
Abstract
Within Enugu and Anambra States, Nigeria, identification of fishes has been based on morphological traits and do not account for existing biodiversity. For DNA barcoding, assessment of biodiversity, conservation and fishery management, 44 fish sampled from Enugu and Anambra States were isolated, amplified and sequenced with mitochondrial cytochrome oxidase subunit I (COI). Twenty groups clustering at 100% bootstrap value including monophyletic ones were identified. The phylogenetic diversity (PD) ranged from 0.0397 (Synodontis obesus) to 0.2147 (Parachanna obscura). The highest percentage of genetic distance based on Kimura 2-parameter was 37.00 ± 0.0400. Intergeneric distances ranged from 15.8000 to 37.0000%. Congeneric distances were 6.9000 ± 0.0140–28.1000 ± 0.0380, with Synodontis as the existing synonymous genus. Confamilial distances in percentage were 16.0000 ± 0.0140 and 25.7000 ± 0.0300. Forty-two haplotypes and haplotype diversity of 0.9990 ± 0.0003 were detected. Nucleotide diversity was 0.7372, while Fu and Li’s D* test statistic was 2.1743 (P < 0.02). Tajima’s D was 0.2424 (P > 0.10) and nucleotide frequencies were C (17.70%), T (29.40%), A (24.82%), G (18.04%) and A + T (54.22%). Transitional mutations were more than transversions. Twenty species (99–100%) were identified with the e-value, maximum coverage and bit-score of 1e−43, 99–100 and 185–1194, respectively. Seventeen genera and 12 families were found and Clariidae (n = 14) was the most dominant among other families. The fish species resolution, diversity assessment and phylogenetic relationships were successfully obtained with the COI marker. Clariidae had the highest number of genera and families. Phylogenetic diversity analysis identified Parachanna obscura as the most evolutionarily divergent one. This study will contribute to fishery management, and conservation of freshwater fishes in Enugu and Anambra States, Nigeria.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fishes are vital aquatic animals of great diversity in morphological appearances and they are more than 35,000 species globally that contribute significantly to the existing vertebrates (Zhang and Hanner 2012; Bingpeng et al. 2018). At a numeric basis, genuine scientific descriptions have been noted for more than 27,977 species of different fishes in approximately 62 orders and 515 families (Nelson 2006). These organisms play significant roles in income generation, protein and mineral dietary supplements for human utilization as well as serving as major components of biodiversity (Ward et al. 2005; Rasmussen et al. 2009; Ugwumba and Ugwumba 2003). Fishes possess characteristics of remarkable morphological features that pose great challenges in identification using only descriptors from morphological and morphometric features (Triantafyllidis et al. 2011; Zhang and Hanner 2011). Furthermore, characteristics of convergence and divergence in fishes are the resultant alterations in the morpho-based features that lead to controversial classification, distinguishability and identification of fishes (Keskin and Atar 2013). Characterization, identification and assessment of biodiversity are the ingredients to fishery investigations and assessment of natural reserves (Ardura et al. 2013; Vartak et al. 2015). The challenges posed by the use of morpho-based identification procedure coupled with dwindling number of experienced taxonomists have necessitated the use of an informative molecular method (Steinke et al. 2009). Unlike the morpho-based method that is faced with inaccuracy of identification due to existence of synonymous external morphological features and variations at different developmental stages, DNA barcoding is free from these barriers and can accurately identify species and also discover cryptic ones (Bingpeng et al. 2018).
Proper identification of fishes which has been traditionally based on morphological attributes requires a better, reliable, sensitive and affordable alternative to understand and obtain basic knowledge of fish identities and species biodiversity enrichment within given geographical locations. Identification of fishes using morphology-based approach poses a great challenge following high diversity in developmental stages and morphological plasticity (Victor et al. 2009). The DNA barcoding identification approach has been developed and noted to be potentially efficacious due to inherent characteristics of sensitivity, reproducibility, reliability and environmental friendliness (Zhang et al. 2004; Comi et al. 2005; Teletchea 2009). This method, if well utilized, will eliminate existing misidentification and the availability of cryptic species that mimic and equally compromise the accuracy of fishes in research, fishery management and conservation (Vecchione et al. 2000; Bortolus 2008). Number of countries including Australia (Ward et al. 2005); Antarctic Scotia Sea (Rock et al. 2008); Alaska and Pacific Arctic (Mecklenburg et al. 2011); Canada (Hubert et al. 2008); Mexico and Guatemala (Valdez-Moreno et al. 2009); Amazon (Ardura et al. 2010); India (Lakra et al. 2011); North America (Aprila et al. 2011); Eastern Nigeria covering only Ebonyi and Anambra States (Nwani et al. 2011) and Japan (Zhang and Hanner 2011) have had DNA barcoding done on some of the fish species sourced from freshwater, sea and marine. Nigeria is a country of above 170 million people with abundant water bodies for fishery. Nigerian fishes need to be studied for adequate knowledge of genetic diversity and possible identification of new species, especially in Enugu and Anambra States that harbor many freshwater bodies. Application of informative molecular markers will provide information on the molecular structure of fish species that will be useful in identification of unique stocks, stock enhancement, breeding programs for sustainable yield and preservation of genetic diversity (Tripathi 2011; Dinesh et al. 1993). For high discriminatory role in fish species from different sources of water, DNA barcoding has been well adjudged including some cryptic ones (Hubert et al. 2008; Carvalho et al. 2011; Pereira et al. 2013; Benzaquem et al. 2015).
Use of COI in DNA barcoding within animal kingdom has become a marker of choice (Hebert et al. 2003). It has been extensively applied for identification of invasive species (Wilson-Wilde et al. 2010), food adulteration analysis (Cohen et al. 2009; Murugaiah et al. 2009; Rojas et al. 2010), in forensic cases (Eaton et al. 2009), ecological discrimination (Berry et al. 2017), biomaterial collections (Cooper et al. 2007) and evaluation and documentation of new species through the use of phylogenetic diversity (PD) (the summation of the phylogenetic tree lengths of all the branches that are members of the corresponding minimum spanning routes for assessing ancestral relationships and conservation) (Faith 1992, 2008). This DNA barcoding technique has also been utilized in the identification of different organisms to their respective species levels as reported in nematodes (Elsasser et al. 2009), fish parasites (Locke et al. 2010), bats (Clare et al. 2007), mosquitoes (Cywinska et al. 2006), fungi (Stockinger et al. 2010), earthworms (Chang et al. 2008), bacteria (Sogin et al. 2006), protists (Chantangsi et al. 2007), spiders (Barret and Hebert 2005), fish (Ward et al. 2005) and crustaceans (Costa et al. 2007). DNA barcoding has become universally important both in animal and plant organisms but plants use chloroplast loci genes (matK, rbcL, rpoB and rpoC1) targeting coding regions and nuclear genes (ITS) (Baldwin and Markos 1998; Mort et al. 2007; Dong et al. 2012), while COI is applied in DNA barcoding of animals due to hyper mutation, maternal inheritance, absence of introns, absence of recombination, high substitution rates and lack of fast nucleotide substitution within the mitochondrial genome where the marker is located (Ballard and Whitlock 2004; Ballard and Rand 2005; Nabholz et al. 2009; Bernt et al. 2013; Hoque et al. 2013). It is a useful tool in different biological studies and has been used by the Barcode of life data Systems (BOLD) as a potential approach for identification of fishes to the species level (Ward et al. 2005; Wong and Hanner 2008). Within the eastern zone of Nigeria, especially, Enugu and Anambra States that maintain abundant freshwater bodies, identification and classification of fish have been based on morphological traits which are prone to errors. Also, there is no record of existing biodiversity of fishes within these States due to the use of only a morpho-based method. Application of modern and informative molecular technique including DNA barcoding has become necessary to address the challenges of inappropriate identification. Therefore, we investigated the utility of COI marker gene for species identification and assessment of genetic diversity within and among fish species collected from different freshwater bodies in Enugu and Anambra States of Nigeria.
Methods
Sample collection
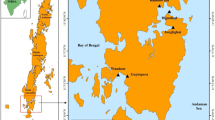
Forty-four (44) fish samples were collected from different locations in Enugu and Anambra States of Nigeria (Fig. 1; Table 1). The freshwater bodies that were easily accessible in the two States included the locations of Nike, Ugwuonwu, Ezu, and Obinna’s farm cutting across different lakes and rivers. Twelve (12), 17, 13, and 2 fishes were respectively collected from Nike Lake, Ugwuonwu Lake, Ezu River, and Obinna’s farm through local farmers who caught the fishes with fishing nets. The collected samples (the cut caudal fin or muscle part from whole fish species already caught by the local farmers) were preserved in 75% ethanol prior to DNA extraction.

Maps of Enugu (a) and Anambra (b) States showing locations of rivers and lakes used for collection of fish samples
DNA extraction
DNA was extracted following the method of Marizzi et al. (2018) with modifications. Briefly, a tissue (caudal fin or muscle) of 0.01 to 0.015 g was cut from each of the ethanol-preserved fish samples and transferred to a sterile 1.5 mL microcentrifuge with addition of 300 μL of lysis solution for homogenization using sterile mortar and pestle. The mixture was incubated in a heat block at 65 °C for 10 min. Next, samples were centrifuged in a balanced configuration at maximum speed (13,000 rev/min) for 1 min to pellet debris followed by transfer of 150 μL of supernatant into new 1.5 mL microcentrifuge tube with care not to disturb the pellet debris. The mixture was well mixed after addition of 3 μL silica resin followed by incubation at 57 °C for 5 min and centrifugation at maximum speed for 30 s to pellet the resin. The supernatant was transferred to new 1.5 mL with addition of 500 μL ice cold wash buffer to the pellet followed by centrifugation at maximum speed for 30 s. After this, the supernatant was transferred with the addition of 500 μL of ice-cold wash buffer, thorough mixture by vortexing, resuspension of the silica resin and centrifugation at maximum speed for 30 s. The wash buffer removes contaminants from the samples while nucleic acids remain bound to the resin. A dry spin step after wash was performed to remove any remnant drops of supernatant with a micropipette. Finally, 100 μL of distilled water was added to the silica resin, mixed well by vortexing and incubated at 57 °C for 5 min. Samples were then centrifuged for 30 s at maximum speed to pellet the resin. Later, 90 μL of the supernatant was transferred to new tubes from the resin. The eluted DNA was stored at − 20 °C prior to PCR step. The extracted was verified by loading 2 μL on 1.0% agarose gel electrophoresis.
Polymerase chain reaction and agarose gel electrophoresis and DNA sequencing
Polymerase chain reaction (PCR) amplification was performed in Ready-To-Go PCR beads in a total volume of 25 µL which consisted of 2 µL of ~ 100 ng DNA and 23 µL of primer/loading dye mix for fish cocktail with pairs of mitochondrial cytochrome oxidase I [(COI) primers forward primer, VF2_t1: 5′-TGTAAAACGACGGCCAGTCAACCAACCACAAAGACATTGGCAC-3′; forward primer, FishF2_t1: 5′-TGTAAAACGACGGCCAGTCGACTAATCATAAAGATATCGGCAC-3′; Reverse primer, FishR2_t1: 5′-CAGGAAACAGCTAGTACACTTCAGGGTGACCGAAGAATCAGAA-3′ and reverse primer, FR1d_t1: 5′-CAGGAAACAGCTATGACACCTCAGGGTGTCCGAARAAYCARAA-3′]. The PCR tubes were placed in a thermal cycler that had been programmed with the appropriate PCR protocol with initial step at 94 °C for 1 min., 35 cycles of 94 °C for 15 s, 54 °C for 15 s, and 72 °C for 30 s., and 8 min final extension at 72 °C was maintained. The PCR products or amplicons were electrophoresed on 1.5% agarose gel containing 0.5 mg/mL ethidium bromide and photographed using UV Transilluminator light (Omega G) to ensure that the PCR was successful and yielded accurate amplicon size. The generated PCR amplicons were prepared and sent to Genewiz LLC, New Jersey, USA, for DNA sequencing. To avoid issues relating to sequencing error, bidirectional sequencing coverage was performed for each sample and also sequenced twice.
Data analyses
A total of 44 sequences out of the 92 samples collected, were validated and used for analyses. The sequencing results generated from the Applied Biosystems Genetic automated sequencer were carefully trimmed, edited, filtered and assembled using DNA Subway (Merchant et al. 2016). Sequences were translated to amino acids and examined for stop codons to ensure there was no pseudogene amplification. Also, multiple and pairwise alignments were done using the ClustalW in BioEdit (Hall 1999; Bousalem et al. 2000; Chenna et al. 2003). The aligned sequences were subjected to phylogenetic trees reconstruction using Maximum Likelihood (ML) and Kimura 2-parameter (K2P) (Kimura 1980) and p-distance procedures with bootstrap test of 1000 replicates (Felsenstein 1981; Nei and Kumar 2000). The tree was drawn to scale, with branch lengths in the same units as those of the genetic distances used to construct the phylogenetic tree with a sequence of Pentalonia nigronervosa as an outgroup. Codon positions included were 1st + 2nd + 3rd + Noncoding. All positions with less than 95% site coverage were eliminated. Also, phylogenetic diversity (PD), a measure of the relative feature diversity of different subsets of taxa from a phylogeny and supports the broad goal of biodiversity, conservation and evolutionary heritage (Faith 2015), was computed using Molecular evolutionary genetic analysis version X (MEGA X) (Kumar et al. 2018). Genetic diversity distances based on K2P were also analysed to obtain intergeneric, congeneric and confamilial genetic distances using MEGA X. Other parameters including haplotype, Fu and Li’s D* test statistics and Tajima’s D analyses were computed using DnaSP version 5.10.01 (Librado and Rozas 2009). Tajima’s D statistics were applied to calculate the neutrality of haplotype. The statistics use the nucleotide diversity (π) and the number of segregating sites (S) observed in a sample of DNA sequences to make two estimates of the scaled mutation rate, θ (S) and θ (π). Tajima’s statistics D < 0 (θ (π) < θ (S) indicates populations that had experienced recent bottleneck effect. Multiple and pairwise alignments for detection of transitions and transversions were done using ClustalW in BioEdit software (Hall 1999; Bousalem et al. 2000; Chenna et al. 2003). Percentage similarity searches were compared with GenBank databases using BLASTn option in NCBI web-based site.
Results
Phylogenetic reconstruction
Phylogenetic reconstruction had a branch length of 1.9896 and percentage replicate trees in which the associated sequences clustered together in the bootstrap test of 1000 replicates were shown next to the branches with 658 positions in the final dataset (Fig. 2). Twenty major groups were identified with each species clustering at 100% bootstrap value followed by an outgroup that was included to ensure accurate and distinct grouping. Group I consisted of upside-down catfish including UpDoCat22, UpDoCat23, CoastalUD24, UpDoCat25, UpDoCat26, UpDoCat27, UpDoCat37 and UpDoCat38. This group had species of fish with 100% bootstrap replications but monophyletic (a group containing the most common ancestor of a given set of sequence taxa and all the descendants of that most recent common ancestor) at a subclade of 74% and clustered with Synodontis obesus. Group II had only RedTailedSy81 that was clearly grouped with a reference sequence of Synodontis clarias at its bootstrap replication of 100%. Groups III, IV and V had AfricanButCf76, WiheadCf83 and UpDoCat79 with their respective reference sequences of Schilbe mystus, Clarote laticeps and Auchenoglanis occidentalis. Each had bootstrap replications of 100%. Group VI had seven fishes including Catfish12, Catfish14, Catfish17, Catfish18, Catfish19, Catfish20, and Catfish21, clustering with Clarias gabonensis at 100% with different subclades of 16%, 63%, 81 and 85%. Group VII also clustered seven fish sequences from AfricanCat5, Catfish10, Catfish11, Catfish46, Catfish87, CfHybrid90, and TropCfish91, with Clarias gariepinus x batrachus and C. gariepinus as reference sequences at 100% bootstrap value. Groups VIII and IX at 100%, contained ElectricF13 and NileTilapia9, clustering together with Malapterurus electricus and Oreochromis aureus, respectively. Groups X, XI and XII with a bootstrap value of 100%, had Tilapia29, Cichlid74 and AfricanJeF73, with respective reference sequences of Tilapia guineensis, Chromidotilapia guntheri and Hemichromis letourneuxi. Group XIII had two fishes (Tilapia1 and Tilapia77), clustering with Hemichromis fasciatus at a replication value of 100%. Also, groups XIV, XV and XVI with 100% bootstraps contained Trunkfish2, Trunkfish56 and AfricanKnF60 that grouped together with their respective reference sequences of Mormrus tapirus, Marcusenius cyprinoides and Xenomystus nigri. In group XVII, five sequences clustered with Hepsetus odoe in which Dogfish84 had 100% replication value, while others such as Dogfish4, Dogfish28, Dogfish30 and Dogfish47 produced a subclade of 85%. Also, groups XVIII, XIX and XX contained ObscureSH6, MoonFish55 and GrassEater72, with each having Parachanna obscura, Citharinus sp and Distichodus rostratus as respective reference sequences.

Phylogenetic reconstruction of 44 fish COI sequences using Maximum Likelihood (ML) method. Gp group
Phylogenetic diversity
Phylogenetic diversity of each group without its respective reference sequences were computed (Fig. 3). The PD ranged from 0.0397 (group I) to 0.2147 (group XVIII). Group I consisted of UpDoCat22, UpDoCat23, CoastalUD24, UpDoCat25, UpDoCat, UpDoCat27, UpDoCat37 and UpDoCat38, with a PD value of 0.0397. In group II, only RedTailedSy81 was detected with 0.0397. Groups III, IV and V comprised of AfricanButCf76, WiheadCf83 and UpDoCat79, with PD values of 0.1089, 0.1058 and 0.1276, respectively. Group VI consisted of seven fish sequences including Catfish12, Catfish14, Catfish17, Catfish18, Catfish19, Catfish20, and Catfish21 at 0.0661. Group VII further clustered seven sequences (AfricanCat5, Catfish10, Catfish11, Catfish46, Catfish87, CfHybrid90, and TropCfish91) that were resolved at a PD value of 0.0628. At 0.13544, ElectricF13 clustered in group VIII, while Tilapia29 and NileTilapia9 clustered in groups IX and X maintaining a similar PD value of 0.0812. In groups XI and XII, Cichlid74 and AfricanJeF73 produced 0.1053, and 0.1169, respectively. In group XIII, Tilapia1 and Tilapia77 had the same value of 0.1053. Groups XIV and XV consisted of Trunkfish2, and Trunkfish56 with the same PD value of 0.1426, while group XVI had AfricanKnF60 with 0.1679. In group XVII, five fishes (Dogfish4, Dogfish28, Dogfish30, Dogfish47 and Dogfish84) had a PD value of 0.1649. Also, groups XIX and XX had MoonFish55 and GrassEater72, with a synonymous PD value of 0.1393, while ObscureSH6 in XVIII had 0.2147. Some of the groups contained similar PD values. For instance, groups I and II had a synonymous PD value of 0.0397, where group I contained Synodontis obesus and Synodontis clarias in group II. In groups IX (Oreochromis aureus) and X (Tilapia guineensis), a PD value of 0.0812 was common to the two groups of species. Phylogenetic diversity value of 0.1053 was identified in groups XI and XIII with Chromidotilapia guntheri and Hemichromis fasciatus, respectively. Groups XIV (Mormyrus tapirus) and XV (Marcusenius cyprinoides) had a similar value of 0.1426, while groups XIX and XX yielded 0.1393. The two groups, XIX and XX, contained Citharinus sp. and Distichodus rostratus, respectively.

Phylogenetic diversity of 44 fish COI sequences using the Maximum Likelihood (ML) method without reference sequences from NCBI database. Gp group
Genetic diversity distances based on Kimura 2-parameter
The highest genetic distances between species computed based on K2P was identified to be 37.00% (standard error, SE = 0.040) between groups 12 and 18 (Additional Table S1). Intergeneric genetic distances ranged from 15.800% to 37.00%. The highest intergeneric genetic divergence (37.00%) was detected between Hemichromis and Parachanna (groups 12 and 18), while the lowest value (15.80%) was between Synodontis and Schilbe (groups 2 and 3). For the congeneric distances, the values ranged from 6.9 ± 0.014 (groups 1 and 2) to 28.1 ± 0.038 (groups 1 and 13) with Synodontis as the existing genus (Table 2). Among the groups having the same genus, there were variations in their respective congeneric genetic distances. For instance, groups 1 and 12 had the same genus, Synodontis but the congeneric distance of 26.10 ± 0.030 was higher than the one (6.90 ± 0.014) obtained from groups 1 and 2 but lower than 28.10 ± 0.032 that was generated from groups 1 and 13. Groups 6 and 7 that had a synonymous genus of Clarias yielded 11.9 ± 0.019, while 12 and 13 possessing Hemichromis as genus produced 18.60 ± 0.025 as congeneric genetic distance. For confamilial genetic distances in percentages, the values ranged from 16.00 ± 0.014 (groups 9 and 10) to 25.7 ± 0.031 (groups 2 and 10) (Table 3). Each of the combined groups had different indices as confamilial genetic distance. Differently combined groups based on their synonymous family of Cichlidae had variable values. For instance, combined groups of 1, 2, 9, 10, 11, 12 and 13 containing the same family of Cichlidae, the highest confamilial value was identified in groups 2 and 10. Groups 4 and 5 had a synonymous family of Claroteidae with a confamilial genetic distance of 21.90 ± 0.027, while groups 14 and 15 possessed Mormyridae and 23.00 ± 0.030, respectively as family and confamilial distance. Mean diversity in entire population was 22.7 ± 0.019.
Haplotype analysis and nucleotide frequencies
A total of 42 haplotypes, H, with haplotype (gene) diversity, Hd of 0.999 ± 0.00003, and 115 mutations were identified among the sequences. Only two sequences (Catfish18 and Catfish21) were found in haplotype 13 (Hap_13), while the remaining sequences had a separate haplotype. Also, 389, 0.73721 and 286.776 were detected as parsimony informative sites, nucleotide diversity, Pi, and average number of pairwise nucleotide difference, K, respectively. Fu and Li’s D* test statistic was 2.17427 and it was found statistically significant (P < 0.02). Computation of Fu and Li’s F* test statistic yielded 1.7450 and was statistically determined (P < 0.005). Also, Tajima’s D of 0.2424 was identified but not statistically significant at P > 0.10.
In the present study, the COI amplified DNA fragments generated sequences with no presence of insertions, deletions or stop codons. Also, there was no identification of nuclear DNA sequences originating from mitochondrial DNA sequences. Average nucleotide frequencies detected were C (17.70%), T (29.40%), A (24.82%), and G (18.04%) with 54.22% of A + T.
Generated fish sequence alignments for identification of variable sites
From the sequence alignments, there were genetic variations at a nucleotide level as determined at different consensus positions of the representative sequences (Additional file Fig. S1). Almost all the fish sequences are diverse and polymorphic at different consensus positions with transitional mutations (C/T, A/G) demonstrating abundance in occurrence compared to transversional types (G/C, A/T, G/T). For instance, transitions occurred at consensus positions of 251 (C/T), 254 (C/T), 257 (A/G), 260 (C/T), 263 (C/T), 266 (A/G), 270 (C/T), 284 (C/T), 288 (C/T), and 294 (C/T), while transversions were identified at positions 245 (A/C), 269 (A/C) and 290 (C/G), respectively.
BLAST analysis
BLAST results demonstrated different species of fish inherent in the samples sequenced. A total of twenty species of fish (Hemichromis fasciatus, Mormyrus tapirus, Hepsetus odoe, Clarias gariepinus, Parachanna obscura, Oreochromis aureus, Clarias gabonensis, Malapterurus electricus, Synodontis obesus, Talapia guineensis, Citharinus sp., Marcusenius cyprinoides, Xenomystus nigri, Distichodus rostratus, Hemichromis letourneuxi, Chromidotilapia guntheri, Schilbe mystus, Auchenoglanis occidentalis, Synodontis clarias, Clarotes laticeps) and a hybrid species of Clarias gariepinus x Clarias batrachus were identified with percentage identity ranging from 99 to 100% across the fish sequences (Table 4). The expected value, e-value, maximum coverage and bit score obtained from the sequences ranged from 1e−43, 99–100 and 185–1194, respectively. A total of twelve families (Cichlidae, Mormyridae, Hepsetidae, Channidae, Malapteruridae, Cichlidae, Citharininae, Mormyridae, Notopteridae, Distichodontidae, Schilbeidae and Claroteidae) were also detected among the fish samples. The highest number of families identified was Clariidae (number, n = 14), followed by Cichlidae (n = 9), Cichlidae (n = 6), Hepsetidae (n = 5), Clarotidae (n = 2), Mormyridae (n = 2), while the smallest in number that had one family each were Channidae, Malapteruridae, Schilbidae, Citharininae, Notopteridae and Distichondontidae. For the total number of genera, 17 of them were found among the fish sequences.
Discussion
Use of DNA barcoding approach with COI gene for species identification has been well acknowledged and documented especially in fishery (Kochzius et al. 2010; Ward 2012; Knebelsberger et al. 2014). The utility of DNA barcoding was demonstrated to be efficient in species identification due to 100% success rate recorded in this study and this corroborates with other reports on DNA barcoding of fishes (Lakra et al. 2015; Shen et al. 2016). Other studies revealed success rates from 90 to 95.60% (Hubert et al. 2008; April et al. 2011; Iyiola et al. 2018). The different species clustered into 12 groups at 100% bootstrap value, thereby, demonstrating the unambiguous resolution and diagnostic utility of COI gene as earlier reported (Shen et al. 2016; Persis et al. 2009). The congeneric and confamilial species were well resolved by the phylogeny. Ward et al. (2009) had earlier pointed out that the COI gene delineates boundaries of different species, and that there was an indication of distinct phylogeny resolution in COI sequences that was linked to the clustering of congeneric and confamilial species. Generally, all the sequences pertaining to all species were correctly grouped together, thereby, demonstrating the potential of COI gene in DNA barcoding for fishery identification and management (Tripathi 2011). Some of the identified fish species in our study have been previously reported in Nigeria (Nwani et al. 2011; Persis et al. 2009; Nwakanma et al. 2015; Falade et al. 2016).
Phylogenetic diversity, which assesses community phylogenetic richness, is obtainable through the summation of the lengths of tree branch lengths or distances that are members of the corresponding minimum traversing species or the sum of branch lengths of the evolutionary trees connecting a set of taxa or individuals, is a crucial diversity index (Faith 1992; Faith and Baker 2006). Applying rbcL DNA barcoding marker, comparison of species abundance for preservation of feature diversity through the use of PD has been documented in plants (Forest et al 2007) and also in the ecology of species to measure their richness using COI gene (Smith and Fisher 2009; Smith et al. 2009; Machac et al. 2011). In the present study, PD ranged from 0.0397 (Synodontis obesus) to 0.2147 (Parachanna obscura). Some of the tree branches of the fish species had similar values of PD, while some yielded variable values. There were different groups of fish species that exhibited identical values of PD (Synodontis obesus and Synodontis clarias, PD = 0.0397; Oreochromis aureus and Tilapia guineensis, PD = 0.0812; Chromidotilapia guntheri and Hemichromis fasciatus, PD = 0.1053; Mormyrus tapirus and Marcusenius cyprinoides, PD = 0.1426; Citharinus sp and Distichodus rostratus, PD = 0.1393), while other groups yielded variable values. This further illustrates the efficacy of this COI marker gene in distinguishing species and identifying relatedness based on their ancestral lineages. In mammals, the PD has been shown to be unevenly distributed across the globe (Davies et al. 2008; Schipper et al. 2008), and that hotspots of species richness might capture more PD than expected by chance (Sechrest et al. 2002; Spathelf and Waite 2007). The detected number of 20 species, 17 genera and 12 families in this study are lower than 70 species, 38 genera and 20 families that were respectively reported in a study involving 363 individuals (Nwani et al. 2011) and these differences could be attributable to the discrepancies in the number of individuals analyzed.
There were variations within the intergeneric, congeneric and confamilial genetic distances thereby exposing the potential effectiveness of this marker in resolving species even within genus and family. We identified different ranges in genetic distances within the genera and families as 6.90–28.1% and 16.00–25.70%, respectively and these values are in agreement with the work of Bingpeng et al. (2018). It has been reported that DNA barcoding is a standardized approach that depends on the assumption that inter-species genetic distance or variability is greater than the one obtainable from intra-species (Hebert et al. 2003; Meyer and Paulay 2005). The highest genetic distance (interspecific divergence) obtained from the sequences was 37.00% and this is ten times higher than the one identified by Bingpeng et al. (2018) but supports the work of Iyiola et al. (2018). This implies that the genetic distance within the species is more than the one obtained from among them. This finding is in agreement with the earlier report where genetic variation within the population was found to be higher (Ren et al. 2017). The obtained congeneric distance range (6.9–28.1%) from this study is higher than the 8% identified in 35 freshwater fishes in Cuban (Lara et al. 2010) and 10.29% from Ebonyi and Anambra States of Southeastern Nigeria (Nwani et al. 2011). The identified K2P intergeneric COI sequence divergence in this study ranged from 15.800–37.00% and this value is slightly higher than the one (0.30–31.40%) reported by Iyiola et al. (2018), but it is in agreement with a previous report from COI (14.6–25.7%) and inter-transcribed sequence, ITS (32.8–35.0%) (Petrov et al. 2016). We obtained the highest intergeneric genetic divergence (37.00%) between Hemichromis and Parachanna, while the lowest value (15.80%) was found between Synodontis and Schilbe. This is in contrast with a previous research that identified the highest value (31.30%) between Parachanna and Malapterurus, while the lowest (0.30%) was between Hyperopisus and Brienomyrus (Iyiola et al. 2018). This difference could possibly be linked to the nature of fish species analyzed in the two separate researches. The identified range of percentage confamilial genetic distances (16.00 ± 0.014–25.7 ± 0.031) corroborates with earlier reports of 20.4% from Cuban freshwater fishes (Lara et al. 2010), 15.38% from Canadian freshwater (Hubert et al. 2008) and 15.46% from Australian freshwater (Ward et al. 2005). Mean diversity of 22.7 ± 0.019 that was generated from the entire population is lower than the one (87.5 ± 0.089) reported by Persis et al. (2009) and this could be due to the heterogeneous nature of the later.
In the present study, a total of 42 haplotypes and 389 polymorphic (variable) sites were obtained. Previous reports yielded 33 haplotypes and 149 polymorphic sites in 83 specimens from four extant Pacifastacus species (Larson et al. 2016); 36 haplotypes and 56 variable sites in 43 sequences of Steindachneridion scriptum (Paixão et al. 2018); and, 44 haplotypes and 76 variable sites in 74 fish (Pappalardo et al. 2015). In this study, the genetic variability (Hd = 0.999) is similar, but nucleotide diversity (Pi = 0.73721) is higher than the ones (Hd = 0.959; Pi = 0.007) reported by Paixão et al. (2018) from Steindachneridion scriptum; and in 68 species of Sicyopus zosterophorum (Hd = 0.885; Pi = 0.0039) by Taillebois et al. (2013). It has been reported that Tajima’s D-neutrality tests are applied to detect evidence of strong selective pressures, while Fu’s Fs-tests are used specifically to identify population expansion (Tajima 1989; Fu 1997). We detected positive values (D* = 0.2424, p > 0.10; Fu and Li’s D* = 2.17427, P < 0.02; Fu and Li’s F* 1.7450, p < 0.005) for all individuals. The positive significant values were estimated for D and this shows that there are strong selective pressure and absence of population expansion within the studied population. Therefore, we accept the hypothesis of selective neutrality. Average nucleotide frequencies detected were similar to those that were previously reported in other studies of 577 fish specimens with base nucleotide compositions of C (27.25%), T (30.74%), A (24.04%), G (17.97%) and A + T = 54.8 (Mabragan et al. 2011); 79 species of fish with C (27.53%), T (29.40%), A (24.82%), G (18.04%) and A + T = 54.22% (Díaz et al. 2016); two species of Hypophthalmichthys molitrix (A = 31·8%, C = 26·9%, G = 15·7%, T = 25·6%, A + T = 57·4%), and H. nobilis (A = 31·6%, C = 27·1%, G = 16·0%, T = 25·3%, A + T = 56·9%) (Li et al. 2009); 89 fish species (A = 24.12%, C = 27·28%, G = 18·2%, T = 30·4%, A + T = 54.52%) (Henriques et al. 2015); and the ones (A = 25.90%, C = 28.90%, G = 17.00% and T = 28.20%, A + T = 54.1) from Iyiola et al. (2018).
At the nucleotide level, the sequence alignment revealed much polymorphism at different consensus positions and this similar degree of variations had been previously observed in 28 species of fish (Persis et al. 2009). The existing variations show a high degree of heterogeneity within the studied fishes. The identified transitional mutations were more abundant than the transversional ones as earlier detected in Snow trout (Ali et al. 2011), Engraulis encrasicolus (Pappalardo et al. 2015) and in Channa striata (Boonkusol and Tongbai 2016). There was no presence of insertions, deletions or stop codons in all the amplified sequences, suggesting composition of functional mitochondrial COI sequences as opined by Shen et al. (2016). Application of genomics is highly informative in differentiating individuals and varieties (Singh et al. 2014). Identification of percentage identity or similarity from 99–100% with the known ones in the GenBank databases was recorded across the fish species and this demonstrates the effectiveness of this genomics tool in ascertaining individual fish relatedness as previously reported (Debenedetti et al. 2014; Bellagamba et al. 2015; Abbas et al. 2017).
Conclusion
This work has successfully demonstrated the utility of COI gene in distinguishing even the closely related species of fishes. The use of phylogeny, PD, BLAST analysis, congeneric, intergeneric and confamilial K2P-based distance computations contributed in identifying and characterizing closely related species with much efficiency. Clariidae had the highest number of genera and families and PD discriminated them on the bases of genetic divergence and ancestral linkages. Parachanna obscura in group XVIII was identified to be most evolutionarily divergent and PD further captured the shared ancestry of the fish species. Our results provided good insights into the phylogeny, genetic diversity, haplotype and nature of identified fish species in Enugu and Anambra States of Nigeria. The results obtained in this present study can facilitate decision makings and selections for biodiversity, breeding and conservation in fishery management.
Data availability
All data generated in this study are included in this published article [and its supplementary information files] and in GenBank (NCBI) [https://www.ncbi.nlm.nih.gov/genbank] where novel sequences with accession numbers MH746731 and MH776981-MH776988 were deposited.
References
Abbas EM, Soliman T, El-Magd MA, Farrag MMS, Ismail RF, Kato M (2017) Phylogeny and DNA barcoding of the family Sparidae inferred from mitochondrial DNA of the Egyptian waters. J Fish Aquat Sci 12:73–81
Ali S, Barat A, Pandey H, Sivaraman GK, Sahoo PK, Sati J (2011) Mitochondrial DNA variation and population genetic structure of Snow trout from Kumaun and Garhwal Himalayan regions of India. J Ecophysiol Occup Hlth 14(1 & 2):23–31
April J, Mayden RL, Hanner RH, Bernatchez L (2011) Genetic calibration of species diversity among North America’s freshwater fishes. Proc Natl Acad Sci USA 108:10602–10607
Aprila J, Mayden RL, Hanner RH, Bernatcheza L (2011) Genetic calibration of species diversity among North America’s freshwater fishes. PNAS 108(26):10602–10607
Ardura A, Linde AR, Moreira J, Garcia-Vazque E (2010) DNA barcoding for conservation and management of Amazonian commercial fish. Biol Conserv 143(6):1438–1443
Ardura A, Planes S, Garcia-Vazquez E (2013) Applications of DNA barcoding to fish landings: authentication and diversity assessment. Zookeys 365:49–65
Baldwin BG, Markos S (1998) Phylogenetic utility of the external transcribed spacer (ETS) of 18S 26S rDNA: congruence of ETS and ITS trees of Calycadenia (Compositae). Mol Phylogenet Evol 10(3):449–463
Ballard JWO, Rand DM (2005) The population biology of mitochondrial DNA and its phylogenetic implications. Annu Rev Ecol Evol Syst 36:621–642
Ballard J, Whitlock M (2004) The incomplete natural history of mitochondria. Mol Ecol 13(4):729–744
Barret RDH, Hebert PDN (2005) Identifying spiders through DNA barcodes. Can J Zool 83:481–491
Bellagamba F, Velayutham D, Cozzi MC, Caprino F, Vasconi M, Busetto ML, Bagnato A, Moretti VM (2015) Cytochrome oxidase-I sequence-based studies of commercially available Pangasius Hypophthalmus in Italy. Ital J Anim Sci 14(3):3928
Benzaquem DC, Oliveira C, da Silva BJ, Zuanon J, Rebelo PJI (2015) DNA barcoding in Pencilfishes (Lebiasinidae: Nannostomus) reveals cryptic diversity across the Brazilian Amazon. PLoS ONE 10:e0112217
Bernt M, Braband A, Schierwater B, Stadler PF (2013) Genetic aspects of mitochondrial genome evolution. Mol Phylogenet Evol 69:328–338
Berry O, Sarre SD, Farrington L, Aitken N (2017) Faecal DNA detection of invasive species: the case of feral foxes in Tasmania. Wildlife Res 34:1–7
Bingpeng X, Heshan L, Zhilan Z, Chunguang W, Yanguo W, Jianjun W (2018) DNA barcoding for identification of fish species in the Taiwan Strait. PLoS ONE 13(6):e0198109
Boonkusol D, Tongbai W (2016) Genetic variation of striped snakehead fish (Channa striata) in river basin of central Thailand inferred from mtDNA COI gene sequences analysis. J Biol Sci 16:37–43
Bortolus A (2008) Error cascades in the biological sciences: the unwanted consequences of using bad taxonomy in ecology. Ambio 37:114–118
Bousalem M, Douzery E, Fargette D (2000) High genetic diversity, distant phylogenetic relationships and intraspecies recombination events among natural populations of Yam mosaic virus: a contribution to understanding potyvirus evolution. J Gen Virol 81:243–255
Cadotte MW, Davies J, Regetz J, Kembel SW, Cleland EE, Oakley TH (2010) Phylogenetic diversity metrics for ecological communities: integrating species richness, abundance and evolutionary history. Ecol Lett 13:96–105
Carvalho DC, Oliveira DAA, Pompeu PS, Leal CG, Oliveira C, Hanner R (2011) Deep barcode divergence in Brazilian freshwater fishes: the case of Sao Francisco River Basin. Mitochondrial DNA 22:71–79
Chang CH, Rougerie R, Chen JH (2008) Identifying earthworms through DNA barcodes: pitfalls and promise. Pedobiologia 52:171–180
Chantangsi C, Lynn DH, Brand MT, Cole JC, Hetrick N, Ikonomi P (2007) Barcoding ciliates: a comprehensive study of 75 isolates of the genus Tetrahymena. Int J Syst Evol Microbiol 57:2412–2425
Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31:3497–3500
Clare LE, Lim BK, Engtrom MD, Ege JL, Hebert PDN (2007) DNA barcoding of neotropical bats: species identification and discovery within Guyana. Mol Ecol Notes 7:184–190
Cohen NJ, Deeds JR, Wong ES, Hanner RH, Yancy HF, White KD, Thompson TM, Wahl M, Pham TD, Guichard FM, Huh I, Austin C, Dizikes G, Gerber SI (2009) Public health response to puffer fish (tetrodotoxin) poisoning from mislabeled product. J Food Prot 72:810–817
Comi G, Iacumin L, Rantsioua K, Cantoni C, Cocolin L (2005) Molecular methods for the differentiation of species used in production of cod-fish can detect commercial frauds. Food Control 16:37–42
Cooper JK, Sykes G, King S, Cottrill K, Ivanova NV, Hanner R, Ikonomi P (2007) Species identification in cell culture: a two pronged molecular approach. In Vitro Cell Dev Biol Animal 43:344–351
Costa FO, deWaard JR, Boutillier J, Ratnasignham S, Dooh RT, Hajibabaei M, Hebert PDN (2007) Biological identifications through DNA barcodes: the case of the Crustacea. Can J Fish Aquat Sci 64:272–295
Cywinska A, Hunter FF, Hebert PDN (2006) Identifying Canadian mosquito species through DNA barcodes. Med Vert Entomol 20:413–424
Davies TJ, Fritz SA, Grenyer R, Orme CD, Bielby J, Bininda-Emonds OR, Cardillo M, Jones KE, Gittleman JL, Mace GM, Purvis A (2008) Phylogenetic trees and the future of mammalian biodiversity. Proc Natl Acad Sci USA 105:11556–11563
Debenedetti F, Dalmasso A, Bottero MT, Gilli M, Gili S, Tepedino V, Civera T (2014) Application of DNA barcoding for controlling of the species from Octopus genus. Ital J Food Safety 3:4521
Díaz J, Villanova GV, Brancolini F, del Pazo F, Posner VM, Grimberg A, Arranz SE (2016) First DNA barcode reference library for the identification of South American freshwater fish from the lower Paraná River. PLoS ONE 11(7):e0157419
Dinesh KR, Lim TM, Chua KL, Chan WK, Phang VP (1993) RAPD analysis: an efficient method of DNA fingerprinting in fishes. Zool Sci 10:849854
Dong W, Liu J, Yu J, Wang L, Zhou S (2012) Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 7(4):e35071
Eaton MJ, Meyers GL, Kolokotronis SO, Leslie MS, Martin AP, Amato G (2009) Barcoding bush meat: molecular identification of Central African and South American harvested vertebrates. Conserv Genet 11:1389–1404
Elsasser SC, Floyd R, Hebert PDN, Schulte-Hostedde AI (2009) Species identification of North American guinea worms (Nematoda: Dracunculus) with DNA barcoding. Mol Ecol Res 9:707–712
Faith DP (1992) Conservation evaluation and phylogenetic diversity. Biol Conserv 61:1–10
Faith DP (2008) Phylogenetic diversity and conservation. In: Carroll SP, Fox C (eds) Conservation biology: evolution in action. Oxford University Press, Oxford
Faith DP (2015) Phylogenetic diversity, functional trait diversity and extinction: avoiding tipping points and worst-case losses. Phil Trans R Soc B 370:20140011
Faith D, Baker A (2006) Phylogenetic diversity (PD) and biodiversity conservation: some bioinformatics challenges. Evol Bioinformatics Online 2:70–77
Falade MO, Opene AJ, Benson O (2016) DNA-barcoding of Clarias gariepinus, Coptedon zillii and Sarotherodon melanotheron from Southwestern Nigeria. F1000Research, 5, 1268.
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368–376
Forest F, Grenyer R, Rouget M, Davis TJ, Cowling RM, Faith DP, Balmford A, Manning JC, Proches S, van der Bank M, Reeves G, Hedderson TA (2007) Preserving the evolutionary potential of floras in biodiversity hotspots. Nature 445:757–760
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking, and background selection. Genet 147:915–925
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser 41:95–98
Hebert PDN, Cywinska A, Ball SL, de Waard JR (2003) Biological identifications through DNA barcodes. Proc R Soc B 270:313–322
Henriques JM, Silva GJC, Ashikaga FY, Hanner R, Foresti F, Oliveira C (2015) Use of DNA barcode in the identification of fish species from Ribeira de Iguape Basin and coastal rivers from São Paulo State (Brazil). DNA Barcodes 3:118–128
Hoque MR, Choi NR, Sultana H, Kang BS, Heo KN, Hong SK, Jo C, Lee JH (2013) Phylogenetic analysis of a privately-owned Korean Native chicken population using mtDNA D-loop variations. Asian-Austr J Anim Sci 26:157–162
Hubert N, Hanner R, Holm E, Mandrak NE, Taylor E, Burridge M, Watkinson D, Dumont P, Curry A, Bentzen P, Zhang J, April J, Bernatchez L (2008) Identifying Canadian freshwater fishes through DNA barcodes. PLoS ONE 3:1–8
Iyiola AO, Nneji LM, Mustapha MK, Nzeh CG, Oladipo CG, Nneji IC, Okeyoyin AO, Nwani CD, Ugwumba OA, Ugwumba AAA, Faturoti EO, Wang YY, Chen J, Wang W-Z, Adeola AC (2018) DNA barcoding of economically important freshwater fish species from north-central Nigeria uncovers cryptic diversity. Ecol Evol 8:6932–6951
Keskin E, Atar HH (2013) DNA barcoding of commercially important fish species of Turkey. Mol Ecol Resour 13(5):788–797
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Knebelsberger T, Landi M, Neumann H, Kloppmann M, Sell AF, Campbell PD, Laak-mann S, Raupach MJ, Carvalho GR, Costa FO (2014) A reliable DNA barcode reference library for the identification of the North European shelf fish fauna. Mol Ecol Resour 14:1060–1071
Kochzius M, Seidel C, Antoniou A, Botla SK, Campo D, Cariani A, Vazquez EG, Hauschild J, Hervet C, Hjorleifsdottir S, Hreggvidsson G, Kappel K, Landi M, Magoulas A, Marteinsson V, Nolte M, Planes S, Tinti F, Turan C, Venugopa MN, Weber H, Blohm D (2010) Identifying fishes through DNA barcodes and microarrays. PLoS ONE 5(9):e12620. https://doi.org/10.1371/journal.pone.0012620
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Lakra WS, Verma MS, Goswami M, Lal KK, Mohindra V, Punia P, Gopalakrishnan A, Singh KV, Ward RD, Hebert P (2011) DNA barcoding Indian marine fishes. Mol Ecol Resour 11:60–71
Lakra WS, Singh M, Goswami M, Gopalakrishnan A, Lal KK, Mohindra V, Ayyappan S (2015) DNA barcoding Indian freshwater fishes. Mitochondrial DNA 27:4510–4517
Lara A, De Leon JLP, Rodriguez R, Casane D, Cote G, Bernatchez L, Garcia-Machado E (2010) DNA barcoding of Cuban freshwater fishes: evidence for cryptic species and taxonomic conflicts. Mol Eco Resour 10:421–430
Larson ER, Castelin M, Williams BW, Olden JD, Abbott CI (2016) Phylogenetic species delimitation for crayfishes of the genus Pacifastacus. Peer J. https://doi.org/10.7717/peerj.1915
Li SF, Xu JW, Yang QL, Wang CH, Chen Q, Chapman DC, Lu G (2009) A comparison of complete mitochondrial genomes of silver carp Hypophthalmichthys molitrix and bighead carp Hypophthalmichthys nobilis: implications for their taxonomic relationship and phylogeny. J Fish Biol 74:1787–1803
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Locke SA, Mclaughlin JD, Macroglose DJ (2010) DNA barcodes show cryptic diversity and a potential physiological basis for host specificity among Diplostomoidea (Platyhelminthes: Digenea) parasitizing freshwater fish in the St Lawrence River canal. Mol Ecol 19:2813–2827
Mabragan E, Dıaz de Astarloa JM, Hanner R, Zhang J, Gonza-lez Castro M (2011) DNA barcoding identifies Argentine fishes from Marine and Brackish waters. PLoS ONE 6(12):e28655. https://doi.org/10.1371/journal.pone.0028655
Machac A, Janda M, Dunn RR, Sanders NJ (2011) Elevational gradients in phylogenetic structure of ant communities reveal the interplay of biotic and abiotic constraints on diversity. Ecography 34:364–371
Marizzi C, Florio A, Lee M, Khalfan M, Ghiban C, Nash B et al (2018) DNA barcoding Brooklyn (New York): a first assessment of biodiversity in Marine Park by citizen scientists. PLoS ONE 13(7):e0199015
Mecklenburg CW, Møller PR, Steinke D (2011) Biodiversity of arctic marine fishes: taxonomy and zoogeography. Mar Biodiver 41:109–140
Merchant N, Lyons E, Goff S, Vaughn M, Ware D, Micklos D et al (2016) The iPlant collaborative: cyberinfrastructure for enabling data to discovery for the life Sciences. PLoS Biol 14(1):e1002342
Meyer CP, Paulay G (2005) DNA barcoding: error rates based on comprehensive sampling. PLoS Biol 3:e422
Mort ME, Archibald JK, Randle CP, Levsen ND, O'Leary TR, Topalov C (2007) Inferring phylogeny at low taxonomic levels: utility of rapidly evolving cpDNA and nuclear ITS loci. Am J Bot 94(2):173–183
Murugaiah C, Noor ZM, Mastakim M, Bilung LM, Selamat J, Radu S (2009) Meat species identification and Halal authentication analysis using mitochondrial DNA. Meat Sci 83(1):57–61
Nabholz B, Glémin S, Galtier N (2009) The erratic mitochondrial clock: variations of mutation rate, not population size, affect mtDNA diversity across birds and mammals. BMC Evol Biol 9:54
Nei M, Kumar S (2000) Molecular evolution and phylogenetics. Oxford University Press, New York
Nelson JS (2006) Fishes of the world. Wiley, Boca Raton
Nwakanma C, Ude G, Unachukwu MN (2015) The use of DNA barcoding in identification of genetic diversity of fish in Ugwu-Omu, Nike river in Enugu. Nigerian J Biotechnol 29:27–33
Nwani CD, Becker S, Braid HE, Ude EF, Okogwu OI, Hanner R (2011) DNA barcoding discriminates freshwater fishes from southeastern Nigeria and provides river system-level phylogeographic resolution within some species. Mitochondrial DNA 22(S1):43–51
Paixão RV, Ribolli J, Zaniboni-Filho E (2018) Genetic variation of the endangered Neotropical catfish Steindachneridion scriptum (Siluriformes: Pimelodidae). Front Genet 9:48
Pappalardo AM, Federico C, Sabella G, Saccone S, Ferrito V (2015) A COI Nonsynonymous mutation as diagnostic tool for intraspecific discrimination in the European anchovy Engraulis encrasicolus (Linnaeus). PLoS ONE 10(11):e0143297. https://doi.org/10.1371/journal.pone.0143297
Pereira LHG, Hanner R, Foresti F, Oliveira C (2013) Can DNA barcoding accurately discriminate megadiverse Neotropical fresh water fish fauna? BMC Genet 14:20–34
Persis M, Reddy ACS, Rao LM, Khedkar GD, Ravinder K, Nasruddin K (2009) COI (cytochrome oxidase-I) sequence-based studies of Carangid fishes from Kakinada coast, India. Mol Biol Rep 36:1733–1740
Petrov NB, Vladychenskaya IP, Drozdov AL, Kedrova OS (2016) Molecular genetic markers of intra- and interspecific divergence within starfish and sea urchins (Echinodermata). Biochem (Moscow) 81(9):972–980
Rasmussen RS, Morrissey MT, Hebert PDN (2009) DNA barcoding of commercially important Salmon and Trout species (Oncorhynchus and Salmo) from North America. J Agric Food Chem 57(18):8379–8385
Ren W, Hua L, Guoa L, Zhanga J, Tanga L, Zhanga E, Zhang J, Luo S, Tanga J, Chena X (2017) Preservation of the genetic diversity of a local common carp in the agricultural heritage rice–fish system. www.pnas.org/cgi/doi/10.1073/pnas. 1709582115.
Rock J, Costa FO, Walker DI, North AW, Hutchinson WF, Carvalho GR (2008) DNA barcodes of fish of the Scotia Sea, Antarctica indicate priority groups for taxonomic and systematics focus. Antarctic Sci 20(3):253–262
Rojas M, Gonzalez I, Pavon MA, Pegels N, Lago A, Hernandez PE, Garcia T, Martin R (2010) Novel TaqMan real-time polymerase chain reaction assay for verifying the authenticity of meat and commercial meat products from game birds. Food Addit Contam 27(6):749–763
Schipper J et al (2008) The status of the world’s land and marine mammals: diversity, threat, and knowledge. Science 322:225–230
Sechrest W, Brooks TM, da Fonseca GAB, Konstant WR, Mittermeier RA, Purvis A, Rylands AB, Gittleman JL (2002) Hotspots and the conservation of evolutionary history. Proc Natl Acad Sci USA 99:2067–2071
Shen Y, Guan L, Wang D, Gan X (2016) DNA barcoding and evaluation of genetic diversity in Cyprinidae fish in the midstream of the Yangtze River. Ecol Evol 6(9):2702–2713
Singh U, Deb R, Alyethodi RR, Alex R, Kumar S, Chakraborty S, Dhama K, Sharma A (2014) Molecular markers and their applications in cattle genetic research: a review. Biomarkers Genom Med 6:49–58
Smith MA, Fisher BL (2009) Invasions, DNA barcodes, and rapid biodiversity assessment using ants of Mauritius. Front Zool 6:31
Smith MA, Fernandez-Triana J, Roughley R, Hebert PDN (2009) DNA barcode accumulation curves for understudied taxa and areas. Mol Ecol Resour 9:208–216
Sogin ML, Morrison HG, Hube JA, Welch DM, Huse SM, Neal PR, Arrieta JM, Herndl GJ (2006) Microbial diversity in the deep sea and the unexpected ‘rare biosphere. Proc Natl Acad Sci USA 103:12115–12120
Spathelf M, Waite TA (2007) Will hotspots conserve extra primate and carnivore evolutionary history? Div Distrib 13:746–751
Steinke D, Zemlak TS, Hebert PDN (2009) Barcoding Nemo: DNA-Based Identifications for the Ornamental Fish Trade. PLoS ONE 4(7):e6300
Stockinger H, Kruger M, Schussler A (2010) DNA barcoding of arbuscular mycorrhizal fungi. New Phytol 187:461–474
Taillebois L, Castelin M, Ovenden JR, Bonillo C, Keith P (2013) Contrasting genetic structure among populations of two amphidromous fish species (Sicydiinae) in the Central West Pacific. PLoS ONE 8(10):e75465. https://doi.org/10.1371/journal.pone.0075465
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genet 123:585–595
Teletchea F (2009) Molecular identification methods of fish species: reassessment and possible applications. Rev Fish Biol Fish 19:265–293
Triantafyllidis A, Bobori D, Koliamitra C, Gbandi E, Mpanti M, Petriki O et al (2011) DNA barcoding analysis of fish species diversity in four north Greek lakes. Mitochondrial DNA 22:37–42
Tripathi SD (2011) Aquaculture: a panacea for the future. Fishing Chimes 31:12–15
Ugwumba A, Ugwumba A (2003) Aquaculture options and the future of fish supply in Nigeria. Zoologist 2(2):96–122
Valdez-Moreno M, Ivanova NV, Elıas-Gutierrez M, ContrerasBalderas S, Hebert PDN (2009) Probing diversity in freshwater fishes from Mexico and Guatemala with DNA barcodes. J Fish Biol 74:377–402
Vartak VR, Narasimmalu R, Annam PK, Singh DP, Lakra WS (2015) DNA barcoding detected improper labelling and supersession of crab food served by restaurants in India. J Sci Food Agric 95(2):359–366
Vecchione M, Mickevich MF, Fauchald K, Collette BB, Williams AB, Munroe TA, Young RE (2000) Importance of assessing taxonomic adequacy in determining fishing effects on marine biodiversity. ICES J Mar Sci 57:677–681
Victor BC, Hanner R, Shivji M, Hyde J, Caldow C (2009) Identification of the larval and juvenile stages of theCubera snapper, Lutjanus cyanopterus, using DNA barcoding. Zootaxa 2215:24–36
Ward R (2012) A case study for DNA barcodes. In: Kress WJ, Erickson DL (eds) DNA barcodes: methods and protocols, methods in molecular biology. Springer, New York
Ward RD, Zemlak TS, Innes BH, Last PR, Hebert PDN (2005) DNA barcoding Australia’s fish species. Philos Trans R Soc B-Biol Sci 360(1462):1847–1857
Ward RD, Hanner R, Hebert PDN (2009) The campaign to DNA barcode all fishes. FISHBOL J Fish Biol 74:329–356
Wilson-Wilde L, Norman J, Robertson J, Sarre S, Georges A (2010) Current issues in species identification for forensic science and the validity of using the cytochrome oxidase 1 (COI) gene. Forensic Sci Med Pathol. https://doi.org/10.1007/s12024-010-9172-y
Wong EHK, Hanner RH (2008) DNA barcoding detects market substitution in North American seafood. Food Res Intern 41:828–837
Zhang JB, Hanner R (2011) DNA barcoding is a useful tool for the identification of marine fishes from Japan. Biochem Syst Ecol 39(1):31–42
Zhang J, Hanner R (2012) Molecular approach to the identification of fish in the South China Sea. PLoS ONE 7(2):e30621. https://doi.org/10.1371/journal.pone.0030621
Zhang JB, Huang LM, Huo HQ (2004) Larval identification of Lutjanus Bloch in Nansha coral reefs by AFLP molecular method. J Exp Mar Biol Ecol 298:3–20
Acknowledgements
The authors are thankful to National Science Foundation (NSF) for the Targeted Infusion HBCU_UP funding that supported this undergraduate students’ research project of Biotechnology Initiatives at Bowie State University.
Funding
Author GNU received research grants (with Grant Number HRD-1438902) from National Science Foundation (NSF) to conduct this study for Targeted Infusion in Historically Black Colleges and Universities Undergraduate Program (HBCU_UP). The funder, NSF, provided fund to principal investigator (PI) and three undergraduate students to travel to Enugu and Anambra States of Nigeria for sample collection and DNA extraction. Also, reagents and DNA sequencing were paid for by the funder.
Author information
Authors and Affiliations
Contributions
All authors were involved in the project design. GNU, DOI, CB, MJ, AB, OO, OCI, OC, MO, CE and VC did the literature search process, extracted data elements, and carried out study compilation. Data analyses were performed by DOI, MU and CO and reviewed by GNU, GA, JO and AD. DOI developed the first draft of the manuscript. All authors read the manuscript and approved the final copy of it.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval and consent to participate
This work does not involve living animals and no ethics approval. The fish samples were obtained dead through the verbal consent of the local fish farmers who are permitted by the local chiefs to use fishing nets to operate within the studied freshwater bodies.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Additional file 1: Table S1
Kimura 2-parameter (K2P) distances with standard error obtained from computations of intergeneric genetic distances of fish species. Supplementary file 1 (DOC 174 kb)
Additional file 2: Fig. S1
Some representatives of consensus sequences generated from 658 bp mitochondrial cytochrome oxidase I (COI) gene. The fish species had their associated consensus points of polymorphisms (variations). Note that the dotted line (…) in the sequence alignment indicates similarity of nucleotides to the nucleotides of Tilapia1. Supplementary file 2 (DOC 195 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ude, G.N., Igwe, D.O., Brown, C. et al. DNA barcoding for identification of fish species from freshwater in Enugu and Anambra States of Nigeria. Conservation Genet Resour 12, 643–658 (2020). https://doi.org/10.1007/s12686-020-01155-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12686-020-01155-7