
Abstract
Advances in high-throughput technologies along with the curation of small-scale experiments has aided in the construction of reference maps of the interactome. These maps are critical to our understanding of genotype-phenotype relationships and disease. However, our knowledge of disease associated genes and the map of the human interactome still remains incomplete. In this study we investigate whether protein–protein interaction networks (PPINs) constructed from either experimental or curated data have an impact upon disease network analysis. An integrative network-driven framework is implemented to integrate diverse heterogeneous data including: gene-expression, PPIN, ontology-based similarity, degree connectivity and betweenness centrality measures to uncover potential Alzhemier disease (AD) candidate genes. Two PPINs have been selected and constructed from (1) experimental high-throughput data and (2) literature-curated sources. Only a marginal overlap of protein pairs between the two PPINs (305 protein pairs) was observed. A total of 17 significant AD gene candidate genes were identified using the literature derived PPIN compared to 20 genes using the PPIN constructed from high-throughput data. Both approaches correctly identified the AD susceptible TRAF1, a critical regulator of cerebral ischaemia–reperfusion injury and neuronal death. Biological process enrichment analysis revealed genes candidates from the literature based PPIN are modulated in AD pathogenesis such as neuron differentiation and involved in KEGG pathways such as neurotrophin signaling pathways. Tissue specific analysis revealed 48 % of AD gene candidates obtained from the literature curated PPIN were expressed in tissues where AD is observed compared to 19 % of gene candidates extracted using the high-throughput PPIN.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
1 Introduction
Both physical and genetic interaction networks have been instrumental in providing valuable insights into complex biological systems. These insights include understanding how different processes communicate through to knowledge of protein function [4]. The advent of high-throughput technologies along with traditional small-scale experiments has aided in the systematic identification of pairwise protein interactions [60] [37] and protein complexes [29] [16]. Public interaction databases including: BioGRID [6], Human Protein Reference Database (HPRD) [28], IntAct [27], Database of Interacting Proteins (DIP) [40] and Kyoto Encyclopedia of Genes and Genomes (KEGG) [26] store many interaction and pathway data across diverse organisms [23]. All these data have been useful as a means to understanding the underlying mechanisms of a cell.
In particular, the construction of protein interaction networks has been beneficial in providing insight into protein function [29]. Protein–protein interactions (PPIs) play an important role in biological processes. Most proteins perform their functions by interacting with other proteins. Furthermore, they aid in the formation of protein complexes and mediate post-translational protein modifications [54]. Systematic efforts have been made over the past few years to map the human protein interaction interactome [collection of all human protein–protein interactions (PPIs)]. These have been performed using high-throughput techniques including: yeast two-hybrid (Y2H) [39], mass spectrometry [13, 50] and co-affinity purification [57]. In addition with curation of small-scale experiments and computational approaches [51], these studies have been advantageous in increasing the coverage of human interactome maps. Furthermore, they have reduced interactome map biases and have been beneficial in providing an estimate of the interactome size [42]. However, these maps still remain incomplete and noisy, which needs to be taken into consideration when applying these PPIs in studies [2]. Literature-curated data sets, although richer in interactions, are prone to investigative biases [60] as they contain more interactions for the more explored disease proteins [56].
With the emergence of the area “network medicine”, further development of protein interaction maps is essential. Network medicine as described by Barabasi et al. [2] aims to explore disease complexity through the systematic identification of disease pathways and modules also taking into consideration molecular relationships between phenotypes. Through the analysis of network topology and network dynamics, key discoveries including identification of novel disease genes and pathways, biomarkers and drug targets for disease are advanced [48]. Key work in the area include the study by Xu et al. [56] who analyzed topological features of a PPI network. This study observed that hereditary disease-genes from the Online Mendelian Inheritance in Man (OMIM) database [21] have a larger degree and tendency to interact with other disease-genes in literature curated networks. These tendencies were not observed in networks constructed from high-throughput experiments. Other studies such as Chuang et al. [9] and Taylor et al. [46] have indicated that the alterations in the physical interaction network may be an indicator of breast cancer prognosis. The paper by Goh et al. [17] illustrated that the majority of disease genes are nonessential and are located in the periphery of functional networks. Research by [14] discovered that genes connected to diseases with similar phenotypes are more likely to interact directly with each other. Network analysis tool such as clustering or graph partitioning have been advantageous in uncovering functional and potential disease modules in the interactome [35]. The study by Vanunu et al. [49] applied a diffusion-based method named PRINCE to prioritize genes in prostate cancer, AD and type 2 diabetes.
What underpins these key research studies and future studies is the reliance on human interactome maps which are critical to the understanding of genotype-phenotype relationships [37]. In this study, we aim into investigate whether experimental data or curated data used to construct a human protein–protein interaction network (PPIN) has an impact upon disease network analysis. Using our previously proposed integrative network-driven pipeline [5] we integrate diverse heterogeneous data including: gene-expression, PPIN, ontology-based similarity, degree connectivity and betweenness centrality measures to uncover potential disease-candidate genes. To investigate the effect of human PPIN selection, a comparison of disease-gene candidates is presented when different human PPINs are integrated into the framework. Two PPINs have been selected for this study: (1) the recently published proteome-scale map of the Human Interactome Network by Rolland et al. [37] which is referred to as PPIN_HTP and (2) a literature curated map obtained from extracting binary PPIs from public databases referred to as PPIN_LIT [51]. To illustrate PPIN impact on disease-gene selection, Alzheimer’s disease (AD) has been selected as a Case Study. AD is a genetically complex disease whereby patients present with progressive dementia [10]. It is the most common form of age cognitive impairment [47]. It is characterized by the loss of neurons along with the presence of axonal dystrophy, mature senile plaques and neurofibrillary tangles [34]. Gene expression profiling studies have been successful in identifying AD affected pathways across different brain areas and tissues including: mitochondrial function, intracellular signaling and neuroinflammation [10]. To evaluate the impact of PPIN selection on the disease-gene selection process we perform biological process enrichment analysis and compare the candidate gene list to a manually curated reference dataset of verified known and susceptible AD disease genes. Furthermore, we investigate the tissues in which AD candidate disease-genes are expressed through incorporation of tissue-specific expression data.
The remainder of the paper is organized as follows, in Sect. 2 the integrative framework is described along with details on datasets and PPINs used in the analysis. Section 3 provides a summary of the results obtained and conclusions along with future work is presented in Sect. 4.
2 Materials and methods
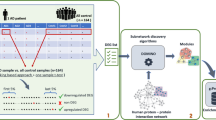
A comparison on the impact human PPINs has on the identification of AD disease-candidate genes is performed via integration of PPIN data with diverse heterogeneous including gene expression and ontology similarity measures. This framework is illustrated in Fig. 1 and detailed in [5]. A literature curated PPIN is obtained by extracting binary interactions from public databases. A PPIN constructed from high-throughput Y2H experiments is obtained from [37].

Methodology overview applied to generate disease candidate genes
2.1 Identification of disease genes
Human AD gene expression data was obtained from the Gene Expression Omnibus (GEO) database. (http://www.ncbi.nlm.nih.gov/geo/). The selected profile GSE4757 was generated using the platform GPL570: Affymetrix Human Genome U133 Plus 2.0 Array. The study by Dunckley et al. [11] examined the transcriptome of entorhinal neurons from six cortical areas with or without neurofibrillary tangles (a histopathology feature of AD) using Laser capture microdissection. The dataset consists of gene expression profiles of NFT-bearing entorhinal cortex neurons from 10 mid-stage AD patients (Disease) compared with 10 histopathologically normal neurons (Control) from the same patients and brain region. These represent the different stages of AD according to the pattern of disease spread. Using the MAS5.0 function in R the CEL files were firstly normalized. Probes in expression profile were then mapped to corresponding NCBI Gene IDs. The average expression value was calculated in cases where the Gene ID related to more than one probe resulting in 20,539 unique Gene IDs.
A total of 10,106 significant genes were obtained using the significance analysis of microarrays (SAM) [48] technique, a regularized t test approach, using the false discovery rate (FDR = 0.98). Differentially expressed (DE) genes are genes whose expression levels are significantly different between two groups of experiments. These may indicate clinical markers for involvement in disease pathology.
2.2 Human protein–protein interaction networks
2.2.1 Literature based PPIN
The PPIN_LIT was obtained from [51]. The dataset consist of 11,045 binary human protein pairs extracted from seven publically available databases including BioGRID [6], DIP [40], Biomolecular Interaction Network Database (BIND) [1], HPRD [28], InACT [27], Protein Data Bank (PDB) [38] and Molecular INTeraction database (MINT) [7]. The set has been filtered to remove large-scale systematic pairs and pairs involving the products: UBC, SUMO1, SUMO2, SUMO3, SUMO4, or NEDD8. To ensure a high level of quality, binary pairs were further filtered on evidence, whereby protein pairs that are only support by two or more pieces of evidence are included.
2.2.2 High-throughput PPIN
Using the high-throughput method Y2H, the PPIN_HTP was obtained from the systematic screening of 15,517 opening read frames (ORFs) from the platform hORFeome v5.1 (Space II) resulting in 13,944 pairwise interactions. A subset of interactions were validated using three assays: (1) reconstituting membrane bound receptor complex in mammalian cells using MAPPIT, (2) reconstituting fluorescent protein in Chinese hamster ovary cells using protein-fragment complementation assay and (3) the well-based protein nucleic acid programmable protein array wNAPPA [37].
2.3 Integration of PPIN and gene expression data
AD gene expression data was mapped to both the PPIN_LIT and PPIN_HTP via NCBI geneIDs using Cytoscape version 3.2.1 [58]. Mapping AD expression data with the PPIN_HTP resulted in an AD disease specific network consisting of 5457 nodes and 10,852 protein pairs. Applying the same approach to the PPIN_LIT resulted in an AD disease network consisting of 4130 nodes and 12,070 protein pairs.
2.4 Identification of disease candidate genes (hubs)
From the AD disease networks, hub genes were defined based on network topological features using Cytoscape version 3.2.1 [58]. The disease networks are represented as an undirected graphs, G = V, E, whereby V represents a set of nodes (proteins) and \( E = \{ \left( {u,v} \right)|u,v \in V\} \), the set of edges connecting the nodes. Two topological analysis measures were applied to the disease networks to obtain hub genes namely (1) node degree (connectivity) and (2) betweenness centrality.
2.4.1 Degree connectivity
Degree is a measure of the number of edges that connects a node. Genes with a high degree of connectivity within a network have large numbers of interacting partners. In PPINs it has been observed that genes with high degrees of connectivity are more likely to be essential as genes. Furthermore, many interacting partners in a network tend to be involved in important cellular processes [2]. Based on this assumption, hub genes for both the PPIN_LIT and PPIN_HTP were chosen by selecting genes, which have high degree distributions as previously applied by Taylor et al. [46]. To determine the degree cut-off threshold for selecting hubs genes, we applied the approach used by Rakshit et al. [36]. The degree cut-off threshold for selecting hubs was defined as \( AVG + 2 \times \left( {Std} \right) \), where AVG is the average degree across all DE genes in the PPINs and Std, the standard deviation.
2.4.2 Betweenness centrality
Betweenness is a topological feature of a network measuring information flow through the network. In biological networks, betweenness measures the paths through which signals can pass through the interaction network Yu et al. [61]. Identified betweenness as an important topological property of a network where nodes with high betweenness control most of the information flow. Betweenness centrality is calculated in Cytoscape. The betweenness of a node is the number of shortest paths that pass through it. It considers node couples such as (v1, v2) and counts the number of shortest paths linking v1 and v2 and passing through a node n. The value is related to the total number of shortest paths linking v1 and v2. The betweenness centrality \( C_{b} \left( n \right) \) of a node n is computed as follows:
where S and t are nodes in the network different from n, \( \theta_{st} \) denotes the number of shortest paths from S to t, and \( \theta_{st} \left( n \right) \) is the number of shortest paths from S to t that n lies on. Using the node betweenness distribution, genes located in the top 50 % are firstly selected as hub genes. For hub genes that have high betweenness but low connectivity (i.e. degree is less than the degree cut-off threshold), additional filtering is applied to include only genes that are directly connected to at least 2 highly connected nodes. A similar approach is applied in [36].
2.4.3 Calculation of network variation of hub genes
For each hub protein in the PPIN_LIT and PPIN_HTP the average of Pearson correlation coefficients between the hub and each of its respective partners was calculated for both disease and control groups. This method has previously been applied by Taylor et al. [46] to measure network variations among candidate genes and their interacting genes. To determine if interactions are varied, the difference of AD gene expression correlations of PPIs in disease and control samples is calculated. Gene co-expression values were mapped to the PPIN nodes via NCBI gene IDs. The average hub difference (\( AvgPCC \)) off correlation [Pearson’s correlation co-efficient (PCC)] values between the disease and control groups was calculated as follows:
where \( D_{i} \) and \( C_{i} \) represent the correlations of a hub and its interactors for the disease and control groups respectively and n the number of i interactors for a given hub.
To identify genes that are significantly different between the disease and control groups we randomly assigned the expression data gene labels to either the disease or control group 1000 times and recalculated the AvgPCC defined in Eq. 2. The P values for each hub was calculated as the frequency of the random AvgPCC being greater than the original AvgPCC divided by 1000. A network of significant hub genes was generated using significant cut-off threshold of P ≥ 0.05. P values are adjusted using Bonferroni correction.
2.4.4 Ontology based semantic similarity
Genes involved in phenotypically similar diseases are often functionally related on the molecular level [41]. Based on this observation, the semantic similarity between hub genes and their interactors has been selected to analyze hub genes based on the gene ontology (GO) [15]. The GO is a controlled vocabulary describing the characteristics of gene products. Semantic similarity measures evaluate information two genes share. The functional similarity between two proteins is estimated using encoded information in the GO hierarchies. In this study Wang’s [53] measure of similarity is applied to the Biological Process hierarchy. This measure determines the semantic similarity of two GO terms based on the locations of terms in the GO graph and their semantic relations with their ancestor terms. Given a GO term A, T A denotes the set of all its ancestor terms including term A itself. \( S_{A} \left( t \right) \) can be defined as the contribution of a term \( t \in T_{A} \) to the semantics of A based on the relative locations of t and A in the graph. Given GO terms A and B respectively, the semantic similarity between these two terms, \( S_{GO} \left( {A,B} \right) \), is defined as:
As one gene may be annotated by many GO terms, similarity between two genes \( Sim\left( {G_{1} ,G_{2} } \right) \), is then calculated by taking the average semantic similarity scores for all pairs of their associated terms. The similarity score can range between (0,1), whereby a value closer to 1 indicates close relatedness of the two genes in biological process. Wang’s measure was implemented using the GOSemSim package in R [59], taking the median semantic similarity between a hub protein and it’s interactors.
2.5 Evaluation
To evaluate the generated list of significant hub genes, three approaches are implemented. Firstly, functional annotation enrichment is performed using DAVID [22] to investigate the biological implications of the gene list. Secondly, prioritized genes were compared to genes associated with AD. A reference dataset containing known and susceptible AD genes was constructed using the OMIM ‘morbid map’ table [21]. Known and recently discovered AD susceptibility genes in detailed in the study by Lamberet et al. [31] were also included. This resulted in a list of 52 AD related genes. Thirdly, analysis was performed on the integrated prioritized disease-gene candidate list using tissue-specific gene expression to evaluate if gene candidates were expressed in tissues where AD is observed including the “whole brain” and prefrontal cortex. Candidate genes were filtered using tissue-specific gene expression data retrieved from BioGPS [55]. This dataset contains the transcription levels of 84 human tissues and cell lines and was processed using the method described by Lopes et al. [32]. Furthermore, a list of 570 housekeeping genes were also included, obtained from [12] as they are believed to be expressed in all tissues.
3 Results and discussion
Using the proposed methodology in Sect. 2, we apply topological, semantic similarity and functional enrichment approaches to analyze the generated candidate AD genes obtained from using the two different PPINs. These results are summarized below.
3.1 Selecting differentially expressed genes
To identify significantly expressed, disease related genes from the AD gene expression data, SAM analysis was performed in R using the SAM 5.0 package from [8]. A total of 10,107 significantly positive DE genes were observed from 20,539 genes in the AD microarray dataset using the T-statistic and two class unpaired (disease and control) response mode. The DE genes were used as input into the construction of the AD specific PPINs. An overview of the top 10 DE genes is presented in Table 1 along with the SAM score based on the T-statistic value.
3.2 Integration of PPIN with gene expression data
Using the 10,107 significant AD genes identified from the SAM analysis, two AD disease networks were constructed from PPIN data described in Sect. 2. The significant genes were mapped to the PPIN nodes via NCBI gene IDs. The PPIN_LIT protein pairs have been derived from small-scale studies described in literature from seven public databases. These binary literature PPIs although high in quality may reflect bias observed in small-scale studies. Based solely on literature-curated binary interactions, the interactome appears restricted to a narrow dense zone [37]. In contrast to the PPIN_LIT, protein pairs in the PPIN_HTP were obtained from large-scale high-throughput Y2H experimentation. Compared to the PPIN_LIT, the study by Rolland et al. [37]. demonstrates a distributed homogeneously of protein pairs across the interactome. An overlap of 305 protein pairs was observed between the two AD networks mapped using the PPIN_LIT (10,852 protein pairs) and PPIN_HTP (12,070 protein pairs) illustrated in Fig. 2.

Venn diagram illustrating the overlap of protein pairs from the AD specific PPIN_LIT and PPIN_HTP
3.3 Analysis of network topology
Topological analysis was performed on the AD specific PPIN_LIT and PPIN_HTP. An overview of the global properties of these networks is presented in Table 2. Studies of model organisms have observed that proteins with high degree of connectivity tend to be encoded by essential genes [24]. Furthermore, detection of these genes leads to larger numbers of phenotypic outcomes compared to genes with lower connectivity [60]. Wachi et al. [52] observed disease gene encoding proteins in lung squamous cell carcinoma tended to have a higher degree of connectivity. However, not all disease genes in humans are essential genes. Goh et al. [17] found that non-essential disease genes tend to be tissue specific located at the functional periphery of the interactome and do not necessarily encode hubs (highly connected genes). Taking this into consideration, we include another indicator of centrality, betweenness. Both Yu et al. [61] and Joy et al. [25] demonstrated how nodes with a low degree of centrality but high betweenness are important in a network (i.e. bottleneck effect).
Using these networks, degree connectivity and betweenness centrality measures were applied to select hub genes. Genes with a high degree of connectivity and genes with low connectivity but high betweenness were selected using the cut-off thresholds defined in Sect. 2. Using this approach, Table 3 presents the number of hubs for both PPIN_LIT and PPIN_HTP networks respectively.
Interestingly, genes such as CDC42P2, SIAH1, STX4 observed in the PPIN_LIT hub list, are enriched in GO Biological Process terms [15] including: neuron differentiation, neuron projection morphogenesis, and involved in KEGG [45] pathways such as neurotrophin signaling pathways and neurotransmitter transportation.
3.4 Gene ontology semantic similarity analysis
Semantic similarity was applied as a quantitative measure of functionality similarity between gene products. The semantic similarity between a gene hub and it’s interacting partners was calculated using the Wang semantic similarity approach [53] detailed in Eq. (4). The R package GOSemSim was used to calculate the similarity between pairs of genes. To obtain the similarity value for the hub and all its interactors, the median similarity was taken across all protein pairs. The semantic similarity values obtained ranged between 0 and 1. The gene hubs were ranked according to their similarity scores whereby 1 represents the highest similarity and 0 the lowest. Figure 3 presents a comparison between the PPIN_LIT and PPIN_HTP in terms of semantic similarity values obtained. Interestingly, hub genes from the PPIN_LIT obtained higher similarity values compared to hub genes in the PPIN_HTP network. This may reflect the quality of the data used to construct the PPIN_LIT, which has been obtained from literature-curated studies. Furthermore, interaction pairs in this network are supported by two or more sources.

Comparison of the semantic similarity values obtained for hubs from the PPIN_LIT and PPIN_HTP networks
3.5 Co-expression analysis of hub genes
Using AD gene expression data integrated with the PPIN_LIT and PPIN_HTP networks we measured the network variation of hub genes and their interactors using the \( AvgPCC \) equation defined in Sect. 2.4.3. For disease and control groups, PCC values between the hubs and their interactors were calculated. Significant hub genes were selecting using the Bonferroni corrected cut-off threshold of P < 0.05. For the PPIN_LIT network, a total of 17 significant hubs were identified, using the PPIN_HTP network, 20 were identified. No overlapping significant hub genes were observed between the two networks. These genes are summarized in Table 4 below.
3.6 Functional annotation enrichment
The DAVID resource [22] was applied to study the most significant GO terms (biological processes, molecular functions, cellular components) and KEGG pathways associated with the hubs and significant hubs obtained using the PPIN_LIT and PPIN_HTP networks outlined in Table 5. Functional annotation was obtained from extracting the most over-representative GO terms (Biological Process, Cellular Component and Molecular Function) for the groups of genes under observation with respect to the whole genome taken as the reference background set (P value <0.05).
Enrichment analysis of PPIN_LIT hub and significant hub genes identified significant biological processes including: (GO:0030182) neuron differentiation, (GO:0048812) neuron projection morphogenesis and (GO:0031175) neuron projection development. Significant KEGG pathways including: (hsa04722) Neurotrophin signaling pathway, (hsa04010) MAPK signaling pathway, (hsa04630) Jak-STAT signaling pathway and (hsa05014) Amyotrophic lateral sclerosis (ALS) signaling pathway were observed. It was noted that fewer GO terms were enriched across the ontologies and pathways using the hub genes obtained from PPIN_HTP analysis. However, for pathway analysis performed on both hub genes from the PPIN_LIT and PPIN_HTP, the KEGG pathway (hsa05200) pathways in cancer was found to be a highly significantly pathway. This is interesting as recent research summarized in [44] has shown that patients with neurodegenerative disorders (such as AD) have a substantially lower overall risk of developing cancer.
3.7 Reference dataset comparison
The lists of hub genes obtained from using the PPIN_LIT and PPIN_HTP networks were compared to the reference dataset consisting of 52 AD related genes. The hubs PSEN1 and TRAF1 were correctly identified as an AD susceptible genes from the list of hub genes identified using PPIN_LIT. Mutations in PSEN1 are the most common cause of early onset of AD. TRAF1, a critical regulator of cerebral ischaemia–reperfusion injury and neuronal death [33]. Interestingly, TRAF1 was also identified using the PPIN_HTP hub proteins along with LZTS2 which has shown associated with late onset AD [3].
3.8 Tissue analysis
Tissue specificity is an important component of network analysis as genetic diseases often target specific tissue(s). Therefore, perturbations of pathways or proteins may have differential effects among diverse tissues [19]. Taking this into consideration, we further analyzed the gene hubs identified from the PPIN_LIT and PPIN_HTP to determine if gene hubs were expressed in tissues in whereby by AD is observed namely the whole brain and prefrontal cortex. Using tissue specific expression data from along with housekeeping genes we identified that 48 genes hub from the PPIN_LIT and 13 gene hubs from PPIN_HTP were located in the whole brain and/or the prefrontal cortex tissues. Further analysis found that the 13 genes observed in the tissue data from the PPIN_HTP the average GO similarity value was 0.1 (greater than the total average of 0.087) and 3 of the hub genes were significant hub genes. Similar analysis performed using the PPIN_LIT noted that the 48 gene hubs observed in the tissue data had an average GO similarity value of 0.41 (same as the total average) and an overlap of 6 significant genes.
4 Conclusions
The development of high-throughput techniques along with the emergence of network medicine is aiding our understanding of disease and the interrelatedness of disease-related genes and protein [2]. Network theory has been useful in the study of complex neurodegenerative diseases such as AD, Parksinson’s Disease [36] and Multiple Sclerosis [48]. In this study we have highlighted AD as a Case Study in disease network analysis. AD is the most common neurodegenerative disease. Presently, AD therapies are only symptomatic, therefore, an important health priority is the development of novel therapies to impede its progress [18]. The integration of PPINs along with disease datasets is an important tool in unraveling the molecular basis of diseases. This integration can provide identification of genes and proteins associated with diseases, an understanding of disease-network properties, identification of subnetworks, and network-based disease gene classification [43]. However, the map of the binary human PPIN is still incomplete. The study by Yu et al. [60] suggested that high-throughput Y2H datasets contained more false positives compared to literature-curated datasets. Whereas, Rolland et al. [37] observed that literature-curated PPINs are highly biased and only cover a small portion of the interactome.
In this study we presented an evaluation between PPINs constructed using data obtained from experimental high throughput experiments compared to curated data and their affect on identifying candidate AD disease genes through network analysis and integration. We firstly observed limited overlap (305 protein pairs) between the AD specific PPIN_LIT and PPIN_HTP. Furthermore, using the integrative framework to identify significant AD gene candidates no overlap between significant AD gene candidate genes identified using the literature derived PPIN compared to the PPIN constructed from high-throughput data were observed. In terms of enrichment analysis, a strong performance was observed for significant gene hubs identified using the PPIN_LIT. Compared to the PPIN_HTP, a larger proportion of terms are enriched in both the GO and KEGG pathways. In addition, gene candidates from the literature based PPIN are modulated in AD pathogenesis such as neuron differentiation and involved in KEGG pathways such as neurotrophin signaling pathways. Interestingly, the AD susceptible TRAF1 gene was identified by both analysis using the PPIN_LIT and PPIN_HTP networks. Through tissue specific expression analysis we observed that 48 % of AD gene candidates obtained from the literature curated PPIN and 19 % of gene candidates extracted using the high-throughput PPIN were found to be expressed in the whole brain and prefrontal cortex tissues. In summary, we could reason that the PPIN_LIT outperforms the PPIN_HTP in terms of enrichment and tissue analysis along with reference dataset comparison. However, it is important to take into consideration the limited availability and coverage of tissue-specific data [20] along with the possibility that significant genes identified by the PPIN_HTP may still be meaningful but have not been identified due to sociological or experimental biases [37].
As more high-throughput experiments are performed such as Y2H, the coverage of the human interactome continues to improve. This increased coverage, quality, and diversity of human PPIN data will provide further opportunities for the molecular characterization and understanding of human disease [2]. In future work we aim to integrate the high quality binary pairs obtained from literature curation with experimental binary interaction maps increasing the coverage of the interactome.
References
Bader GD, Betel D, Hogue CWV (2003) BIND: the biomolecular interaction network database. Nucleic Acids Res 31:248–250
Barabási A-L, Gulbahce N, Loscalzo J (2011) Network medicine: a network-based approach to human disease. Nat Rev Genet 12:56–68
Beecham GW, Martin ER, Li YJ et al (2009) Genome-wide association study implicates a chromosome 12 risk locus for late-onset Alzheimer disease. Am J Hum Genet 84:35–43
Beltrao P, Cagney G, Krogan NJ (2010) Quantitative genetic interactions reveal biological modularity. Cell 141:739–745. doi:10.1016/j.cell.2010.05.019
Browne F, Wang H, Zheng H (2014) An integrative network-driven pipeline for the prioritization of Alzheimer’s disease genes. In: 2014 IEEE international conference on bioinformatics and biomedicine (BIBM). IEEE, pp 385–390
Chatr-Aryamontri A, Breitkreutz BJ, Heinicke S, et al (2013) The BioGRID interaction database: 2013 Update
Chatr-aryamontri A, Ceol A, Palazzi LM et al (2007) MINT: the molecular INTeraction database. Nucleic Acids Res 35:D572–D574. doi:10.1093/nar/gkl950
Chu G, Li J, Narasimhan B, et al (2011) SAM: significance analysis of microarrays—users guide and technical document. Policy 1–42
Chuang H-Y, Lee E, Liu Y-T et al (2007) Network-based classification of breast cancer metastasis. Mol Syst Biol 3:140. doi:10.1038/msb4100180
Cooper-Knock J, Kirby J, Ferraiuolo L et al (2012) Gene expression profiling in human neurodegenerative disease. Nat Rev Neurol 8:518–530. doi:10.1038/nrneurol.2012.156
Dunckley T, Beach TG, Ramsey KE et al (2006) Gene expression correlates of neurofibrillary tangles in Alzheimer’s disease. Neurobiol Aging 27:1359–1371. doi:10.1016/j.neurobiolaging.2005.08.013
Eisenberg E, Levanon EY (2003) Human housekeeping genes are compact. Trends Genet 19:362–365
Ewing RM, Chu P, Elisma F et al (2007) Large-scale mapping of human protein–protein interactions by mass spectrometry. Mol Syst Biol 3:89
Gandhi TKB, Zhong J, Mathivanan S et al (2006) Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet 38:285–293
Gene T, Consortium O (2000) Gene ontology: tool for the unification of biology. Nat Genet 25:25–29. doi:10.1038/75556
Gingras A-C, Gstaiger M, Raught B, Aebersold R (2007) Analysis of protein complexes using mass spectrometry. Nat Rev Mol Cell Biol 8:645–654
Goh K-I, Cusick ME, Valle D et al (2007) The human disease network. Proc Natl Acad Sci USA 104:8685–8690. doi:10.1073/pnas.0701361104
Goñi J, Esteban FJ, de Mendizábal NV et al (2008) A computational analysis of protein–protein interaction networks in neurodegenerative diseases. BMC Syst Biol 2:52. doi:10.1186/1752-0509-2-52
Guan Y, Gorenshteyn D, Burmeister M et al (2012) Tissue-specific functional networks for prioritizing phenotype and disease genes. PLoS Comput Biol 8:e1002694. doi:10.1371/journal.pcbi.1002694
Guan Y, Gorenshteyn D, Burmeister M, et al (2012b) Tissue-specific functional networks for prioritizing phenotype and disease genes. PLoS Comput Biol 8(9):e1002694
Hamosh A, Scott AF, Amberger JS et al (2005) Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 33:D514–D517. doi:10.1093/nar/gki033
Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Ideker T, Krogan NJ (2012) Differential network biology. Mol Syst Biol 8:565. doi:10.1038/msb.2011.99
Jeong H, Mason SP, Barabási AL, Oltvai ZN (2001) Lethality and centrality in protein networks. Nature 411:41–42
Joy MP, Brock A, Ingber DE, Huang S (2005) High-betweenness proteins in the yeast protein interaction network. J Biomed Biotechnol 2005:96–103
Kanehisa M (2002) The KEGG database. Novartis Found Symp 247: 91–101; discussion 101–103, 119–128, 244–252
Kerrien S, Aranda B, Breuza L, et al (2012) The IntAct molecular interaction database in 2012
Keshava Prasad TS, Goel R, Kandasamy K et al (2009) Human protein reference database: 2009 update. Nucleic Acids Res 37:D767–D772
Krogan NJ, Cagney G, Yu H et al (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440:637–643
Lage K, Hansen NT, Karlberg EO et al (2008) A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proc Natl Acad Sci USA 105:20870–20875. doi:10.1073/pnas.0810772105
Lambert JC, Ibrahim-Verbaas CA, Harold D et al (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45:1452–1458
Lopes TJS, Schaefer M, Shoemaker J et al (2011) Tissue-specific subnetworks and characteristics of publicly available human protein interaction databases. Bioinformatics 27:2414–2421
Lu Y-Y, Li Z-Z, Jiang D-S et al (2013) TRAF1 is a critical regulator of cerebral ischaemia–reperfusion injury and neuronal death. Nat Commun. doi:10.1038/ncomms3852
Mattson MP (2004) Pathways towards and away from Alzheimer’s disease. Nature 430:631–639
Navlakha S, Kingsford C (2010) The power of protein interaction networks for associating genes with diseases. Bioinformatics 26:1057–1063
Rakshit H, Rathi N, Roy D (2014) Construction and analysis of the protein–protein interaction networks based on gene expression profiles of Parkinson’s disease. PLoS One 9:e103047. doi:10.1371/journal.pone.0103047
Rolland T, Taşan M, Charloteaux B et al (2014) A proteome-scale map of the human interactome network. Cell 159:1212–1226. doi:10.1016/j.cell.2014.10.050
Rose PW, Prlić A, Bi C et al (2014) The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res 43:D345–D356. doi:10.1093/nar/gku1214
Rual JF, Venkatesan K, Hao T et al (2005) Towards a proteome-scale map of the human protein–protein interaction network. Nature 437:1173–1178. doi:10.1038/nature04209
Salwinski L, Miller CS, Smith AJ et al (2004) The database of interacting proteins: 2004 update. Nucleic Acids Res 32:D449–D451
Schlicker A, Lengauer T, Albrecht M (2010) Improving disease gene prioritization using the semantic similarity of gene ontology terms
Schwartz AS, Yu J, Gardenour KR et al (2009) Cost-effective strategies for completing the interactome. Nat Methods 6:55–61
Sevimoglu T, Arga KY (2014) The role of protein interaction networks in systems biomedicine. Comput Struct Biotechnol J 11:22–27. doi:10.1016/j.csbj.2014.08.008
Tabarés-Seisdedos R, Rubenstein JL (2013) Inverse cancer comorbidity: a serendipitous opportunity to gain insight into CNS disorders. Nat Rev Neurosci 14:293–304. doi:10.1038/nrn3464
Tanabe M, Kanehisa M (2012) Using the KEGG database resource
Taylor IW, Linding R, Warde-Farley D et al (2009) Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat Biotechnol 27:199–204. doi:10.1038/nbt.1522
Tiraboschi P, Hansen LA, Thal LJ, Corey-Bloom J (2004) The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology 62:1984–1989
Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98:5116–5121. doi:10.1073/pnas.091062498
Vanunu O, Magger O, Ruppin E et al (2010) Associating genes and protein complexes with disease via network propagation. PLoS Comput Biol 6:e1000641. doi:10.1371/journal.pcbi.1000641
Vasilescu J, Figeys D (2006) Mapping protein–protein interactions by mass spectrometry. Curr Opin Biotechnol 17:394–399
Venkatesan K, Rual J-F, Vazquez A et al (2009) An empirical framework for binary interactome mapping. Nat Methods 6:83–90
Wachi S, Yoneda K, Wu R (2005) Interactome-transcriptome analysis reveals the high centrality of genes differentially expressed in lung cancer tissues. Bioinformatics 21:4205–4208. doi:10.1093/bioinformatics/bti688
Wang JZ, Du Z, Payattakool R et al (2007) A new method to measure the semantic similarity of GO terms. Bioinformatics 23:1274–1281. doi:10.1093/bioinformatics/btm087
Wang S, Xing J (2013) A primer for disease gene prioritization using next-generation sequencing data. Genomics Inform 11:191–199. doi:10.5808/GI.2013.11.4.191
Wu C, Orozco C, Boyer J et al (2009) BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol 10:R130
Xu J, Li Y (2006) Discovering disease-genes by topological features in human protein–protein interaction network. Bioinformatics 22:2800–2805
Xu X, Song Y, Li Y et al (2010) The tandem affinity purification method: an efficient system for protein complex purification and protein interaction identification. Protein Expr Purif 72:149–156
Yeung N, Cline MS, Kuchinsky A et al (2008) Exploring biological networks with cytoscape software. Curr. Protoc, Bioinforma
Yu G, Li F, Qin Y et al (2010) GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 26:976–978
Yu H, Braun P, Yildirim MA et al (2008) High-quality binary protein interaction map of the yeast interactome network. Science 322:104–110. doi:10.1126/science.1158684
Yu H, Kim PM, Sprecher E et al (2007) The importance of bottlenecks in protein networks: correlation with gene essentiality and expression dynamics. PLoS Comput Biol 3:713–720
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Browne, F., Wang, H. & Zheng, H. Investigating the impact human protein–protein interaction networks have on disease-gene analysis. Int. J. Mach. Learn. & Cyber. 9, 455–464 (2018). https://doi.org/10.1007/s13042-016-0503-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13042-016-0503-5