for errors, e.g., as those reported by Speksnijder et al. (2001) and Acinas et al. (2005), against the alignment of the approved direct sequences. Clones with unique nucleotides or gaps in the conservative sites were disregarded. Polymorphism in the remaining clones was regarded as natural. Strict consensuses of the clones with the polymorphic sites encoded with IUPAC symbols were employed in the main phylogenetic analyses. Altogether we submitted 75 CO1 sequences and 236 sequences with the three regions of 28S rDNA including clone libraries (different versions from the same individuals) to GenBank (Table 1).
Alignments
All alignments were performed in SeaView 4.3.4 (Galtier et al. 1996; Gouy et al. 2010). CO1 sequences comprising exactly 658 bp each were aligned manually. The sequences of 28S rDNA varied in length. B10–C1 regions were 799–822 bp long, D1–D19 649–653 bp, and D20–E19 646–648 bp. Sequences of different regions were concatenated with 19 overlapping nucleotides, and their preliminary alignment was performed with the MUSCLE algorithm (Edgar 2004) implemented in SeaView. The initial alignment was further refined manually under consideration of the RNA secondary structure (Erpenbeck et al. 2007a, b, 2008). The GenBank sequence of Polymastia pachymastia de Laubenfels, 1932 (accession AY561924), being the longest (3550 bp) polymastiid sequence of 28S rDNA published so far, was used as a template for adapting the secondary structures reconstructed from other families to Polymastiidae. A 90 % consensus of all sequences employed in our study was then adjusted to this template. The resulting alignment was 2155 sites long. Because no secondary structure was proposed for the highly variable C-region (Erpenbeck et al. 2007a, b, 2008) comprising positions 406–813 in our alignment, we treated all sites flanked by the C1-helix strands as single stranded. Main length variation occurred in the C-region (from 366 to 396 bp) and in the terminal loop on helix D5 (alignment positions 940–948, variation from 4 to 8 bp). Search for unambiguously aligned sites was initially performed in GBlocks 0.91b (Castresana 2000) as implemented in SeaView. That excluded 51 sites. However, the resulting set was manually extended to exclude in total only 43 sites, because some obviously homologous sites were neglected by the algorithm. All alignments (CO1 matrix, 28S rDNA complete matrix, and 28S rDNA matrix reduced by 43 sites) were deposited at TreeBase and are available at http://purl.org/phylo/treebase/phylows/study/TB2:S18487. Some descriptive statistics of the alignments was explored with MEGA7 (Nei and Kumar 2000; Kumar et al. 2016).
Selection of substitution models and phylogenetic analyses
For all computing procedures, identical sequences were collapsed into one sequence that is indicated by the sequence labels of the taxa represented in the trees (Figs. 1, 2, and 3). Five datasets, CO1 data alone (35 unique polymastiid sequences), two 28S rDNA matrices (49 sequences), the complete one and the matrix reduced by 43 ambiguously aligned sites, and two corresponding concatenated matrices (47 sequences), CO1 + complete 28S rDNA and CO1 + reduced 28S rDNA, were analyzed. Search for the best fitting substitution model for the CO1 dataset carried out with both hierarchical likelihood ratio tests and Akaike information criterion (AIC) in MrModeltest 2.0 (Nylander 2004) selected GTR+G+I. In all analyses, the CO1 data were split into two partitions, codon positions 1 + 2 and codon position 3. For RNA-specific models for the 28S rDNA datasets, we applied the model selection procedure implemented in PHASE 3.0 (Allen and Whelan 2014), a recent modification of PHASE 2.0 (Gowri-Shankar and Jow 2006), that included running the script “model_selection.pl” (Allen and Whelan 2014). As an input tree topology for this procedure, we used a ML tree calculated in PhyML (Guindon et al. 2010) under the best fitting standard model, GTR+G, determined with AIC in JModelTest 2.1.6 (Guindon and Gascuel 2003; Darriba et al. 2012). The mixed model RNA16C+G for helix positions and REV+G for loop positions was selected as the best fit. Analyses of the concatenated datasets CO1 + 28S rDNA were run under the mixed model comprising the models selected for the single gene matrices. All datasets were analyzed in a Bayesian inference framework, with MrBayes 3.2 (Ronquist et al. 2011) for the CO1 matrix and with PHASE 3.0 (Allen and Whelan 2014) for the 28S rDNA matrices and the concatenated matrices CO1 + 28S rDNA, and in a maximum likelihood framework (ML) with RAxML 8.1.24 (Stamatakis 2014).

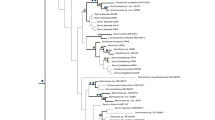
Bayesian consensus tree reconstructed from the reduced 28S rDNA matrix. Inset: a fragment of the comparable Bayesian consensus tree reconstructed from the complete 28S rDNA matrix, displaying better resolution inside Clade III. Nodal supports: upper values—Bayesian posterior probabilities, lower values—ML bootstrap supports in percents. Expansion of the branch labels denoting multiple specimens: Quasillina brevis (five specimens)—ZMBN 98049, ZMBN 98067, ZMBN 98082, ZMBN 98084, ZMBN 98090; Polymastia boletiformis (seven specimens)—BELUM: MC5014, GNM 901:1, ZMBN 98047, ZMBN 98048, ZMBN 98081, ZMBN 98088, ZMBN 98089; Polymastia euplectella (four specimens)—ZMBN 98044, ZMBN 98085, ZMBN 98086, ZMBN 98087; Polymastia penicillus (four specimens)—BELUM: MC5284, BELUM: MC6505, GNM 460:1, GNM 460:2

Bayesian consensus tree reconstructed from the CO1 matrix. Nodal supports: upper values—Bayesian posterior probabilities, lower values—ML bootstrap supports in percents. Expansion of the branch labels denoting multiple specimens: Polymastia arctica (five specimens)—ZMBN 98060, ZMBN 98062, ZMBN 98063, ZMBN 98065, ZMBN 98068; Polymastia andrica (five specimens)—ZMBN 98055, ZMBN 98057, ZMBN 98074, ZMBN 98102, ZMBN 98108; Polymastia grimaldii (three specimens)—ZMBN 98064, ZMBN 98110, ZMBN 98112; Radiella hemisphaerica (five specimens)—ZMBN 98043, ZMBN 98058, ZMBN 98069, ZMBN 98071, ZMBN 98077; Polymastia thielei (five specimens)—ZMBN 98052, ZMBN 98053, ZMBN 98070, ZMBN 98107, ZMBN 98109; Sphaerotylus capitatus (three specimens)—GNM 902, ZMBN 98042, ZMBN 98075; Quasillina brevis (five specimens)—ZMBN 98049, ZMBN 98067, ZMBN 98082, ZMBN 98084, ZMBN 98090; Radiella sarsi (two specimens)—ZMBN 98039, ZMBN 98098; Radiella sp. (three specimens)—ZMBN 98038, ZMBN 98040, ZMBN 98041; Spinularia spinularia (four specimens)—ZMBN 98037, ZMBN 98050, ZMBN 98076, ZMBN 98079

Bayesian consensus tree reconstructed from the concatenated dataset CO1 + reduced 28S rDNA of Polymastiidae. Inset: a fragment of the comparable Bayesian consensus tree reconstructed from the concatenated dataset CO1 + complete 28S rDNA, displaying better resolution inside Clade III. Nodal supports: upper values—Bayesian posterior probabilities, lower values—ML bootstrap supports in percents. Expansion of the branch label denoting five specimens of Quasillina brevis—ZMBN 98049, ZMBN 98067, ZMBN 98082, ZMBN 98084, ZMBN 98090
MrBayes 3.2 was run on the CIPRES (Cyberinfrastructure for Phylogenetic Research) Science Gateway V. 3.3 (https://www.phylo.org/) and on the Lifeportal at the University of Oslo using the high-performance computing cluster Abel (https://lifeportal.uio.no/). In the MrBayes 3.2 session, the model parameters were optimized independently for each partition. Two runs with eight chains each were launched under the default chain “temperatures” and flat Dirichlet distributions for the model parameter priors. The chains were sampled each 100 generations. The initial 2.5 million of the samples were disregarded in the burn-in. The convergence of the runs was controlled with the average standard deviation of split frequencies in MrBayes 3.2, while the sufficiency of the number of generations was estimated with the effective sample size (ESS) for all parameters in Tracer 1.5 (Rambaut and Drummond 2009). The convergence was reached and the ESSs exceeded 200 after ten million generations had been run.
PHASE analyses were performed on a desktop computer. Ten million iterations with sampling period 200 iterations after a burn-in of one million iterations were initially run. Each analysis was repeated twice, specifying a different random seed. After the output files had been transformed with the Perl script phase2tracer.pl (Voigt et al. 2012, modified from the script of Matt Yoder (https://github.com/mjy/phase-utils/blob/master/phase2tracer.pl)), the stabilization of all parameters was monitored in Tracer 1.5. If stabilization had not been achieved, the computations were repeated under optimized settings and with extra 5–30 million iterations.
RAxML 8.1.24 was run on the CIPRES. Search for the best scoring ML-tree along with rapid bootstrapping (1000 replicates) was performed. Because the model RNA16C is not implemented in RAxML 8.1.24, the more exhaustive model RNA16A was invoked for helix positions of the 28S rDNA data.
Bayesian analyses of the single-gene datasets revealed some incongruence between the CO1 and 28S rDNA phylogenies. To illustrate the conflicts, we repeated the analyses on the matrices with the identical set of 47 taxa for both CO1 and 28S rDNA (reduced matrix) and, based on the resulting consensus trees, computed a rooted galled network (Huson et al. 2009) with Dendroscope 3 (Huson and Scornavacca 2012) (Fig. 4). To explore these conflicts, we performed an incongruence length difference test (ILD, Farris et al. 1994) on the concatenated dataset CO1 + reduced 28S rDNA with all parsimony uninformative sites excluded running 500 replicates in PAUP* 4.0b10 (Swofford 2002). Furthermore, we used Bayes factor comparisons of the model likelihoods to test the conflicting topological hypotheses on the single-gene datasets following Kass and Raftery (1995). To obtain more accurate model likelihoods, stepping-stone samplings were performed in MrBayes 3.2. The monophyly of the congruent clades was constrained as recommended by Bergsten et al. (2013). Two runs with two chains each were launched. Four million generations on the CO1 data and ten million generations on the 28S rDNA were run to reach the convergence of the runs.

Rooted galled network compiled from the Bayesian consensus trees reconstructed from CO1 alone and 28S rDNA alone with identical sets of taxa. Bold curves indicate discrepancies in the topology. Expansion of the branch labels denoting multiple specimens: Spinularia spinularia (three specimens)—ZMBN 98037, ZMBN 98050, ZMBN 98076; Radiella sp. (three specimens)—ZMBN 98038, ZMBN 98040, ZMBN 98041; Polymastia euplectella (three specimens)—ZMBN 98044, ZMBN 98085, ZMBN 98086; Quasillina brevis (five specimens)—ZMBN 98049, ZMBN 98067, ZMBN 98082, ZMBN 98084, ZMBN 98090
Additionally, in order to examine the intragenomic polymorphism of the D1–D19 region of 28S rDNA, a dataset comprising all versions of this region in three species of Polymastia, Polymastia andrica de Laubenfels, 1949, Polymastia arctica (Merejkowsky, 1878), and Polymastia grimaldii (Topsent, 1913), with Polymastia mamillaris (Müller, 1806) as the outgroup taxon, was analyzed with Minimum-spanning network algorithm (Bandelt et al. 1999) implemented in PopArt 1.7 (http://popart.otago.ac.nz) and in a ML framework with PhyML (Guindon et al. 2010). Consensus trees resulting from the Bayesian analyses along with the ML-tree illustrating the intragenomic polymorphism were deposited at TreeBase and are available at http://purl.org/phylo/treebase/phylows/study/TB2:S18487.
Tracing of the evolution of morphological characters
The consensus tree resulting from the Bayesian analysis of the concatenated dataset CO1 + reduced 28S rDNA was chosen for tracing of the morphological evolution. Branches corresponding to different individuals of the same species or OTU were collapsed. A matrix with 21 morphological characters of the respective 30 polymastiid taxa and two outgroup taxa was built based on the dataset for phylogenetic scenario 3 from Plotkin et al. (2012), but with two emendations: the multistate character “Growth pattern” was assigned by the amalgamation of four binary characters and the character “Longitudinal tracts of principal monactines in the cortex” (presence/absence) was excluded since in the matrix used in this study the state “present” was autapomorphy of Quasillina brevis (Bowerbank, 1866) (Online Resources 1–3). The ancestral state reconstruction with parsimony criterion for each character was performed in Mesquite 3.04 (Maddison and Maddison 2015), while the consistency indices were computed in PAUP* 4.0b10 (Swofford 2002).
Results
Statistics of alignments
Parsimony-informative sites comprised almost 25 % of all sites in the CO1 matrix and about 15 % of all sites in the 28S rDNA matrices. Sites with intragenomic polymorphisms comprised ca. 1 and 0.7 % of all sites in the 28S rDNA complete matrix and reduced matrix, respectively (Table 2).
Congruent aspects of the CO1 and 28S rDNA phylogenies
Separate analyses of CO1 and 28S rDNA resulted in similar overall phylogenies (Figs. 1 and 2) except for few conflicts (see the section “Incongruence between the CO1 and 28S rDNA phylogenies” below). The phylogenies were not affected by intraspecific or intragenomic polymorphisms except for the relationships within three small terminal subclades (see the section “Intraspecific and intragenomic polymorphism” below). All analyses supported the monophyly of the polymastiids against the two outgroups (Figs. 1, 2, and 3). At the same time, the polymastiid genera Polymastia, Radiella, Sphaerotylus Topsent, 1898, and Tentorium Vosmaer, 1887 were nonmonophyletic. Polymastia spp. were scattered over different clades, Radiella hemisphaerica (Sars, 1872) fell distantly from other Radiella spp., Sphaerotylus borealis (Swarczewsky, 1906) lay remotely from its congeners, and Tentorium papillatum (Kirkpatrick, 1908) fell on a long branch as the sister group to a clade of the remaining polymastiids. Moreover, in the 28S rDNA tree, the type species of Tentorium, Ten. semisuberites, and Ten. cf. semisuberites did not group together, although the support for their nonmonophyly was very weak (Fig. 1). Unfortunately, no CO1 data from Ten. cf. semisuberites were obtained. Three clades of species (highlighted in Figs. 1, 2, 3, and 4) were recovered by all analyses.
Clade I comprised Polymastia andrica, P. arctica, P. grimaldii, P. mamillaris (type species of Polymastia), P. thielei, P. uberrima, and Radiella hemisphaerica. The support for this clade in the 28S rDNA tree was slightly weaker (Fig. 1) than in the CO1 tree (Fig. 2). Analyses of the 28S rDNA alone and the concatenated datasets CO1 + 28S rDNA supported the sister relationships between the pair Polymastia sp. 1 + Polymastia sp. 2 and Clade I (Figs. 1 and 3). In the CO1 tree, Polymastia sp. 1 and Polymastia sp. 2 grouped with Polymastia sp. 3, and the position of this trio as the sister to Clade I had very weak support (Fig. 2). Unfortunately, no 28S rDNA was obtained from Polymastia sp. 3. Inside Clade I, P. uberrima was the sister to the subclade of the remaining six species strongly supported by all analyses (Figs. 1, 2, and 3). In its turn, this subclade split up into two groupings, P. mamillaris + P. thielei + R. hemisphaerica and P. andrica + P. arctica + P. grimaldii. The pair P. thielei + R. hemisphaerica was supported by all analyses (Figs. 1, 2, and 3). But the grouping of P. mamillaris as the sister to this pair was strongly supported only by the analyses of 28S rDNA alone (Fig. 1) and the concatenated datasets (Fig. 3), while the analysis of CO1 alone demonstrated just a very weak support for this relationship (Fig. 2). The grouping P. andrica + P. arctica + P. grimaldii was supported in all analyses (Figs. 1, 2, and 3), but the relationships between these three species were unresolved in the 28S rDNA tree (Fig. 1) because of the intraspecific and intragenomic polymorphism (see the respective section below) and resolved with just a low support for P. andrica + P. arctica in the CO1 tree (Fig. 2).
Clade II comprised Sphaerotylus capitatus (Vosmaer, 1885) (type species of Sphaerotylus), Sphaerotylus sp. 1 and Sphaerotylus sp. 2. This clade was strongly supported in all trees (Figs. 1, 2, and 3). Sph. capitatus and Sphaerotylus sp. 1 had identical CO1 and were sisters in the 28S rDNA tree with strong support (Fig. 1).
Clade III comprised Radiella sarsi, Radiella cf. sarsi, Radiella sp., and Spinularia spinularia (Bowerbank, 1866) (type species of Spinularia Gray, 1867). This clade was strongly supported in all trees (Figs. 1, 2, and 3). In the CO1 tree, Clade III and Tentorium semisuberites were sisters with strong support (Fig. 2), but this was not confirmed by the analyses of 28S rDNA alone (Fig. 1). R. sarsi, Radiella sp., and Spi. spinularia had identical CO1 and formed a strongly supported subclade in the 28S rDNA tree (Fig. 1). In the same tree, there was also some support for the sister relationships between Radiella sp. and Spi. spinularia. Spi. spinularia was represented by two groups of individuals that differed from each other by two nucleotides in 28S rDNA and each of them differed from Radiella sp. (identical sequences from three individuals) by 11 nucleotides. Three of these 11 nucleotides were located in the 43 sites excluded as ambiguously aligned in the matrix as a whole. The exclusion of these sites from the analyses led to a polytomy formed by Radiella sp. and the two groups of individuals of Spi. spinularia (main trees in Figs. 1 and 3). However, within Clade III, these excluded sites could be aligned unambiguously and provided a sufficient phylogenetic signal to resolve the polytomy (insets in Figs. 1 and 3).
Furthermore, all analyses strongly supported the pair Polymastia boletiformis (Lamarck, 1815) + Q. brevis (type species of Quasillina) (Figs. 1, 2, and 3) and also revealed the grouping Sphaerotylus borealis + Polymastia cf. conigera Bowerbank, 1874 + Weberella bursa (Müller, 1806), although the latter was strongly supported only by the analyses of the concatenated data CO1 + 28S rDNA (Fig. 3), while its support in the single-gene trees was very weak (Figs. 1 and 2). Within this grouping, P. cf. conigera and W. bursa were sisters with a strong support in the CO1 tree (Fig. 2), but a much weaker support in the 28S rDNA tree (Fig. 1). At the same time, the analyses of the 28S rDNA alone strongly supported the pair Polymastia euplectella Rezvoj, 1927 + Polymastia penicillus (Montagu, 1818) (Fig. 1), while the support for this pair in the CO1 tree was negligible (Fig. 2).
Incongruence between the CO1 and 28S rDNA phylogenies
ILD test of the concatenated dataset CO1 + 28S rDNA rejected the hypothesis of congruent data with a p value of 0.002. The conflicts between the single gene phylogenies are visualized as reticulations in the galled network (Fig. 4). One conflict concerned dissimilarity in the relationships between Polymastia boletiformis + Quasillina brevis and other taxa. In the CO1 tree, this pair was the sister to Polymastia invaginata Kirkpatrick, 1907 (Fig. 2), whereas in the 28S rDNA tree, it was the sister to the grouping Clade I + Polymastia sp. 1 + Polymastia sp. 2 (Fig. 1). Bayesian support for the indicated relationships was strong in each gene tree, and they were not affected by polymorphism in any of the species. Bayes factor tests revealed no support for the alternative hypothesis in either of the two topologies (Table 3). Two conflicts were due to the low resolution of the CO1 tree. In this tree, five clades formed an unresolved polytomy with Polymastia corticata, while Sphaerotylus antarcticus Kirkpatrick, 1907 was the sister to Clade II, although with very weak Bayesian support (Fig. 2). Conversely, in the 28S rDNA tree, P. corticata was the sister to Clade II, and Sph. antarcticus in its turn was the sister to P. corticata + Clade II with strong support (Fig. 1). The conflicts between the CO1 and 28S rDNA phylogenies caused by the gene polymorphism were revealed within three small terminal subclades, the trio Polymastia andrica + P. arctica + P. grimaldii in Clade I, the pair P. boletiformis + Q. brevis, and the group of three individuals of P. invaginata (Fig. 4). These conflicts are considered below.
Intraspecific and intragenomic polymorphism
The most conspicuous intraspecific polymorphism was revealed in four sites of the B10–C1 region (positions 578–580 and 583 in the complete matrix) and in seven sites of the D1–D19 region (positions 941–943, 947–948, and 1294–1295) of 28S rDNA in Polymastia andrica, P. arctica, and P. grimaldii. The variation within B10–C1 was estimated on the direct sequences. The sequences of this region from three P. andrica were identical, while P. arctica displayed a polymorphism—individual ZMBN 98063 differed from P. andrica just by one ambiguity, individual ZMBN 98068 by three nucleotides, and two individuals, ZMBN 98060 and ZMBN 98062, by four nucleotides. P. grimaldii ZMBN 98064 differed from the latter two individuals of P. arctica just by one ambiguity. The variation within D1–D19 was estimated on four direct sequences (two from P. andrica and two from P. arctica) and 18 clones from five individuals (one of P. andrica, three of P. arctica, and one of P. grimaldii). Of the seven polymorphic sites, five were parsimony-informative when the sequences were aligned with the corresponding DNA region of P. mamillaris as the outgroup (see the respective alignment at http://purl.org/phylo/treebase/phylows/study/TB2:S18487). Here an intragenomic polymorphism was discovered. Seven versions of D1–D19 were spread among the individuals of different species (Fig. 5):

Minimum-spanning network reconstructed from the dataset of the clones and direct sequences of the D1–D19 region, 28S rDNA from Polymastia andrica, P. arctica, and P. grimaldii with P. mamillaris as outgroup. Numbers denote gene versions described in the text. Hatch marks on the branches indicate mutations
-
Version 1 (in three P. andrica, three P. arctica, and the only P. grimaldii employed in this analysis)
-
Version 2 (in one P. andrica and one P. arctica)
-
Version 3 (in one P. arctica and P. grimaldii)
-
Version 4 (only in P. grimaldii)
-
Version 5 (in one P. andrica, two P. arctica, and P. grimaldii)
-
Version 6 (in one P. andrica, two P. arctica, and P. grimaldii)
-
Version 7 (in one P. andrica and one P. arctica)
While the 28S rDNA sequences of P. andrica, P. arctica, and P. grimaldii displayed intraspecific and intragenomic polymorphism, the CO1 data from these species were consistent, i.e., the sequences from the individuals of the same species were identical. Based on these data, P. andrica and P. arctica were sisters, although the support for this relationship was weak (Fig. 2).
Among other species, intraspecific polymorphism was revealed in Polymastia boletiformis and P. invaginata. All seven P. boletiformis studied had identical 28S rDNA and grouped with Quasillina brevis (Fig. 1). CO1 was obtained only from two individuals of P. boletiformis, of which one, ZMBN 98047, differed from Q. brevis just by one nucleotide in this gene, while the other, ZMBN 98089, differed from Q. brevis by six nucleotides, that resulted in a grouping of ZMBN 98047 with Q. brevis instead of the conspecific ZMBN 98089 (Fig. 2). Cloning of the PCR products confirmed these relationships. Two individuals of P. invaginata, ZMBN 98093 and ZMBN 98094, had identical 28S rDNA, whereas individual ZMBN 98046 differed from them by two nucleotides (Fig. 1). Conversely, CO1 of ZMBN 98046 and ZMBN 98093 were identical, while ZMBN 98094 differed from them by 19 nucleotides (Fig. 2).
Homoplasy of the morphological characters
Only two characters were fully consistent—character 5, “Exhalant papillae”, and character 9, “Oscula on the body surface” (Online Resources 1–3). All polymastiid taxa studied possess exhalant papillae and lack oscula on the surface, i.e., these features are the synapomorphies of the polymastiid clade compared with the given outgroups. All other characters were more or less homoplastic (Online Resources 1–3).
Most polymastiids possess one or several well-developed exhalant papillae (characters 6 and 7 in Online Resources 1–3, Fig. 6a–c). Weakly developed papillae (Fig. 6d–e) are present in the taxa from three remote groupings (Fig. 6b)—in all species from Clade III + Tentorium semisuberites, in Sphaerotylus capitatus from Clade II and in Quasillina brevis from the pair Polymastia boletiformis + Q. brevis. Among these taxa, only Q. brevis and three species of Radiella, R. sarsi, R. cf. sarsi, and Radiella sp., always possess just single papillae (Fig. 6d), whereas Spinularia spinularia, Ten. semisuberites, and Sph. capitatus may have several exhalant papillae as all other polymastiids studied (Fig. 6e).

Key morphological characters of Polymastiidae: depiction of states and tracing of evolution along the Bayesian consensus tree reconstructed from the concatenated dataset CO1 + reduced 28S rDNA (the same as in Fig. 3, but with the branches corresponding to different individuals of the same species collapsed). a Evolution of the character “Number of exhalant papillae” (N 6 in Online Resources 1–3); b evolution of the character “Development of exhalant papillae” (N 7 in Online Resources 1–3); c numerous normally developed papillae in Polymastia bartletti ZMBN 98111 (University Museum of Bergen); d single weakly developed papilla of Radiella sp. ZMBN 98040 (sampled from the Norwegian Sea); e three weakly developed papillae in Spinularia spinularia (not deposited); f evolution of the character “Exotyles” (N 14 in Online Resources 1–3); g evolution of the character “Marginal spicule fringe” (N 16 in Online Resources 1–3); h exotyles projecting above the cortex, histological section through the body of Sphaerotylus capitatus BMNH 10.1.1.1199 (paralectotype, Natural History Museum, London); i distal ornamentation of an exotyle, SEM image, preparation from Sphaerotylus capitatus ZMB 10855 (Museum für Naturkunde, Berlin); j prominent marginal spicule fringe bordering the body of Radiella hemisphaerica NHMO-B862 (holotype, Natural History Museum, University of Oslo); k evolution of the character “Main choanosomal skeleton” (N 17 in Online Resources 1–3); l evolution of the character “Arrangement of the regular choanosomal skeleton” (N 18 in Online Resources 1–3); m irregular and reduced choanosomal skeleton, histological section through the body of Quasillina brevis (not deposited); n regular radial choanosomal skeleton, histological section through the body of Polymastia arctica ZMBN 98068 (University Museum of Bergen); o regular reticulate choanosomal skeleton, histological section through the body of Weberella bursa (not deposited). Scale bars c–e: 1 cm, h: 0.1 mm; i: 0.01 mm; j: 1 cm; m–o: 1 mm
Exotyles, i.e., extraordinary spicules protruding above the sponge surface (character 14 in Online Resources 1–3), are present in three remote groupings (Fig. 6f). Within the Sphaerotylus borealis + Polymastia cf. conigera + Weberella bursa group, the first two species possess exotyles, whereas W. bursa does not. All three species of Clade II have exotyles (Fig. 6h–i). Of the two species grouping with Clade II in the 28S rDNA tree, one species, Sphaerotylus antarcticus, possesses exotyles, while the other, Polymastia corticata, has no exotyles. Among the species of Clade I, P. andrica possesses exotyles.
The presence of a marginal fringe of extra-long spicules (character 16 in Online Resources 1–3, Fig. 6g) is shared by all species of Clade III, but also recorded in two species falling into two different subclades of Clade I, Radiella hemisphaerica (Fig. 6j) and Polymastia grimaldii.
Most polymastiids share the presence of a well-developed and regular choanosomal skeleton with one of the outgroup species, Tethya citrina (character 17 in Online Resources 1–3, Fig. 6k). The only exception is Quasillina brevis (Fig. 6m), which shares the presence of an irregular and reduced choanosomal skeleton with the other outgroup taxon, Suberites ficus.
The main choanosomal skeleton in most polymastiids as well as in Tet. citrina is radial (character 18 in Online Resources 1–3, Fig. 6l, n). Reticulate choanosomal skeleton (Fig. 6o) is recorded in taxa from four remote groupings—in Weberella bursa (Sphaerotylus borealis + Polymastia cf. conigera + W. bursa), in Polymastia corticata (P. corticata + Clade II), in Polymastia boletiformis (P. boletiformis + Quasillina brevis), and in Polymastia thielei (Clade I).
Discussion
Monophyly of Polymastiidae and taxa of uncertain family affiliation
Our analyses demonstrated the monophyly of the clade formed by all polymastiid taxa studied when using a suberitid and a tethyid as outgroups, and these results are congruent with most previous studies (Morrow et al. 2012, 2013; Redmond et al. 2013; Vargas et al. 2015). However, it should be noted that due to the lack of reliable molecular data, our analyses did not consider the two taxa with uncertain family affiliation, the genus Pseudotrachya Hallmann, 1914 and the species Aaptos papillata, which seem to be important for understanding of the polymastiid early evolution. A. papillata is usually regarded as a suberitid (Van Soest et al. 2015), despite that it displays strong morphological similarities with Polymastiidae, and in the trees reconstructed from a morphological dataset by Plotkin et al. (2012), it formed a polytomy with Tentorium semisuberites and the clade of other polymastiids. Pseudotrachya was commonly regarded as a polymastiid genus (Boury-Esnault 2002). But the type species of this genus, P. hystrix, did not group with other polymastiids in the phylogenies based on morphological data (Plotkin et al. 2012), and similarly Pseudotrachya sp. fell outside the main polymastiid clade in the tree reconstructed from a 28S rDNA dataset (Nichols 2005). Meanwhile, the taxonomic identification in the latter study raised some doubts because in the same 28S rDNA tree, another polymastiid, Polymastia sp. 1, also did not group with the main polymastiid clade, while in the CO1 trees, Pseudotrachya sp., Polymastia sp. 1, and Polymastia sp. 2 appeared in different clades. Additionally, the 28S rDNA sequences of these three species recovered by Nichols (2005) were much shorter than those used in our study, and therefore, we did not include Nichols’ sequences in the analyses.
Molecular phylogenies contradict the morphology-based classification of polymastiids
Our most important outcome is the inapplicability of morphological characters, the majority of which has appeared to be highly homoplastic, for the phylogenetic reconstruction and hence for the natural classification of Polymastiidae. Homoplasy is a general problem in morphological taxonomy of the demosponges (e.g., Cárdenas et al. 2011; Morrow et al. 2013). Meanwhile, inside Polymastiidae, our study has recovered three clades strongly supported by the data from both genes studied. Each clade includes the type species of the certain genus, Clade I—the type species of Polymastia, P. mamillaris; Clade II—the type species of Sphaerotylus, Sph. capitatus; and Clade III—the type species of Spinularia, Spi. spinularia, and hence, these clades may be used as reference points in future classification of the family. However, no morphological synapomorphies can at present be defined for the clades revealed. Moreover, about 58 % of the species studied do not fall into any of these clades. Thus, for the time being, we cannot propose a satisfactory classification of Polymastiidae.
Abandonment of Radiella
We can, however, propose to transfer Radiella hemisphaerica to Polymastia since this species groups with the type species of Polymastia and five other Polymastia spp. forming Clade I. Likewise, Radiella sarsi, Radiella cf. sarsi, and Radiella sp. can be allocated to Spinularia since these three species and the type species of Spinularia form a monophyletic group, Clade II. The status of the genus Radiella is controversial (Boury-Esnault 2002; Plotkin and Janussen 2008; Plotkin et al. 2012), and the affinities of its type species, R. sol Schmidt, 1870, still remain ambiguous. Type material is lost, fresh material is not available, and the age-old nontype specimen identified as R. sol by Schmidt (1880) and redescribed by Boury-Esnault (2002) displays similarity to R. hemisphaerica, that does not match the drawing in the original description (Schmidt, 1870), which rather shows similarity between R. sol and R. sarsi (Plotkin and Janussen 2008; Plotkin et al. 2012). Nevertheless, no matter whether R. sol is related to R. hemisphaerica or R. sarsi, we propose to abandon Radiella since R. hemisphaerica is placed in Polymastia, R. sarsi is placed in Spinularia, and both Polymastia Bowerbank, 1862 and Spinularia Gray, 1867 are older names than Radiella Schmidt, 1870.
Disjunct distributions of polymastiid species are questioned
Our study questions the bipolar distribution of two polymastiid species, Tentorium semisuberites and Radiella sarsi. In the 28S rDNA tree, morphologically very similar Ten. semisuberites from the North Atlantic and Ten. cf. semisuberites from the Antarctic (see Table 1 for details on geography) did not group together. The type locality of Ten. semisuberites is Greenland (Schmidt 1870), and hence, we assume that the Antarctic sponges may be a separate species. Likewise, the pair of morphologically similar R. sarsi from the Norwegian Sea and Radiella cf. sarsi from Mozambiquean Coast was nonmonophyletic in both CO1 and 28S rDNA trees that questioned the allocation of these two to the same species.
Another example calling for reflection on disjunct distributions of sponge species is the pair Polymastia euplectella and Polymastia bartletti de Laubenfels, 1942, which display strong morphological similarities. The former species was recorded from the Barents Sea and adjacent areas (Rezvoj 1927; Plotkin 2004), whereas the latter was, before our study, known only from the type locality in the Baffin Sea (de Laubenfels 1942). We got genetic data from four Norwegian individuals identified as P. euplectella, one Canadian individual identified as P. bartletti, and a juvenile sponge from Sweden considered as P. cf. bartletti. These sponges were very similar in morphology, but in all phylogenetic trees, the Canadian individual and the Swedish sponge fell quite distantly from the Norwegian P. euplectella. All P. euplectella had identical CO1 and 28S rDNA except for one individual, for which no CO1 was obtained. P. bartletti and P. cf. bartletti slightly differed in both genes, but still were sisters. These results argue for that P. euplectella and P. bartletti are valid species. However, without studying more material, we cannot judge whether the small genetic differences between the Canadian P. bartletti and the Swedish P. cf. bartletti is just an intraspecific polymorphism, or these two are different, recently diverged species. Consequently, we cannot conclude whether P. bartletti is geographically isolated from P. euplectella or not.
Possible reasons for inconsistence between the single-gene phylogenies
Our analyses and tests supported dissimilar relationships of Polymastia boletiformis + Quasillina brevis with other clades and taxa in the single-gene trees. In the CO1 tree, this pair was the sister to Polymastia invaginata, whereas in the 28S rDNA tree, it was the sister to Clade I + Polymastia sp. 1 + Polymastia sp. 2. This may be due to real differences in genealogical histories of the mitochondrial and nuclear genes. However, on the other hand, the position of P. boletiformis + Q. brevis in the CO1 tree may be affected by very low resolution leading to Clade I. Likewise, unresolved relationships of Polymastia corticata along with weakly supported grouping of Sphaerotylus antarcticus with Clade II in the CO1 tree are obviously due to low resolution and hence to weak phylogenetic signal provided by our CO1 data.
Intraspecific polymorphism indicates incomplete lineage sorting?
Several inconsistencies between the CO1 and 28S rDNA phylogenies within three small terminate subclades were caused by intraspecific polymorphism. Our study could not resolve the relationships between three sibling species of Polymastia, P. andrica, P. arctica, and P. grimaldii. The CO1 data on these species were congruent with the morphological differences between them—individuals of the same species possessed identical CO1 and morphologically similar P. andrica and P. arctica grouped together against morphologically more distinct P. grimaldii, although the Bayesian and ML bootstrap support for this relationship was rather weak. At the same time, the analysis of 28S rDNA failed to resolve the relationships between these three species because of the intraspecific and intragenomic polymorphism. Intraspecific polymorphism was also revealed in P. boletiformis and P. invaginata. Two individuals of P. boletiformis possessed identical 28S rDNA, but fairly different CO1. In three individuals of P. invaginata, the identity by the mitochondrial gene mismatched that by the nuclear gene.
The ascertained cases of intraspecific polymorphism may indicate incomplete lineage sorting in the closely related polymastiid species and their populations. For example, each lineage may carry one unique version of CO1, but several versions of 28S rDNA, if its ancestor was polymorphic by this gene, and vice versa. When further divergence of the lineages takes place, some gene versions inherited from the polymorphic ancestor may be lost owing to genetic drift or selection (Rogers and Gibbs 2014). Another explanation of the revealed polymorphism may be a gene flow through hybridization between different species, but this assumption requires more thorough studies.
Insufficient variability in the 5′-end barcoding region of CO1
To reconstruct the CO1 phylogeny of Polymastiidae we used “Folmer’s” barcoding region, which was successfully applied to recover phylogenies of many invertebrate taxa (Folmer et al. 1994), in particular two large sponge families, Geodiidae (Cárdenas et al. 2010) and Halichondriidae (Erpenbeck et al. 2012). Our results revealed the insufficiency of variation of this region in Polymastiidae that might cause some inconsistencies between the CO1 and 28S rDNA phylogenies (see above) and also hindered the separation of the species in Clades II and III based on CO1 alone, while these species were otherwise successfully separated by the 28S rDNA data. The similar problem with the “Folmer’s” region was reported for some other sponge families, e.g., Lubomirskiidae (Schröder et al. 2002), Clionaidae (Ferrario et al. 2010), and Irciniidae (Pöppe et al. 2010). To solve this problem, sequencing of an additional downstream region of the CO1 gene providing more variability was recommended (Erpenbeck et al. 2006, Sponge Barcoding Project at http://www.palaeontologie.geo.uni-muenchen.de/SBP/). The analyses of the extended CO1 barcoding region may probably reduce the inconsistence between the CO1 and 28S rDNA phylogenies of Polymastiidae and resolve the relationships between sibling polymastiid species.
Conclusions
Our study presents the first comprehensive phylogenetic reconstruction of the family Polymastiidae based on molecular data. Our results show that its classification based on morphology is in a strong conflict with molecular phylogenies. Accordingly, the majority of previously assumed morphological synapomorphies appear to be highly homoplastic, and a natural classification of Polymastiidae will require a thorough and comprehensive taxonomic revision. Here we have set up a sound molecular framework for this task and recovered several strongly supported clades. In order to determine the morphological synapomorphies of these clades, a reinterpretation of the currently used characters and a selection of additional characters are needed. Furthermore, we have reported evidence for that sorting of lineages of different genes may follow different ways under the evolutionary divergence of sponge species and that the gene flow between populations of recently diverged species may also take place. Finally, we have demonstrated that the standard 5′-end barcoding region of CO1 provides insufficient data that may result in some inconsistence between the CO1 and 28S rDNA phylogenies and failure to reconstruct the relationships between some polymastiid species, which are otherwise recovered with 28S rDNA data. Hence, we argue once again for the advantages of multigene datasets and extended barcoding regions for reconstructing of phylogenies at the family and generic level.
References
Acinas, S. G., Sarma-Rupavtarm, R., Klepac-Ceraj, V., & Polz, M. F. (2005). PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Applied and Environmental Microbiology, 71(12), 8966–8969.
Allen, J. E., & Whelan, S. (2014). Assessing the state of substitution models describing non-coding RNA evolution. Genome Biology and Evolution, 6(1), 65–75.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215, 403–410.
Bandelt, H.-J., Forster, P., & Röhl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution, 16(1), 37–48.
Bergsten, J., Nilsson, A. N., & Ronquist, F. (2013). Bayesian tests of topology hypotheses with an example from diving beetles. Systematic Biology, 62(5), 660–673.
Boury-Esnault, N. (1987). The Polymastia species (Demosponges, Hadromerida) of the Atlantic area. In J. Vacelet & N. Boury-Esnault (Eds.), Taxonomy of Porifera from the N.E. Atlantic and Mediterranean Sea, NATO ASI Series, G 13 (pp. 29–66). Berlin: Springer.
Boury-Esnault, N. (2002). Family Polymastiidae Gray, 1867. In J. N. A. Hooper & R. W. M. Van Soest (Eds.), Systema Porifera, a guide to the classification of sponges (Vol. 1, pp. 201–219). New York: Kluwer Academic/Plenum.
Boury-Esnault, N., & Bézac, C. (2007). Morphological and cytological descriptions of a new Polymastia species (Hadromerida, Demospongiae) from the North-West Mediterranean Sea. In M. R. Custódio, G. Lôbo-Hajdu, E. Hajdu, & G. Muricy (Eds.), Porifera research: biodiversity, innovation and sustainability (pp. 23–30). Rio de Janeiro: Museu Nacional, Série Livros 28.
Boury-Esnault, N., & Rützler, K. (1997). Thesaurus of sponge morphology. Smithsonian Contributions to Zoology, 596, 1–55.
Bowerbank, J. S. (1862). On the anatomy and physiology of the Spongiadae. Part III. On the generic characters, the specific characters, and on the method of examination. Philosophical Transactions of the Royal Society, 152(2), 1087–1135.
Bowerbank, J. S. (1866). A monograph of the British Spongiadae (Vol. 2). London: Ray Society.
Bowerbank, J. S. (1874). A monograph of the British Spongiadae (Vol. 3). London: Ray Society.
Cárdenas, P., Rapp, H. T., Schander, C., & Tendal, O. S. (2010). Molecular taxonomy and phylogeny of the Geodiidae (Porifera, Demospongiae, Astrophorida)—combining phylogenetic and Linnean classification. Zoologica Scripta, 39, 89–106.
Cárdenas, P., Xavier, J. R., Reveillaud, J., Schander, C., & Rapp, H. T. (2011). Molecular phylogeny of the Astrophorida (Porifera, Demospongiae p) reveals an unexpected high level of spicule homplasy. PLoS ONE, 6(4), e18318. doi:10.1371/journal.pone.0018318.
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution, 17(4), 540–552.
Darriba, D., Taboada, G. L., Doallo, R., & Posada, D. (2012). JModelTest 2: more models, new heuristics and parallel computing. Nature Methods, 9(8), 772.
de Laubenfels, M. W. (1932). The marine and fresh-water sponges of California. Proceedings of the United States National Museum, 81(2927), 1–140.
de Laubenfels, M. W. (1942). Porifera from Greenland and Baffinland collected by Capt. Robert A. Bartlett. Journal of the Washington Academy of Sciences, 32(9), 263–269.
de Laubenfels, M. W. (1949). The sponges of Woods Hole and adjacent waters. Bulletin of the Museum of Comparative Zoology at Harvard College, 103(1), 1–55.
De Rijk, P., Robbrecht, E., de Hoog, S., Caers, A., Van de Peer, Y., & De Wachter, R. (1999). Database on the structure of large subunit ribosomal RNA. Nucleic Acids Research, 27(1), 174–178.
De Rijk, P., Wuyts, J., Van de Peer, Y., Winkelmans, T., & De Wachter, R. (2000). The European large subunit ribosomal RNA database. Nucleic Acids Research, 28(1), 177–178.
Dendy, A. (1888). Studies on the comparative anatomy of sponges. I. On the genera Ridleia, n. gen., and Quasillina, Norman. Quarterly Journal of Microscopical Science, 282, 513–529.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32(5), 1792–1797.
Eilertsen, M. H., & Malaquias, M. A. E. (2013). Systematic revision of the genus Scaphander (Gastropoda, Cephalaspidea) in the Atlantic Ocean, with a molecular phylogenetic hypothesis. Zoological Journal of the Linnean Society, 167, 389–429.
Erpenbeck, D., Hooper, J. N. A., & Wörheide, G. (2006). CO1 phylogenies in diploblasts and the “barcoding of life”—are we sequencing a suboptimal partition? Molecular Ecology Notes, 6, 550–553.
Erpenbeck, D., Cleary, D. F. R., Voigt, O., Nichols, S. A., Degnan, B. M., Hooper, J. N. A., & Wörheide, G. (2007a). Analysis of evolutionary, biogeographical and taxonomic patterns of nucleotide composition in demosponge rRNA. Journal of the Marine Biological Association of the United Kingdom, 87(6), 1607–1614.
Erpenbeck, D., Nichols, S. A., Voigt, O., Dohrmann, M., Degnan, B. M., Hooper, J. N. A., & Wörheide, G. (2007b). Phylogenetic analyses under secondary structure-specific substitution models outperform traditional approaches—case studies with diploblast LSU. Journal of Molecular Evolution, 64, 543–557.
Erpenbeck, D., Voigt, O., Gültas, M., & Wörheide, G. (2008). The Sponge Genetree Server providing a phylogenetic backbone for poriferan evolutionary studies. Zootaxa, 1939, 58–60.
Erpenbeck, D., Hall, K., Alvarez, B., Büttner, G., Sacher, K., Schätzle, S., Schuster, A., Vargas, S., Hooper, J. N. A., & Wörheide, G. (2012). The phylogeny of halichondrid demosponges: past and present re-visited with DNA-barcoding data. Organisms Diversity and Evolution, 12(1), 57–70.
Farris, J. S., Källersjö, M., Kluge, A. G., & Bult, C. (1994). Testing significance of congruence. Cladistics, 10, 315–319.
Ferrario, F., Calcinai, B., Erpenbeck, D., Galli, P., & Wörheide, G. (2010). Two Pione species (Hadromerida, Clionaidae) from the Red Sea: a taxonomical challenge. Organisms Diversity and Evolution, 10(4), 275–285.
Folmer, O., Black, M., Hoeh, W., Lutz, R., & Vrijenhoek, R. (1994). DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Molecular Marine Biology and Biotechnology, 3, 294–299.
Galtier, N., Gouy, M., & Gautier, C. (1996). SeaView and Phylo_Win: two graphic tools for sequence alignment and molecular phylogeny. Computer Applications in the Biosciences, 12(6), 543–548.
Geller, J., Meyer, C. P., Parker, M., & Hawk, H. (2013). Redesign of PCR primers for mitochondrial cytochrome c oxidase subunit I for marine invertebrates and application in all-taxa biotic surveys. Molecular Ecology Resources, 13, 851–861.
Gouy, M., Guindon, S., & Gascuel, O. (2010). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Molecular Biology and Evolution, 27(2), 221–224.
Gowri-Shankar, V., & Jow, H. (2006). PHASE: a software package for phylogenetics and sequence evolution. Version 2.0 Available from http://www.bioinf.man.ac.uk/phase/.
Gray, J. E. (1848). List of the specimens of British sponges in the collection of the British Museum (London). London: British Museum Publication.
Gray, J. E. (1867). Notes on the arrangement of sponges, with the descriptions of some new genera. Proceedings of the Zoological Society of London, 1867, 492–558.
Guindon, S., & Gascuel, O. (2003). A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Systematic Biology, 52, 696–704.
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., & Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Systematic Biology, 59(3), 307–321.
Hallmann, E. F. (1914). A revision of the monaxonid species described as new in Lendenfeld’s ‘Catalogue of the Sponges in the Australian Museum’. Part I, II, III. Proceedings of the Linnean Society of New South Wales, 39, 263–315, 327–376, 398–446
Hooper, J. N. A., & Van Soest, R. W. M. (2002). Class Demospongiae Sollas, 1885. In J. N. A. Hooper & R. W. M. Van Soest (Eds.), Systema Porifera, a guide to the classification of sponges (Vol. 1, pp. 15–18). New York: Kluwer Academic/Plenum.
Huson, D. H., & Scornavacca, C. (2012). Dendroscope 3—an interactive viewer for rooted phylogenetic trees and networks. Systematic Biology. doi:10.1093/sysbio/sys062.
Huson, D. H., Rupp, R., Berry, V., Gambette, P., & Paul, C. (2009). Computing galled networks from real data. Bioinformatics, 25(12), i85–i93.
Johnston, G. (1842). A history of British sponges and lithophytes. Edinburgh: W.H. Lizars.
Kass, R. E., & Raftery, A. E. (1995). Bayes factors. Journal of the American Statistical Association, 90, 773–795.
Keller, C. (1880). Neue Coelenteraten aus dem Golf von Neapel. Archiv für mikroskopische Anatomie und Entwicklungsmechanik, 18, 271–280.
Kelly-Borges, M., & Bergquist, P. R. (1997). Revision of Southwest Pacific Polymastiidae (Porifera: Demospongiae: Hadromerida) with descriptions of new species of Polymastia Bowerbank, Tylexocladus Topsent, and Acanthopolymastia gen. nov. from New Zealand and the Norfolk Ridge, New Caledonia. New Zealand Journal of Marine and Freshwater Research, 31, 367–402.
Kirkpatrick, R. (1907). Preliminary report on the Monaxonellida of the National Antarctic Expedition. Annals and Magazine of Natural History, 20, 271–291.
Kirkpatrick, R. (1908). Porifera (Sponges). II. Tetraxonida, Dendy. National Antarctic Expedition, 1901–1904, Natural History, 4, Zoology, 1–56.
Koltun, V. M. (1964). Sponges (Porifera), collected in the Greenland Sea and in the North area off Spitzbergen and Frantz Josef Land by r/v “F. Litke” in 1955, r/v “Ob” in 1956 and r/v “Lena” in 1957–1958. Oceanographic expeditions to the northern part of the Greenland Sea and the adjacent Arctic Basin. Publications Arctic and Antarctic Scientific Institute, 259, 143–166.
Kumar, S., Stecher, G., & Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution. doi:10.1093/molbev/msw054.
Lamarck, J. B. P. De Monet, Comte De (1815). Suite des polypiers empâtés. Mémoires du Muséum d’Histoire naturelle, 1. Paris, 69–80, 162–168, 331–340.
Maddison, W. P., & Maddison, D. R. (2015). Mesquite: a modular system for evolutionary analysis. Version 3.04 Available from http://mesquiteproject.org.
Merejkowsky, C. (1878). Preliminary account on sponges of the White Sea. Proceedings of the Imperial Saint-Petersburg Society of Naturalists, 9, 249–270.
Meyer, C. P. (2003). Molecular systematics of cowries (Gastropoda: Cypraeidae) and diversification patterns in the tropics. Biological Journal of the Linnean Society, 79, 401–459.
Montagu, G. (1818). An essay on sponges, with descriptions of all the species that have been discovered on the coast of Great Britain. Memoirs of the Wernerian Natural History Society, 2(1), 67–122.
Morrow, C. C., & Boury-Esnault, N. (2000). Redescription of the type species of the genus Polymastia Bowerbank, 1864 (Porifera, Demospongiae, Hadromerida). Zoosystema, 22, 327–335.
Morrow, C. C., & Cárdenas, P. (2015). Proposal for a revised classification of the Demospongiae (Porifera). Frontiers in Zoology, 12, 7.
Morrow, C. C., Picton, B. E., Erpenbeck, D., Boury-Esnault, N., Maggs, C. A., & Allcock, A. L. (2012). Congruence between nuclear and mitochondrial genes in Demospongiae: a new hypothesis for relationships within the G4 clade (Porifera: Demospongiae). Molecular Phylogenetics and Evolution, 62, 174–190.
Morrow, C. C., Redmond, N. E., Picton, B. E., Thacker, R. W., Collins, A. G., Maggs, C. A., Sigwart, J. D., & Allcock, A. L. (2013). Molecular phylogenies support homoplasy of multiple morphological characters used in the taxonomy of Heteroscleromorpha (Porifera: Demospongiae). Integrative and Comparative Biology, 53, 428–446.
Müller, O. F. (1806). Zoologia danica seu animalium Daniae et Norvegiae rariorum ac minus notorum. Descriptiones et Historia. 4. (N. Christensen: Hauniae).
Nei, M., & Kumar, S. (2000). Molecular evolution and phylogenetics. New York: Oxford University Press.
Nichols, S. A. (2005). An evaluation of support for order-level monophyly and interrelationships within the class Demospongiae using partial data from the large subunit rDNA and cytochrome oxidase subunit I. Molecular Phylogenetics and Evolution, 34, 81–96.
Norman, A. M. (1869). Notes on a few Hebridean sponges, and on a new Desmacidon from Jersey. Annals and Magazine of Natural History, 3, 296–299.
Nylander, J. A. A. (2004). MrModeltest v2. Program distributed by the author. Uppsala: Evolutionary Biology Centre, Uppsala University.
Plotkin, A. (2004). Biodiversity and distribution of Polymastiidae (Demospongiae, Hadromerida) in the Arctic area. In M. Pansini, M. R. Pronzato, G. Bavestrello, & R. Manconi (Eds.), Sponge sciences in new millennium. Bollettino dei Musei e degli Instituti Biologici dell’Universita di Genova (Vol. 68, pp. 535–547).
Plotkin, A., & Janussen, D. (2007). New genus and species of Polymastiidae (Demospongiae: Hadromerida) from the Antarctic deep sea. Journal of the Marine Biological Association of the United Kingdom, 87(6), 1395–1401.
Plotkin, A., & Janussen, D. (2008). Polymastiidae and Suberitidae (Porifera: Demospongiae: Hadromerida) of the deep Weddell Sea, Antarctic. In P. Martínez Arbizu & S. Brix (Eds.), Bringing light into deep-sea biodiversity, zootaxa (Vol. 1866, pp. 95–135).
Plotkin, A., Rapp, H. T., & Gerasimova, E. (2012). Phylogenetic reconstruction of Polymastiidae (Demospongiae: Hadromerida) based on morphology. Hydrobiologia, 687, 21–41.
Pöppe, J., Sutcliffe, P., Hooper, J. N. A., Wörheide, G., & Erpenbeck, D. (2010). CO I barcoding reveals new clades and radiation patterns of Indo-Pacific sponges of the family Irciniidae (Demospongiae: Dictyoceratida). PLoS ONE, 5(4), e9950. doi:10.1371/journal.pone.0009950.
Rambaut, A., & Drummond, A. J. (2009). Tracer. MCMC trace analysis tool. Version v1.5.0. Available from www.http://tree.bio.ed.ac.uk/software/tracer/.
Redmond, N. E., Morrow, C. C., Thacker, R. W., Diaz, M. C., Boury-Esnault, N., Cárdenas, P., Hajdu, E., Lôbo-Hajdu, G., Picton, B. E., Pomponi, S. A., Kayal, E., & Collins, A. G. (2013). Phylogeny and systematics of Demospongiae in light of new small subunit ribosomal DNA (18S) sequences. Integrative and Comparative Biology, 53, 388–415.
Rezvoj, P. D. (1927). A new species of sponges, Polymastia euplectella, from the Murman Coast. Comptes Rendus de l’Académie des Sciences USSR, 18, 301–302.
Ridley, S. O., & Dendy, A. (1886). Preliminary report on the Monaxonida collected by H.M.S. “Challenger”. Annals and Magazine of Natural History, 18, 325–351, 470–493.
Rogers, J., & Gibbs, R. A. (2014). Comparative primate genomics: emerging patterns of genome content and dynamics. Nature Reviews. Genetics, 15(5), 347–359.
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D., Darling, A., Höhna, S., Larget, B., Liu, L., Suchard, M. A., & Huelsenbeck, J. P. (2011). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology, 61(3), 539–542.
Sarà, M., & Melone, N. (1965). Una nuova specie del genere Tethya, Tethya citrina sp. n. dal Mediterraneo (Porifera Demospongiae). Attidella Società Peloritana di Scienze Fisiche, Matematiche e Naturali, 11(Supplement), 123–138.
Sars, G. O. (1872). Spongiae. In Kongelige Norske Universitet (Ed.), On some remarkable forms of animal life from the great depths off the Norwegian coast. I. Partly from posthumous manuscripts of the late professor Dr. Michael Sars (pp. 62–82). Christiania, Norway: Brøgger & Christie.
Schmidt, O. (1870). Grundzüge einer spongien-fauna des Atlantischen gebietes. Leipzig: Wilhelm Engelmann.
Schmidt, O. (1880). Die Spongien des Meerbusen von Mexico (und des caraibischen Meeres). Heft II. Abtheilung II. Hexactinelliden. Abtheilung III. Tetractinelliden. Monactinelliden und Anhang. Nachträge zu Abtheilung I (Lithistiden). In Reports on the dredging under the supervision of Alexander Agassiz, in the Gulf of Mexico, by the USCSS ‘Blake’ (pp. 33–90). Jena: Gustav Fischer.
Schröder, H. C., Efremova, S. M., & Itskovich, V. B. (2002). Molecular phylogeny of the freshwater sponges in Lake Baikal. Journal of Zoological Systematics and Evolutionary Research, 41, 80–86.
Sollas, W. J. (1885). A classification of the sponges. Scientific Proceedings of the Royal Dublin Society (new series), 5, 112.
Speksnijder, A. G. C. L., Kowalchuk, G. A., De Jong, S., Kline, E., Stephen, J. R., & Laanbroek, H. J. (2001). Microvariation artifacts introduced by PCR and cloning of closely related 16S rRNA gene sequences. Applied and Environmental Microbiology, 67(1), 469–472.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30(9), 1312–1313.
Swarczewsky, B. A. (1906). Beiträge zur Spongien-Fauna des Weissen Meeres. Memoires de la Société des Naturalistes de Kiew, 20, 307–371.
Swofford, D. L. (2002). PAUP*: phylogenetic analysis using parsimony (*and other methods). Sunderland: Sinauer Associates.
Topsent, E. (1890). Notice préliminaire sur les spongiaires recueillis durant les campagnes de l’Hirondelle. Bulletin de la Société zoologique de France, 15(26–32), 65–71.
Topsent, E. (1894). Une réforme dans la classification des Halichondrina. Mémoires de la Société zoologique de France, 7, 5–26.
Topsent, E. (1898). Eponges nouvelles des Açores. Première serie. Mémoires de la Société zoologique de France, 11, 225–255.
Topsent, E. (1913). Spongiaires provenant des campagnes scientifiques de la Princesse “Alice” dans les Mers du Nord (1898–1899, 1906–1907). Résultats des campagnes scientifiques accomplies par le Prince Albert I de Monaco, 45, 1–67.
Van Soest, R. W. M., Boury-Esnault, N., Hooper, J. N. A., Rützler, K., de Voogd, N. J., Alvarez de Glasby, B., Hajdu, E., Pisera, A. B., Manconi, R., Schoenberg, C., Janussen, D., Tabachnick, K. R., Klautau, M., Picton, B. E., Kelly, M., Vacelet, J., Dohrmann, M., Díaz, M.-C., & Cárdenas, P. (2015). World Porifera database. Accessed at http://www.marinespecies.org/porifera on 2015-08-11.
Vargas, S., Schuster, A., Sacher, K., Büttner, G., Schätzle, S., Läuchli, B., Hall, K., Hooper, J. N. A., Erpenbeck, D., & Wörheide, G. (2012). Barcoding sponges: an overview based on comprehensive sampling. PLoS ONE, 7(7), e39345. doi:10.1371/journal.pone.0039345.
Vargas, S., Kelly, M., Schnabel, K., Mills, S., Bowden, D., & Wörheide, G. (2015). Diversity in a cold hot-spot: DNA-barcoding reveals patterns of evolution among Antarctic Demosponges (class Demospongiae, phylum Porifera). PLoS ONE, 10(6), e0127573. doi:10.1371/journal.pone.0127573.
Voigt, O., Wülfing, E., & Wörheide, G. (2012). Molecular phylogenetic evaluation of classification and scenarios of character evolution in calcareous sponges (Porifera, class Calcarea). PLoS ONE, 7(3), e33417. doi:10.1371/journal.pone.0033417.
Vosmaer, G. C. J. (1885). The sponges of the “Willem Barents” expedition 1880 and 1881. Bijdragen tot de Dierkunde, 12, 1–47.
Vosmaer, G. C. J. (1887). Klassen und Ordnungen der Spongien (Porifera). In H. G. Bronn (Ed), Die Klassen und Ordnungen des Thierreichs, 2 (pp. 1–496). Leipzig–Heidelberg.
Wuyts, J., Van de Peer, Y., & De Wachter, R. (2001). Distribution of substitution rates and location of the insertion sites in the tertiary structure of ribosomal RNA. Nucleic Acids Research, 29(24), 5017–5028.
Acknowledgments
We would like to express our special gratitude to all staff of the BioDiversity Laboratories at the Department of Biology, University of Bergen and personally to Kenneth Meland for his inestimable help with the optimization of PCR protocols and with the digital processing and alignment of sequences, to Solveig Thorkildsen for the implementation of cloning of PCR products that has brought us extremely precious data, and to Louise Lindblom who has secured the convenience and safety of our lab work.
Great thanks also come to the scientists who have sampled the material for us or donated the material from their institutions for this study—Jon Anders Kongsrud (Natural History Collections, University Museum of Bergen) for the material from Norway and the Nordic Seas; Christine Morrow (Department of Zoology, Ryan Institute, National University of Ireland, Galway) and Bernard Picton (Department of Natural Sciences, National Museums Northern Ireland, Belfast) for the material from the British Isles; Megan Best, Vonda Wareham, Ellen Kenchington, and Javier Murillo (Department of Fisheries and Oceans Canada, Ottawa) for the material from Newfoundland and Nova Scotia; Dorte Janussen (Senckenberg Naturmuseum, Frankfurt am Main) and Christoph Noever (Department of Biology, University of Bergen) for the sponges from the Weddell Sea, Antarctic; Natalia Chervyakova (Moscow State University) for sampling from the Bellingshausen Sea, Antarctic and the White Sea, Russia; Kennet Lundin and Carola Azurduy Högström (Gothenburg Natural History Museum) for the samples from Sweden; and Javier Cristobo and Pilar Rios (Spanish Institute of Oceanography, Madrid) for the samples from Mozambique.
AP would also like to appreciate the people who organized his field work in Norway and assisted him during SCUBA diving—Prof. Bjørn Gulliksen (University of Tromsø) for the organization of the sampling around Svalbard and giving precious tips about the sponge localities in other areas, Bjørn Tore Dragnes (OMNIMAR Dragnes, Tromsø) for the arrangement of diving in Northern Norway, Erling Svensen (OceanPhoto/Dalane Tidende AS, Egersund) for the arrangement of diving in South-Western Norway and valuable tips, and Christoph Noever (Department of Biology, University of Bergen) for the assistance during diving around Bergen.
We are also much grateful to Elena Gerasimova (Rådgivende Biologer AS, Bergen) who has done most of the work with the histological preparation of the samples for taxonomic identification.
Phylogenetic computing under our study has exploited the excellent online services provided by the international Cyberinfrastructure for Phylogenetic Research (CIPRES) Science Gateway (https://www.phylo.org/) and the Lifeportal at the University of Oslo (https://lifeportal.uio.no/) using the high performance computing cluster Abel.
The last but not the least: we would like to thank two reviewers for their helpful comments.
Authors’ contributions
Conceiving the study: AP and HTR. Sampling the material: AP and HTR. Primary treatment and taxonomic identification of the samples: AP. DNA extraction, amplification and sequencing: AP. Contributing reagents/consumables/tools: HTR. Reconstruction of the secondary structure, 28S rDNA: OV. Alignments and phylogenetic analyses: OV, AP and EW. Tracing morphological characters along the molecular tree: EW and AP. Writing the manuscript: AP. Revising the manuscript: OV, EW and HTR.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Funding
This study was supported by the Norwegian Biodiversity Information Centre (grant to HTR, project number 70184219), the Norwegian Academy of Science and Letters (grant to HTR), the Research Council of Norway (through contract number 179560), and Horizon 2020, the European Union Framework Programme for Research and Innovation (grant agreement No 679848).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Plotkin, A., Voigt, O., Willassen, E. et al. Molecular phylogenies challenge the classification of Polymastiidae (Porifera, Demospongiae) based on morphology. Org Divers Evol 17, 45–66 (2017). https://doi.org/10.1007/s13127-016-0301-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13127-016-0301-7