
Abstract
Hypothyroidism is the most frequent endocrine pathology. Although clinical or overt hypothyroidism has been traditionally associated to low T3 / T4 and high thyrotropin (TSH) circulating levels, other forms exist such as subclinical hypothyroidism, characterized by normal blood T3 / T4 and high TSH. In its different forms is estimated to affect approximately 10% of the population, especially women, in a 5:1 ratio with respect to men. Among its consequences are alterations in cardiac electrical activity, especially in the repolarization phase, which is accompanied by an increased susceptibility to cardiac arrhythmias. Although these alterations have traditionally been attributed to thyroid hormone deficiency, recent studies, both clinical trials and experimental models, demonstrate a fundamental role of TSH in cardiac electrical remodeling. Thus, both metabolic thyroid hormones and TSH regulate cardiac ion channel expression in many and varied ways. This means that the different combinations of hormones that predominate in different types of hypothyroidism (overt, subclinic, primary, central) can generate different forms of cardiac electrical remodeling. These new findings are raising the relevant question of whether serum TSH reference ranges should be redefined.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although not as famous as diabetes, hypothyroidism (HT) is actually the most prevalent endocrine disease in Western countries. In Spain, almost half of the endocrinology patients are treated for thyroid diseases [23]. Primary hypothyroidism, the most frequent thyroid disease, is characterized by a decrease in circulating levels of the thyroid hormones thyroxine (T3) and triiodothyronine (T4). This is caused by a deficient function of the thyroid gland, accompanied by a compensatory elevation of pituitary thyroid-stimulating hormone (thyrotropin or TSH) (Table 1). Its prevalence is higher in women, with a female-to-male ratio of 5:1 in European countries [6, 23, 37]. The prevalence of HT in industrialized countries is not very different: around 0.6% in Europe, 0.35% in the USA, and 0.7% in Korea [22, 32, 39].
Subclinical hypothyroidism is a silent form of HT in which the thyroid gland is damaged, but normal T3 and T4 levels are maintained at the expense of a compensatory elevation of TSH levels. Since blood T3 levels are normal, most patients remain undiagnosed. According to the Spanish Society of Endocrinology and Nutrition [9], only 600,000 people are diagnosed with subclinical hypothyroidism (1.3% of the population), but the estimated amount could be close to 3 million patients (around 6%) [11]. In Europe, the prevalence is around 4.5%; in the USA, between 4.3 and 8.5%; and in Korea 3.1% [22, 32, 39]. Very importantly, patients with subclinical HT have an estimated probability of progressing to clinical HT between 5–20% at 1 year, and 63% at 10 years. In any case, this progression seems to be proportional to elevated TSH levels, advanced age, and the presence of antithyroid antibodies [5, 21, 27].
Finally, central hypothyroidism is an infrequent form of HT caused by insufficient stimulation of the thyroid gland due to low levels of thyroid-stimulating hormone (Table 1). Central HT can be secondary, whose origin is pituitary, or tertiary, with hypothalamic origin. In children, it is usually caused by craniopharyngiomas and anterior cranial irradiation of brain tumors or hematologic malignancies. In adults, it is usually due to macroadenomas, pituitary surgery, or post-irradiation. The estimated prevalence of central HT is 1/80,000 to 1/120,000 worldwide [24].
One of the main targets of thyroid hormones is the cardiovascular system [30]. The main cardiac symptoms of HT are poor exercise tolerance and increased fatigue [13]. In more advanced stages, ventricular and atrial fibrosis, myocardial edema, and reduced cardiac output usually appear [10, 50, 58]. The effects of thyroid dysfunction on cardiac and cardiovascular mechanical function, such as heart failure and coronary disease, have recently been reviewed [67]. However, it should be noted that the first signs of cardiac dysfunction are often found on the electrocardiogram. It is known that cardiac arrhythmias can be elicited or maintained by circulatory factor such as inflammatory cytokines or by structural remodeling such as myocardial infarction or interstitial fibrosis, and their involvement in arrhythmogenesis in hypothyroidism has also been recently reviewed [57]. Therefore, through a historical perspective, this review aims to delve specifically into the causes of the cardiac electrical remodeling that occurs in patients with hypothyroidism and in experimental models of this disease.
Cardiac electrical remodeling in hypothyroidism
Primary overt hypothyroidism
The electrocardiograms of hypothyroid patients often show significant sinus bradycardia, atrial fibrillation, and, mainly, prolonged ventricular repolarization (Fig. 1). Thus, there is a lengthening of the QT interval (indicator of the duration of ventricular depolarization), and of the rate-corrected QT (QTc). In addition, HT increases QT dispersion or QTd (which is the difference between the longest and shortest QT in all leads), and causes a prolongation of the duration of the T-peak to T-end (Tpeak–Tend), reflecting an increase in ventricular electrical heterogeneity. Either QTc prolongation, QTd increase, or Tpeak–Tend lengthening is independently associated with an elevated incidence of polymorphic ventricular tachyarrhythmias, such as torsades de pointes (TdP). Although TdP usually extinguish spontaneously, if they persist, they can ultimately lead to ventricular fibrillation and sudden cardiac death [3, 4, 10, 20, 60]. Animal models of HT consistently reproduce the electrocardiographic alterations observed in patients [15, 16, 69].

Original electrocardiographic recordings in experimental animals. Healthy (control), central (cHT), and primary hypothyroidism (pHT). The recording of the animal with central HT is very similar to the control recording, while that of the animal with primary HT presents severe alterations, such as atrial fibrillation and a lengthened QT interval. The recordings correspond to lead II. P, R, S, and T waves are indicated in the control recording. The Q wave is not visible in any recording. RR, RR interval; QT, QT interval
Subclinical hypothyroidism
Individuals affected by subclinical HT are usually asymptomatic but show electrocardiographic abnormalities similar to those seen in patients with overt hypothyroidism. Evidence of this is that the patients who have overcome Hasimoto’s thyroiditis and maintain normal T3/T4 levels at the expense of an elevated TSH have prolonged QTc and increased QTd. These patients, like those with overt HT, show a higher incidence of atrial and ventricular premature beats [21].
Central hypothyroidism
Because the prevalence of central hypothyroidism is very low and patients usually have other hormonal deficits, the effects on the ECG have been poorly studied. However, experiments in animal models reveal that central HT does not cause significant electrocardiographic alterations (Fig. 1) [15].
The distinctive feature of central HT, what makes it different from primary clinical and subclinical HT, is that low levels of T3 and T4 are accompanied by low levels of TSH.
Role of TSH in cellular cardiac electrical remodeling in hypothyroidism
Given that in subclinical HT thyroid hormone levels are still normal, the coincidence in electrocardiographic manifestations between established and subclinical HT suggests that cardiac electrical remodeling is not only due to thyroid hormone deficiency, but also to some other factor. In fact, incubation of human-induced stem cell-derived cardiac myocytes (hiPS-CMs) with low concentrations of T3 (0.01 nM) and T4 (0.0002 and 0.02 nM) did not cause lengthening of the field potential duration (FPD), which is the in vitro equivalent of the QT interval [59].
It is important to remember that overt and subclinical HT have in common high TSH levels. In this regard, several groups have described that in patients with HT, QTc lengthening and QTd increase reverse after treatment, when TSH levels normalize. Therefore, based on correlation analyses performed in healthy individuals and in hypothyroid patients, with or without treatment, several groups proposed that QTc and QTd alterations are caused by elevated TSH levels [21, 38, 60]. In the same line of research, a correlation between TSH concentration and the duration of the Tpeak–Tend has recently been described [4].
Obviously, for this to be true, the TSH receptor must be expressed in cardiac muscle. In the last decades, TSH receptor expression has been described in several extrathyroidal tissues such as skin, kidney, liver, bone, blood vessels, or the immune system, although its physiological relevance is difficult to establish and is still a matter of debate [62]. In the heart, the TSH receptor was first described in mouse atria and human cardiac muscle in 1995 [14]. Later studies in cardiac ventricles of rodents confirmed its expression at the mRNA and protein level and, more importantly, found that TSH receptors in the heart activate the cAMP and PKA pathway [12, 26]. More recently, experiments in isolated rat ventricular cardiomyocytes performed by our research group demonstrated that incubation with TSH regulates the expression of Kv4.3, Kv4.2, and Kir2.1 K+ channels and the accessory protein KCHIP2, whereas blockade of the TSH receptor with a specific antibody prevented the effect [2]. Similarly, in adult human atrial myocytes, PKA inhibition suppresses the TSH-induced effect [15]. This demonstrates that TSH regulates cardiac electrical activity directly, through activation of its receptor and its canonical signaling pathway.
Hormones responsible for cellular cardiac electrical remodeling in hypothyroidism
In the human heart, the action potential (AP) is the result of a balance between depolarizing and repolarizing currents. The fast inward Na+ current (INa), carried by Nav1.5 channels, which are codified by the SCN5A gene, depolarizes the phase 0 of the cardiac AP, whereas the inward L-type Ca2+ current (ICa-L), carried by Cav1.2 channels, which are the product of the CACNA1C gene, depolarizes the plateau phase. On the other hand, the main four repolarizing K+ currents are as follows: the transient outward (Ito), the rapid delayed rectifier (IKr), the slow delayed rectifier (IKs), and the ultrarapid delayed rectifier (IKur). These currents are generated by the outflow of potassium through Kv4.3; Kv11.1 or hERG; Kv7.1 or KCNQ1, together with KCNE1; and Kv1.5 channels, which are codified by the KCND3, KCNH2, KCNQ1/KCNE1, and KCNA5 genes, respectively. Therefore, the HT-induced lengthening of the ECG QT interval can be caused by an increase in depolarizing currents, a decrease in repolarizing currents, or a combination of both.
In the 1990s, the role of thyroid hormones in the regulation of cardiac ionic currents began to be studied intensively. However, the results obtained were in many cases contradictory, due to differences in the animal models and the techniques used between the different studies. After 30 years of research, the picture is beginning to become clearer.
Na+ and Ca2+ currents
Patch-clamp experiments in hypothyroid guinea pigs showed that HT had no effect on INa [8]. This result was confirmed by microarray analysis in hypothyroid rats, where sodium channel subunit transcripts were unaffected [33].
Although that work in guinea pigs also found no effect on the ICa-L [8], subsequent research has consistently contradicted this result. Thyroid hormones regulate ICa-L at both the transcriptional and post-transcriptional levels. On the one hand, T3 acutely increases the calcium current by acting directly on rat cardiac myocytes and activating the cAMP cascade [64]. On the other hand, it inhibits the expression of L-type Ca2+ channels. Consequently, animals with primary or central hypothyroidism have higher expression of CACNA1C gene and higher ICa-L amplitude compared with euthyroid animals [15, 33, 64]. This increase in ICa-L may contribute to the lengthening of cardiac repolarization observed in hypothyroid patients. Last, regarding the role of TSH, our group has found no effect on ICa-L amplitude or CACNA1C channel expression [2, 15].
Repolarizing K+ currents
Transient outward K+ current
Since Ito in rodents has a very large current amplitude, it has been extensively studied. Pioneering studies in neonatal cardiac myocytes demonstrated that thyroid hormones were necessary for correct transient outward current development, that is, for normal channel protein expression and normal current amplitude [52, 53, 65]. These findings led to several studies in rats with myocardial infarction who showed reduced Ito, and found that treatment with thyroid hormones and hormone analogues like 3,5-diiodothyropropionic acid (DITPA) restored Ito density and expression of Kv4.2 and Kv1.4 channels [45, 66]. In addition, treatment with DITPA also restored cardiac Ito in diabetic animals with hypothyroidism [17]. In fact, HT per se, reduces the amplitude of several potassium currents, including Ito. Our group found that the decrease in Ito amplitude is not homogeneous along the ventricular wall, which, in turn, explains the alterations in QT dispersion and Tpeak–Tend duration [16] observed in hypothyroid animals.
Interestingly, unlike its crucial role for normal Ito development, it has also been consistently demonstrated that T3 has no effect on this current in adult cardiac myocytes, either healthy or hypothyroid [53, 56]. Therefore, the absence of T3 does not explain the reduction of Ito observed in adult hypothyroidism.
Conversely, we found a direct relationship between TSH and reduced Ito in hypothyroidism [2, 15]. Both current amplitude and Kv4.3 channel expression were reduced in cardiac myocytes isolated from animals with primary HT (with high TSH), but not in those isolated from animals with central HT (with low TSH). We also observed that incubation with TSH reduced Ito in myocytes from control and central hypothyroid animals, but not in myocytes from animals with primary HT (in which the current was already reduced due to high plasma TSH levels). Furthermore, incubation with TSH reduced KCND3 gene expression in human heart samples in vitro. Taken together, these results grant a novel role for TSH in cardiac electrical remodeling and support the hypothesis that QTc and QTd alterations are caused by elevated TSH levels.
Delayed rectifying currents IKr, IKs, and IKur
In the human ventricle, the main repolarizing current is the rapid delayed rectifier or IKr that flows through the hERG channel. In human-induced stem cell-derived cardiac myocytes (hiPS-CMs), treatment with T3 stimulated myocytic maturation in terms of increased cell size, sarcomere length, and contractile force [68]. In a recent work in hIPS-CMs, combined T3+Dexamethasone treatment increased IKr expression and therefore improved the electrophysiological maturation of these cells [63]. However, neither hypo- nor hyperthyroidism modify the expression of the KCNH2 gene, which encodes the IKr channels, in adult murine cardiomyocytes [33]. These results suggest that, as with Ito, thyroid hormones are necessary for the normal development of IKr in neonatal myocytes, and that the effect is reduced throughout development until it disappears in adult cardiac myocytes. Finally, regarding the role of the thyroid-stimulating hormone, our group recently reported that incubation with TSH in human heart samples did not affect the expression of KCNH2 gene [15].
The slow delayed rectifier K+ current, IKs, is primarily responsible for the adaptation of repolarization duration to changes in heart rate and sympathetic stimulation. Again, T3+Dexamethasone-treated hIPS-CMs developed increased IKs expression [63], consistent with an important role for T3 in assisting electrophysiological maturation of cardiac cells. However, to our knowledge, the direct effect of thyroid hormones on IKs in adult cardiac myocytes has not been studied. In the context of hypothyroidism, the results on the IKs are contradictory. A first study reported a reduction of IKs amplitude in guinea pigs with primary hypothyroidism [65]. In contrast, later work in hypothyroid mice found that the expression of the IKs-generating genes KCNQ1 and KCNE1 and the current amplitude were increased [33]. Further experiments would be needed to confirm the effect of thyroid hormones on adult cardiac myocytes, but in light of the results described above on ICa-L, the guinea pig may not to be a good model for studying the electrophysiological effects of thyroid hormones [8, 15, 33, 64, 65]. On the other hand, we have observed that incubation with TSH reduces the expression of the KCNQ1 gene in control adult human heart samples. This could explain, at least in part, the higher incidence of cardiac arrhythmias in hypothyroid patients under conditions of sympathetic stimulation, observed both in vivo and in silico [15].
Last, the ultrarapid outward current, IKur, has little physiological relevance in the human ventricle but is essential for atrial repolarization. Both primary (with high TSH) and central HT (with low TSH) reduce Kv1.5 channel expression and IKur amplitude [15, 33, 47]. This finding, together with the absence of effect after incubation with TSH, indicates that IKur is directly regulated by T3 [2]. This could also explain the high incidence of atrial arrhythmias in hypothyroid patients.
Cardiac electrical remodeling is different depending on the combination of hormones
Since the expression of ionic currents is not homogeneous in the heart, action potential waveform is different in sinoatrial and atrioventricular nodes, atria, Purkinje fibers, and ventricular endocardium, midmyocardium, and epicardium. In addition, plasmatic levels of T3 and TSH are different in each type of HT, and these hormones selectively regulate ionic currents. Ultimately, several combinations of hormone levels and hormone-induced effects on almost all cardiac ionic currents have been extensively studied. These results can be introduced in mathematical models of atrial and ventricular action potentials [43, 44] to predict global effects in the heart (Fig. 2).

Human atrial and ventricular action potentials (AP) simulated in conditions of primary or central HT. Atrial and ventricular electrical remodeling caused by central hypothyroidism (cHT; low TSH) and primary hypothyroidism (pHT; high TSH). cHT significantly prolongs the duration of atrial AP, but only slightly that of ventricular AP. In pHT, repolarization in the atrium fails, which explains the appearance of atrial fibrillation on the ECG. Similarly, in pHT, the ventricular action potential also shows a significant alteration of repolarization, which explains the incidence of early afterpotentials (EAD) and extrasystoles. Atrial and ventricular action potentials were simulated using the Nygren-Firek-Clark-Lindblad-Clark-Giles and O’Hara-Rudy dynamic models, respectively
T3 hormone is necessary for the postnatal development of potassium currents such as Ito, IKr, and IKs. Subsequently, the effect of this hormone on Ito and IKr currents is reduced throughout development and finally disappears in adult cardiomyocytes. In contrast, T3 hormone increases the expression of CACNA1, KCNQ1, and KCNE1 genes and the amplitude of ICa-L and IKs currents in adult cardiac myocytes [33, 52, 53, 56, 63, 64, 68].
On the other hand, TSH has also direct effects on cardiac ion channel expression. Whereas TSH has no effect on the expression or amplitude of ICa-L or IKr, it does reduce the expression and amplitude of Ito, IKs, and IKur through the activation of its receptor and the cAMP/PKA pathway [2, 15].
Clinical implications
Arrhythmogenic mechanisms
An accepted paradigm in cardiology is a substrate and a trigger are necessary for an arrhythmia to occur. Probably, the most common pro-arrhythmic substrate is QT prolongation [31, 41]. As explained, hypothyroidism modifies the functional expression of cardiac ionic currents, and this electrical remodeling often results in QT prolongation. Then, the main triggers of arrhythmia, which are early afterdepolarizations (EAD) and late afterdepolarizations (DAD), could act on this substrate.
DADs are often driven by spontaneous calcium release during diastole when intracellular Ca2+ overload increases the activity of the Na+/Ca2+ exchanger (NCX). Research from different groups demonstrated that, in hypothyroidism, the elevation of circulating TSH levels increases the expression of the NCX at the mRNA and protein level [15, 16, 33]. Surprisingly, although there are more NCX, its activity is inhibited by TSH [15]. In addition, hypothyroidism does not induce Ca2+ overload and even reduces Ca2+ transients and sarcoplasmic reticulum Ca2+ release [40]. Taken together, these results rule out DAD as the main trigger of arrhythmia in HT.
On the other side, in patients with prolonged QT interval, arrhythmias are often triggered by EADs [36]. EADs are voltage oscillations during the phase 2 or 3 of the action potential caused by the reactivation of depolarizing currents. The increase of ICa-L and the decrease of K+ currents prolong AP duration and can induce EADs due to the activation of the late sodium current, INa-L [25]. Our group has used the O’Hara-Rudy-dynamic model of ventricular AP [44] in in silico populations of control and hypothyroid patients and found higher incidence of EADs in the modeled hypothyroid patients [15]. In agreement with these simulations, a very recent paper demonstrated a high incidence of EADs in hypothyroid mouse hearts, which were abolished by the INa-L-specific blocker ranolazine [54].
Treatment of hypothyroidism
Hypothyroidism was treated with crude thyroid extract since the end of the nineteenth century. Later, a mixture of T3 and T4 began to be used, which continued to be used until the third quarter of the twentieth century. From then on, the use of T3 began to be eliminated because it was discovered that the organism transforms T4 into T3 and because exogenous T3 caused adverse effects. Although originally one of the reasons for the decline in the use of T3 in the treatment of HT was that it caused ventricular extrasystoles, it was demonstrated that treatment with either T4 or T3 improves cardiac electrical remodeling and reduces the incidence of arrhythmias in animal models of myocardial infarction and diabetes [17, 49, 65]. Currently, the standard treatment of hypothyroidism is daily levothyroxine (LT4) at the dose that normalizes TSH levels. However, it has been shown that this treatment does not eliminate symptoms in up to 20% of patients, and therefore, the introduction of T3 is being discussed again [9, 29, 51], although its effect on arrhythmogenesis in humans still needs to be studied in depth.
Relevance of TSH levels
Serum TSH reflects thyroid status in a more sensitive way than free thyroxine, but it is only a valid measure when the hypothalamic-pituitary axis is intact. Although serum TSH measurement alone is not sufficient for the diagnosis of central hypothyroidism, currently, it is the best test to screen for primary hypothyroidism [48, 61].
The Framingham study [46] assessed whether TSH could affect left ventricular structure and function. No significant associations were observed between this hormone and left ventricular mass, wall thickness, or systolic function. However, TSH affects cardiac electrical behavior. In this sense, several clinical studies indicated that elevated serum TSH level played an essential role in QT prolongation and dispersion in patients with hypothyroidism [7, 21]. Experimental findings from our laboratory are consistent with this, since TSH directly modulates cardiac currents, prolongs repolarization, and increases the susceptibility to cardiac arrhythmias [2, 15].
A very recent study has revealed that hypothyroidism is an independent predictor of progression from paroxysmal AF to persistent AF after AF ablation, even after treatment and normalization of thyroxine levels. This supports the hypothesis that TSH could be involved in the remodeling, either electrical or structural, responsible for the appearance or maintenance of the AF substrate [35].
In addition, increased mortality and incidence of major adverse cardiovascular endpoints (MACE) have been reported among individuals with both high and low TSH levels, even within the range of values considered normal [1, 28, 42]. In the clinical setting, this raises the question of whether the reference ranges of serum TSH should be re-redefined [18, 19, 34, 55].
Conclusion
Metabolic thyroid hormones are important in the development and maturation of potassium currents such as Ito and IKr, but their effect on these currents vanishes during development. In adult cardiac myocytes, thyroid hormones modulate the expression and behavior of ICa-L and IKs.
Thyroid-stimulating hormone has also important effects in the regulation of cardiac electrical activity. Through the activation of its receptor in the heart, TSH modulates the expression of the repolarizing currents Ito, IKs, and IKur. The important role that this hormone plays in cardiac electrical remodeling and the appearance of cardiac arrhythmias that occur in hypothyroidism have recently been demonstrated (Fig. 3).

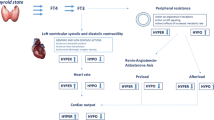
Both T3 and TSH are essential for the regulation of cardiac electrical activity. T3 regulates the expression of Ca2+ and K+ channels in the cardiac myocyte membrane in healthy conditions. TSH, in addition to modulating the release of T3 from the thyroid, has a direct effect on cardiac myocytes, as it regulates the expression of K+ channels. The balance between these hormones determines the shape and duration of the action potential in isolated cardiomyocytes, and the characteristics of the electrocardiogram of patients. Low T3 and high TSH increase Ca2+ channels and decrease K+ channel expression, and this deregulation leads to arrhythmia. (Healthy, black traces; HT, red traces; RA/LA, right/left atria; RV/LV, right/left ventricle; Ao/PA, aorta/pulmonar artery)
References
Akirov A, Gimbel H, Grossman A, Shochat T, Shimon I (2017) Elevated TSH in adults treated for hypothyroidism is associated with increased mortality. Eur J Endocrinol 176:57–66
Alonso H, Fernandez-Ruocco J, Gallego M, Malagueta-Vieira LL, Rodríguez-de-Yurre A, Medei E, Casis O (2015) Thyroid stimulating hormone directly modulates cardiac electrical activity. J Mol Cel Cardiol 89:280–286
Altun A, Altun G, Ozbay G (1999) QT dispersion in hypothyroidism. Int J Cardiol 72:93–95
Aweimer A, Schiedat F, Schöne D, Landgrafe-Mende G, Bogossian H, Mügge A, Patsalis PC, Gotzmann M, Akin I, El-Battrawy I, Dietrich JW (2021) Abnormal cardiac repolarization in thyroid diseases: results of an observational study. Front Cardiovasc Med 8:738–517. https://doi.org/10.3389/fcvm.2021.738517
Ayala A, Mark D, Ladenson MD (2000) When to treat mild hypothyroidism. Endocrinol Metabol Clin North Am 29:399–415
Bagchy N, Brown T, Parish F (1990) Thyroid dysfunction in adults over age 55 years. A study in an urban US community. Arch Intern Med 150:785–787
Bakiner O, Ertorer M, Haydardedeoglu F, Bozkirli E, Tutuncu N, Demirag N (2008) Subclinical hypothyroidism is characterized by increased QT interval dispersion among women. Med Princ Pract 17:390–394
Bosch RF, Wang Z, Li GR, Nattel S (1999) Electrophysiological mechanisms by which hypothyroidism delays repolarization in guinea pig hearts. Am J Physiol 277:211–220
Casula S, Ettleson MD, Bianco AC (2023) Are we restoring thyroid hormone signaling in levothyroxine-treated patients with residual symptoms of hypothyroidism? Endocr Pract 29(7):581–588. https://doi.org/10.1016/j.eprac.2023.04.003
Ciulla M, Paliotti R, Cortelazzi D, Tortora G, Barelli M, Buonamici V, Magrini F, Beck-Peccoz P (2001) Effects of thyroid hormones on cardiac structure: a tissue characterization study in patients with thyroid disorders before and after treatment. Thyroid 7:613–619
Chacón I, Barrio V (2013) Nota de prensa de la Sociedad Española de Endocrinología y Nutrición
Davies T, Marians R, Latif R (2002) The TSH receptor reveals itself. J Clin Invest 110:161–164
Dillmann WH (1989) Diabetes and thyroid-hormone-induced changes in cardiac function and their molecular basis. Annu Rev Med 40:373–394
Drvota V, Janson A, Norman C, Sylven C, Haggblad J, Bronnegard M, Marcus C (1995) Evidence for the presence of functional thyrotropin receptor in cardiac muscle. Biochem Biophys Res 211:426–431
Fernandez-Ruocco J, Gallego M, Rodriguez-de-Yurre A, Zayas-Arrabal J, Echeazarra L, Alquiza A, Fernandez-Lopez V, Rodriguez-Robledo JM, Brito O, Schleier Y, Sepulveda M, Oshiyama NF, Vila-Petroff M, Bassani RA, Medei EH, Casis O (2019) High thyrotropin is critical for cardiac electrical remodeling and arrhythmia vulnerability in hypothyroidism. Thyroid 29:934–945
Ferrer T, Arín R, Casis E, Torres-Jacome J, Sanchez-Chapula JA, Casis O (2012) Mechanisms responsible for the altered cardiac repolarization dispersion in experimental hypothyroidism. Acta Physiol (Oxf) 204:502–512
Ferrer T, Gallego M, Madrigal R, Torres J, Navarro R, Casis O, Sánchez-Chapula JA (2006) DITPA restores the repolarizing potassium currents Itof and Iss in cardiac ventricular myocytes of diabetic rats. Life Sci 79:883–889
Fitzgerald SP, Bean NG, Falhammar H, Tuke J (2020) Clinical parameters are more likely to be associated with thyroid hormone levels than with thyrotropin levels: a systematic review and meta-analysis. Thyroid 30(12):1695–1709. https://doi.org/10.1089/thy.2019.0535
Fitzgerald SP, Falhammar H (2022) Redefinition of successful treatment of patients with hypothyroidism. Is TSH the best biomarker of euthyroidism? Front Endocrinol (Lausanne) 13:854–920. https://doi.org/10.3389/fendo.2022.920854
Galetta F, Franzoni F, Fallahi P, Tocchini L, Braccini L, Santoro G, Antonelli A (2008) Changes in heart rate variability and QT dispersion in patients with overt hypothyroidism. Eur J Endocrinol 158:85–90
Galetta F, Franzoni F, Fallahi P, Tocchini L, Graci F, Gaddeo C, Rossi M, Cini G, Carpi A, Santoro G, Antonelli A (2010) Changes in autonomic regulation and ventricular repolarization induced by subclinical hyperthyroidism. Biomed Pharmacother 64:546–549
Garber JR, Cobin RH, Gharib H, Hennessey JV, Klein I, Mechanick JI, Woeber KA (2012) Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Endocrine Practice 18(6):989–1028
Gascó E, Serna MC, Vázquez A, Peremiquel M, Ibarz M, Serra L (1999) The prevalence of thyroid functional disorders in the province of Lleida. Aten Primaria 24:460–475
Gupta V, Lee M (2011) Central hypothyroidism. Indian. J. Endocrinol Metab 15:S99–S106
Horváth B, Szentandrássy N, Almássy J, Dienes C, Kovács ZM, Nánási PP, Banyasz T (2022) Late sodium current of the heart: where do we stand and where are we going? Pharmaceuticals (Basel) 15(2):231. https://doi.org/10.3390/ph15020231
Huang W, Xu J, Jing F et al (2014) Functional thyrotropin receptor expression in the ventricle and the effects on ventricular BNP secretion. Endocrine 46:328–339
Huber G, Staub J, Meier C, Mitrache C, Guglielmetti M, Huber P, Braverman LE (2002) Prospective study of the spontaneous course of subclinical hypothyroidism: prognostic value of thyrotropin, thyroid reserve and thyroid antibodies. J Clin Endocrinol Metab 87:3221–3226
Inoue K, Tsujimoto T, Saito J, Sugiyama T (2016) Association between serum thyrotropin levels and mortality among euthyroid adults in the United States. Thyroid 10:1457–1465
Jonklaas J, Bianco AC, Cappola AR, Celi FS, Fliers E, Heuer H, McAninch EA, Moeller LC, Nygaard B, Sawka AM, Watt T, Dayan CM (2021) Evidence-based use of levothyroxine/liothyronine combinations in treating hypothyroidism: a consensus document. Thyroid 31(2):156–182. https://doi.org/10.1089/thy.2020.0720
Kahaly GJ, Dillmann WH (2005) Thyroid hormone action in the heart. Endocr Rev 26:704–728
Keating MT, Sanguinetti MC (2001) Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104:569–580
Kim WG, Kim WB, Woo G, Kim H, Cho Y, Kim TY et al (2017) Thyroid stimulating hormone reference range and prevalence of thyroid dysfunction in the Korean population: Korea National Health and Nutrition Examination Survey 2013 to 2015. Endocrinol Metab (Seoul) 32:106–114
Le-Bouter S, Demolombe S, Chambellan A, Bellocq C, Aimond F, Toumaniantz G, Lande G, Siavoshian S, Baró I, Pond AL, Nerbonne JM, Léger JJ, Escande D, Charpentier F (2003) Microarray analysis reveals complex remodeling of cardiac ion channel expression with altered thyroid status: relation to cellular and integrated electrophysiology. Circ Res 92:234–242
Leung AM (2016) Mortality risks among individuals with high-normal serum TSH concentrations were increased as compared with those with midnormal TSH concentrations. Clin Thyroidol 28:287–289
Li GY, Elimam AM, Lo LW, Lin YJ, Chang SL, Hu YF, Chung FP, Chao TF, Lin CY, Liu CM, Liao JN, Ton AK, Yugo D, Lin L, Tuan TC, Kao PH, Liu SH, Chhay C, Kuo L et al (2023) Factors predicting the progression from paroxysmal to persistent atrial fibrillation despite an index catheter ablation. J Cardiovasc Electrophysiol. https://doi.org/10.1111/jce.16100
Liu GX, Choi BR, Koren G (2012) Differential conditions for early after-depolarizations and triggered activity in cardiomyocytes derived from transgenic LQT1 and LQT2 rabbits. J Physiol 590:1171–1180
Lorenzo Gómez T, Cardelle Pérez F, De Las Heras Liñero E (2010) The high prevalence of thyroid dysfunction in psychiatric inpatients. Rev Psiquiatr Salud Ment 3:23-26
McDermott MT, Ridgway EC (2001) Subclinical hypothyroidism is mild thyroid failure and should be treated. J Clin Endocrinol Metab 86:4585–4590
Mendes D, Alves C, Silverio N, Batel Marques F (2019) Prevalence of undiagnosed hypothyroidism in Europe: a systematic review and meta-analysis. European Thyroid Journal 8(3):130–143
Montalvo D, Pérez-Treviño P, Madrazo-Aguirre K, González-Mondellini FA, Miranda-Roblero HO, Ramonfaur-Gracia D, Jacobo-Antonio M, Mayorga-Luna M, Norma L, Gómez-Víquez N, García N, Altamirano J (2018) Underlying mechanism of the contractile dysfunction in atrophied ventricular myocytes from a murine model of hypothyroidism. Cell Calcium 72:26–38
Moss AJ, Kass RS (2005) Long QT syndrome: from channels to cardiac arrhythmias. J. Clin. Invest 115:2018–2024
Müller P, Leow MK, Dietrich JW (2022) Minor perturbations of thyroid homeostasis and major cardiovascular endpoints-physiological mechanisms and clinical evidence. Front Cardiovasc Med 9:942–971. https://doi.org/10.3389/fcvm.2022.942971
Nygren A, Fiset C, Firek L, Clark JW, Lindblad DS, Clark RB, Giles WR (1998) Mathematical model of an adult human atrial cell: the role of K+ currents in repolarization. Circ Res 82(1):63–81. https://doi.org/10.1161/01.res.82.1.63
O'Hara T, Virág L, Varró A, Rudy Y (2011) Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol 7(5):e1002061. https://doi.org/10.1371/journal.pcbi.1002061
Ojamaa K, Kenessey A, Shenoy R, Klein I (2000) Thyroid hormone metabolism and cardiac gene expression after acute myocardial infarction in the rat. Am J Physiol Endocrinol Metab 279:E1319–E1324
Pearce EN, Yang Q, Benjamin EJ, Aragam J, Vasan RS (2010) Thyroid function and left ventricular structure and function in the Framingham Heart Study. Thyroid 20(4):369–373
Pereira L, Bare DJ, Galice S, Shannon TR, Bers DM (2017) β-Adrenergic induced SR Ca2+ leak is mediated by an Epac-NOS pathway. J Mol Cell Cardiol 108:8–16
Persani L, Ferretti E, Borgato S et al (2000) Circulating thyrotropin bioactivity in sporadic central hypothyroidism. J Clin Endocrinol Metab 85:3631–3635. https://doi.org/10.1210/jcem.85.10.6895
Rajagopalan V, Zhang Y, Ojamaa K, Chen YF, Pingitore A, Pol CJ, Saunders D, Balasubramanian K, Towner RA, Gerdes AM (2016) Safe oral triiodo-L-thyronine therapy protects from post-infarct cardiac dysfunction and arrhythmias without cardiovascular adverse effects. PLoS One 11(3):151–413. https://doi.org/10.1371/journal.pone.0151413
Roberts CG, Ladenson PW (2004) Hypothyroidism. Lancet 363:793–803
Shakir MKM, Brooks DI, McAninch EA, Fonseca TL, Mai VQ, Bianco AC, Hoang TD (2021) Comparative effectiveness of levothyroxine, desiccated thyroid extract, and levothyroxine+liothyronine in hypothyroidism. J Clin Endocrinol Metab 106(11):4400–4413. https://doi.org/10.1210/clinem/dgab478
Shimoni Y, Fiset C, Clark RB, Dixon JE, McKinnon D, Giles WR (1997) Thyroid hormone regulates postnatal expression of transient K+ channel isoforms in rat ventricle. J Physiol 500:65–73
Shimoni Y, Severson DL (1995) Thyroid status and potassium currents in rat ventricular myocytes. Am J Physiol 268:H576–H583
Souza DS, Marques LP, Costa AD, Cruz JS, Rhana P, Santos-Miranda A, Joviano-Santos JV, Durço AO, Vasconcelos CML, Roman-Campos D (2022) Experimental hypothyroidism induces cardiac arrhythmias and ranolazine reverts and prevents the phenotype. Life Sci 308:120–945. https://doi.org/10.1016/j.lfs.2022.120945
Spaulding SW (2017) Sequential TSH determinations may help in assessing the adequacy of treatment for overt hypothyroidis in older patients. Clin Thyroidol 29:48–51
Sun Z, Ojamaa K, Coetzee W, Artman M, Klein I (2000) Effects of thyroid hormone on action potential and repolarizing currents in rat ventricular myocytes. Am J Physiol Endocrinol Metab 278:E302–E307
Takawale A, Aguilar M, Bouchrit Y, Hiram R (2022) Mechanisms and management of thyroid disease and atrial fibrillation: impact of atrial electrical remodeling and cardiac fibrosis. Cells 11(24):40–47. https://doi.org/10.3390/cells11244047
Tang Y, Kuzman J, Said S, Anderson B, Wang X, Gerdes A (2005) Low thyroid function leads to cardiac atrophy with chamber dilatation, impaired myocardial blood flow, loss of arterioles, and severe systolic dysfunction. Circulation 112:3122–3130
Ulivieri A, Lavra L, Magi F et al (2022) Thyroid hormones regulate cardiac repolarization and QT-interval related gene expression in hiPSC cardiomyocytes. Sci Rep 12:568
Unal O, Erturk E, Ozkan H, Kiyici S, Guclu M, Ersoy C, Yener F, Imamoglu S (2007) Effect of levothyroxine treatment on QT dispersion in patients with subclinical hypothyroidism. Endocr Pract 13:711–715
Van Uytfanghe K, Ehrenkranz J, Halsall D, Hoff K, Ping Loh T, Spencer CA, Köhrle J, ATA Thyroid Function Tests Writing Group (2023) Thyroid stimulating hormone and thyroid hormones (triiodothyronine and thyroxine): an American Thyroid Association-commissioned review of current clinical and laboratory status. Thyroid 33:1013–1028
Vieira IH, Rodrigues D, Paiva I (2022) The Mysterious Universe of the TSH Receptor. Front Endocrinol (Lausanne) 13:715–944
Wang L, Wada Y, Ballan N, Schmeckpeper J, Huang J, Rau CD, Wang Y, Gepstein L, Knollmann BC (2021) Triiodothyronine and dexamethasone alter potassium channel expression and promote electrophysiological maturation of human-induced pluripotent stem cell-derived cardiomyocytes. J Mol Cell Cardiol 161:130–138
Watanabe H, Washizuka T, Komura S, Yoshida T, Hosaka Y, Hatada K, Aizawa Y, Chinushi M, Yamamoto T, Ma M, Watanabe K (2005) Genomic and non-genomic regulation of L-type calcium channels in rat ventricle by thyroid hormone. Endocr Res 31:59–70
Wickenden AD, Kaprielian R, Parker TG, Jones OT, Backx PH (1997) Effects of development and thyroid hormone on K+ currents and K+ channel gene expression in rat ventricle. J Physiol 504:271–286
Wickenden A, Kaprielian R, You X, Backx P (2000) The thyroid hormone analog DITPA restores I(to) in rats after myocardial infarction. Am J Physiol Heart Circ Physiol 278:H1105–H1116
Yamakawa H, Kato TS, Noh JY, Yuasa S, Kawamura A, Fukuda K, Aizawa Y (2021) Thyroid hormone plays an important role in cardiac function: from bench to bedside. Front. Physiol 12:606–931. https://doi.org/10.3389/fphys.2021.606931
Yang X, Rodriguez M, Pabon L, Fischer KA, Reinecke H, Regnier M, Sniadecki NJ, Ruohola-Baker H, Murry CE (2014) Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. J Mol Cell Cardiol 72:296–304. https://doi.org/10.1016/j.yjmcc.2014.04.005
Zhang Y, Dedkov EI, Teplitsky D, Weltman NY, Pol CJ, Rajagopalan V, Lee B, Gerdes AM (2013) Both hypothyroidism and hyperthyroidism increase atrial fibrillation inducibility in rats. Circ Arrhythm Electrophysiol 6(5):952–959. https://doi.org/10.1161/CIRCEP.113.000502
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. The work has been financed by the Basque Government (IT1707-22) and the Ministerio de Ciencia e Innovación, MICINN (PID2020-118814RB-I00). BS-D is a predoctoral fellow of the UPV/EHU (PIF21/313).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declares no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key points
• Both overt and subclinical hypothyroid patients suffer from cardiac arrhythmias
• T3 regulates the expression and function of some cardiac ion channels
• TSH regulates the expression of some other cardiac ion channels
• Different hormone combinations induce different cardiac remodeling
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Casis, O., Echeazarra, L., Sáenz-Díez, B. et al. Deciphering the roles of triiodothyronine (T3) and thyroid-stimulating hormone (TSH) on cardiac electrical remodeling in clinical and experimental hypothyroidism. J Physiol Biochem 80, 1–9 (2024). https://doi.org/10.1007/s13105-023-01000-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13105-023-01000-z