
Abstract
Background
Lung adenocarcinoma (LUAD) is still one of the most prevalent malignancies. Interleukin factors are closely associated with the initiation and progression of cancer. However, the relationship between interleukin factors and LUAD has not been fully elucidated. This study aimed to use Mendelian randomization (MR) and RNA sequencing (RNA-seq) analyses to identify the interleukin factors associated with the onset and progression of LUAD.
Methods
Exposure-related instrumental variables were selected from interleukin factor summary datasets. The LUAD summary dataset from FINGENE served as the outcome. MR and sensitivity analyses were conducted to screen for interleukin factors associated with LUAD occurrence. Transcriptome analyses revealed the role of interleukin factors in lung tissues. The results were validated through Western blotting and further confirmed with driver gene-negative patients from multiple centers. Potential mechanisms influencing LUAD occurrence and development were explored using bulk RNA-seq and single-cell RNA-seq data.
Results
MR analysis indicated that elevated plasma levels of IL6RB, IL27RA, IL22RA1, and IL16 are causally associated with increased LUAD risk, while IL18R1 and IL11RA exhibit the opposite effect. Transcriptome analyses revealed that IL11RA, IL18R1, and IL16 were downregulated in tumor tissues compared with normal lung tissue, but only higher expression of IL11RA correlated with improved prognosis in patients with LUAD from different centers and persisted even in driver-gene negative patients. The IL11RA protein level was lower in various LUAD cell lines than in human bronchial epithelial cells. The genes co-expressed with IL11RA were enriched in the Ras signaling pathway and glycosylation processes. Fibroblasts were the primary IL11RA-expressing cell population, with IL11RA+fibroblasts exhibiting a more immature state. The genes differentially expressed between IL11RA+and IL11RA- fibroblasts were involved in the PI3K-Akt/TNF signaling pathway.
Conclusion
According to the MR and transcriptome analyses, the downregulation of IL11RA was closely related to the occurrence and development of LUAD.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Background
Lung cancer is currently one of the most prevalent malignancies and is a primary cause of cancer-related mortality [1]. Lung adenocarcinoma (LUAD) constitutes a significant proportion of all lung cancer cases (approximately 40%), predominantly affects females, is prone to metastasis, and ranks among the foremost pathologies in thoracic surgery [2]. Presently, the primary treatment modality for nonmetastatic lung adenocarcinoma is surgical intervention, often complemented by neoadjuvant or postoperative adjuvant therapies to increase patient survival [3, 4]. In contrast, late-stage lung cancer is predominantly managed through pharmacological approaches. EGFR, KRAS, ALK, etc., are considered driver genes [5] closely associated with the onset and progression of lung cancer. Targeted therapies focusing on driver genes, particularly EGFR, using tyrosine kinase inhibitors (TKIs) have significantly improved the survival prospects of lung cancer patients. However, a subset of lung adenocarcinoma patients lacks mutations in driver genes such as EGFR or KRAS, rendering them ineligible for targeted therapies directed at these specific targets; in some cases, patients exhibit pan-driver gene negativity, precluding the use of any targeted therapies [6]. Although the advent of immune checkpoint inhibitors such as PD1/PDL1 inhibitors has benefited these patients, the emergence of drug resistance has limited their utility. As a result, the exploration of biomarkers influencing the onset and progression of lung adenocarcinoma is highly important.
The interleukin family, recognized as crucial mediators in cellular communication, is generally associated with inflammatory responses, and chronic inflammation is widely regarded as a significant driving factor in the initiation and progression of solid tumors [7]. Indeed, recent research has indicated a close correlation between interleukins and related factors and the incidence and development of cancer [8]. In LUAD patients, interleukins may also play a pivotal role. For example, Zhiwen Luo and colleagues reported that the IL-15 concentration can predict the survival outcomes of LUAD patients [9]. Qi Tan and others have shown that IL-1β can stimulate glycolysis in LUAD cells [10]. Additionally, IL6R expression is positively correlated with overall survival (OS) in LUAD patients [11]. Numerous studies suggest that interleukins and related factors can either promote the proliferation of lung adenocarcinoma cells or exert opposing effects. However, even with these findings, previous research has not fully elucidated the causal relationship between these interleukin factors and the occurrence of LUAD.
Mendelian randomization (MR), a tool utilizing instrumental variables for causal analyses, effectively mitigates reverse causality and the influence of potential confounding factors [12]. Currently, MR has extensive applications in disease-related research, particularly for the selection of drug targets. Furthermore, transcriptome sequencing has continually matured in recent years, advancing from traditional bulk RNA sequencing to higher-resolution single-cell RNA-seq, thereby contributing significant value to the molecular exploration of diseases.
In this study, MR was used to determine the causal association between interleukin factors and LUAD. Transcriptomic analyses were subsequently performed to elucidate the role of these interleukin factors in the development of LUAD.
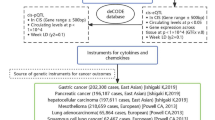
2 Methods
2.1 Mendelian randomization
2.1.1 Data source
The Genome-Wide Association Study (GWAS) data utilized in this Mendelian randomization study were obtained from the IEU GWAS Project [13]. The datasets used to extract instrumental variables related to interleukin factors were sourced from seven studies [14,15,16,17,18,19,20], encompassing a total of 189 datasets and spanning 79 unique interleukin factors. For comprehensive information, please refer to Table S1. The Lung Adenocarcinoma GWAS dataset was derived from the Fingene database [21]. This constitutes the most recent GWAS data about LUAD, encompassing the largest quantity of SNPs to date, totaling 16,380,303. This expansive coverage of the genome enhances the capacity to capture a broader spectrum of genetic variations.
2.1.2 Instrumental variable extraction and Mendelian randomization
Due to the limited availability of plasma protein-related instrumental variables (IVs), we established a threshold for IVs extraction at 5*10^-6. To overcome the bias caused by weak instrumental variables, the included SNPs were required to have an F value greater than 10 [22]. Additionally, we amalgamated multiple datasets containing the same interleukin factors to ensure the utmost reliability of our findings. After removing linkage disequilibrium (clump distance was 10,000 kb and r2 > 0.001), the SNPs were used to calculate the MR effect. Three classical methods, namely, inverse-variance weighted (IVW), MR‒Egger, and median-weighted analyses, were employed to calculate the causal effect between interleukin factors and LUAD. IVW served as the main estimate result when pleiotropy was not apparent [23]. The effect estimates obtained by the three methods are considered reliable only if they are in the same direction.
2.1.3 Sensitivity analyses
We employed the Egger intercept to investigate the presence of the instrumental variable horizontal pleiotropy. A p value ≥ 0.05 was considered indicative of the absence of pronounced horizontal pleiotropy. Further scrutiny was conducted using Mendelian randomization pleiotropy residual sum and outlier (MR-PRESSO) to detect and remove abnormal SNPs to minimize the effects of pleiotropy. [24] To assess heterogeneity, Cochran’s Q test and Rucker's Q test were employed [25]. A p value < 0.05 was regarded as evidence of heterogeneity, leading to the adoption of the IVW random effects model in such cases. Additionally, leave-one-out analyses were conducted to test the reliability of the MR results.
2.2 Bulk RNA sequencing analyses
2.2.1 Sample acquisition and processing
Sixty driver gene-negative lung adenocarcinoma patients who underwent surgery at the First Affiliated Hospital of Sun Yat-sen University (FAH) between 2003 and 2015 were randomly selected for this study; 15 patients were from each of the stages I, II, III, and IV. Unfortunately, RNA degradation affected the tissue samples of eight patients, resulting in a final cohort of 52 patients from whom both tumor and adjacent normal tissue samples were collected for bulk RNA sequencing. This project received approval from the Ethical Committee and Institutional Review Board of Sun Yat-sen University (No. 2021‐531), and written informed consent was obtained from all participants. In this study, RNA sequencing was conducted according to the protocol provided by Agilent Technologies. A 4 × 44 K whole human genome expression microarray was used to assess the expression of 27,958 genes within patient tissues. The probe nomenclature annotation platform used was GPL13497. After quality control and ID conversion, a total of 21,754 genes were examined and subsequently subjected to further analyses. We conducted follow-up assessments of these patients and gathered prognostic data up to 2021 for survival analyses. Notably, three patients were excluded from our subsequent survival analyses due to loss to follow-up, which resulted in the unavailability of prognostic data. The detailed survival data can be found in Table S2.
2.2.2 Publicly available bulk RNA-seq
The publicly available LUAD datasets utilized in this study encompass five distinct cohorts, The Cancer Genome Atlas Program-Lung Adenocarcinoma (TCGA-LUAD), GSE31210, GSE116959, GSE102511, and GSE72094, which were derived from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) platforms [26, 27]. The TCGA-LUAD dataset comprises 515 tumor samples and 59 normal samples. Of these, 504 patients had comprehensive clinical survival data and were subsequently subjected to survival analyses [26]. The GSE31210 dataset, generated by Hirokazu Okayama and colleagues, includes data from patients with lung adenocarcinoma. The dataset included transcriptomic sequencing data from 226 tumor tissues and 20 normal tissues, along with information on driver gene mutations and survival outcomes [28, 29]. The GSE116959 dataset, led by Laura Moreno Leon and colleagues, consists of transcriptomic sequencing data from 57 LUAD tissues and 11 adjacent normal tissues [30]. The GSE102511 dataset, which originated from Chui E Wong and collaborators, was used to explore the differences among LUAD, atypical adenomatous hyperplasia (AAH), and normal lung tissues. The dataset included sequencing data from 16 LUAD tissues, 17 AAH tissues, and 15 normal lung tissues. We utilized sequencing data from LUAD tissues and normal tissues [31]. The GSE72094 dataset, curated by Eschrich SA and colleagues, involved sequencing tumor tissues from 442 LUAD patients. This dataset includes information on driver gene mutations (EGFR/KRAS) as well as complete prognostic data [32]. The detail clinical information of TCGA-LUAD, GSE31210, and GSE72094 cohorts are shown in Table S2.
2.2.3 Differential analyses and survival analyses
The proteins that exhibited a statistically significant impact on lung adenocarcinoma, as identified through MR screening, were selected for further analyses. Datasets from the TCGA-LUAD, GSE31210, GSE116959, and GSE102511 cohorts were utilized for differential analyses to assess the differences in gene expression between cancerous and normal tissues. Survival analyses of the high- and low-gene expression groups were performed using the TCGA-LUAD, GSE31210, and GSE72094 cohorts. To verify whether the gene expression pattern was conserved in driver gene-negative patients, three cohorts were used: EGFR/KRAS/ALK-negative patients in the GSE31210 cohort, EGFR/KRAS-negative patients in the GSE72094 cohort, and pandriver gene-negative patients in the FAH cohort.
2.2.4 Clinical correlation analyses
We compared IL11RA expression in patients with various clinical characteristics using data from FAH and the TCGA-LUAD dataset. A correlation analysis was performed between IL11RA expression and the circulating tumor cell count.
2.2.5 Cell infiltration analyses
Three different methods were utilized to score cellular infiltration in the four cohorts of patients. The estimated scores of immune cells and stromal cells were calculated using the R package “estimate”. Cell-type Identification by Estimating Relative Subsets of RNA Transcripts (CIBERSORT) was applied to 22 immune cell type scores. Additionally, Xcell was used to calculate scores for 64 stromal and immune cell types within tumor tissue. The correlation between IL11RA expression and these cell infiltration scores was assessed and visualized with a bubble diagram.
2.2.6 Gene co-expression and enrichment analyses
To identify conserved genes associated with IL11RA in LUAD, we separately computed the correlation between the expression levels of each gene and IL11RA in the 4 cohorts. Genes with r > 0.1 and p < 0.05 were considered co-expressed with IL11RA. The intersection of co-expressed genes obtained from the 4 cohorts was considered to indicate highly conservative co-expressed genes. Functional enrichment analyses of these genes were performed using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses.
2.3 Cell culture and Western blot
The experiments conducted in this study involved five distinct cell lines: HBEC, H1299, H441, A549, and PC-9. These cell lines were procured from Guangzhou Hechuang Biological Technology Company. Primary cells were obtained from The Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The anti-IL11RA antibody (dilution 1:1000) was obtained from ABclonal (catalog No. A9365).
Culturing procedures were initiated for all five cell lines, after which protein extractions were performed. Subsequently, SDS‒PAGE electrophoresis was carried out, followed by protein transfer and hybridization with the IL11RA antibody and visualization. GAPDH was utilized as an internal reference protein. The protein expression levels of the four types of LUAD cells and HBECs were subsequently compared.
2.4 Single-cell RNA sequencing analyses
2.4.1 scRNA-seq datasets
Single-cell RNA-seq data were also obtained from the GEO database. [27] The GSE131907 cohort comprises 58 samples, including tumor tissue, normal tissue, lymph node tissue, and brain metastasis tissue [33] data. We extracted 11 tumor samples and 11 normal tissue samples and merged them with tumor samples from the GSE189357 and GSE171145 cohorts [34, 35]. This process resulted in a merged dataset containing 29 tumor samples, 11 normal tissue samples, and a total of 154,476 cells.
2.4.2 Dimensionality reduction, clustering, and trajectory analyses
Data preprocessing was performed using the Seurat package, and quality control was performed by filtering out cells with fewer than 50 detected genes and those with mitochondrial gene proportions exceeding 5%. Principal component analysis (PCA) was applied for dimensionality reduction, and batch effects were corrected using the Harmony function. Clustering was performed using the FindCluster function, which helps identify marker genes for each cluster. Visualization was carried out using UMAP plots in accordance with the methods of Diether Lambrechts and cell marker databases for cell annotation [36, 37]. Subsequently, a subpopulation of fibroblasts was extracted for further clustering analyses. Trajectory analyses were conducted using the Monocle package based on clustering-specific marker genes.
2.5 Statistical analysis
The Wilcoxon rank sum test was used to assess the statistical significance of differences in RNA expression between the tumor and normal groups. Since the patient samples from the FAH cohort were paired, a paired-sample rank sum test was used to compare expression differences between the two groups.
For survival analyses, the R package maxstat was used to compute the optimal cutoff value, with the minimum group sample size set above 25% and the maximum group sample size set below 75%. Patients were then divided into high- and low-expression groups based on the optimal cutoff value. Further survival analyses were conducted using the R package survival to evaluate the prognostic differences between the two groups, and the log-rank test was employed to assess the significance of differences in survival outcomes between different groups.
Gene expression correlation analyses were performed using Pearson correlation analyses. Unless otherwise specified, a p value less than 0.05 was considered to indicate statistical significance.
3 Results
3.1 Six interleukin factors in plasma are associated with the occurrence of LUAD
A total of 189 datasets related to interleukin factors were collected from the IEU database, comprising 79 interleukin factors. (Table S1). The heterogeneity analyses (Table S3), and horizontal pleiotropy analyses (Table S4) showed that the heterogeneity and horizontal pleiotropy were not significant. The causal effects of each protein on LUAD were calculated. Among these 79 proteins, elevated IL6RB (OR: 1.35, 95% CI 1.03 − 1.77, p-value = 0.03), IL27RA (OR: 1.17, 95% CI 1.03 − 1.34, p-value = 0.02), IL22RA1 (OR: 1.21, 95% CI 1.02 − 1.43, p-value = 0.03), and IL16 (OR: 1.14, 95% CI 1.02 − 1.28, p-value = 0.02) in plasma were risk factors for LUAD. Conversely, the plasma concentrations of IL18R1 (OR = 0.92, 95% CI 0.86–0.99, p-value = 0.04) and IL11RA (OR = 0.73, 95% CI 0.57–0.92, p-value = 0.007) were protective factors against LUAD. (Fig. 1). The effects of other interleukin factors on LUAD did not reach statistical significance. (Table S5). Leave one out test showed that the MR results obtained by us are reliable, but rs35026308 is strongly affected when calculating the effect of IL27RA on LUAD, which may cause unrobust results. (Figure S1).

The effects of interleukin factors on lung adenocarcinoma
The figure showed six interleukin factors with significant causal effects on LUAD onset, the association effects between each interleukin factor and LUAD were calculated by three methods. Inverse variance weighted effect is considered as the main estimate effect. IL11RA and IL18R1 are considered protective factors for lung adenocarcinoma (shown in blue), whereas IL16, IL6RB, IL22RA1, and IL27RA are considered risk factors (shown in red).
3.2 IL18R1, IL11RA and IL16 were downregulated in LUAD tissue
Differential expression analyses were also conducted on the aforementioned 6 mRNAs across the 4 different datasets. The results revealed that in all four datasets, IL18R1, IL11RA, and IL16 were significantly downregulated in tumor tissues. For IL6RB, IL22RA1, and IL27RA, the differential expression results varied across different cohorts. (Fig. 2A–D).

Differential analyses and survival validation of six interleukins/interleukin receptors via transcriptomics. A–D Differential expression of the six interleukins/interleukin receptors in the (A) TCGA-LUAD cohort. B GSE31210 cohort. C GSE116959 cohort. D GSE102511 cohort. The red column indicates the expression level of tumor tissue, and the blue column indicates the expression level of normal tissue. The p value of the difference between tumor and normal tissue expression was above the column. E Survival differences in the TCGA-LUAD cohort based on IL11RA expression. F Differences in survival in the GSE31210 cohort stratified according to IL11RA expression. G Survival differences in the GSE72094 cohort based on IL11RA expression. H Survival differences in the TCGA-LUAD cohort based on IL18R1 expression. I Differences in survival in the GSE31210 cohort stratified according to IL18R1 expression. J Differences in survival in the GSE72094 cohort stratified according to IL18R1 expression. K Survival differences in the TCGA-LUAD cohort stratified according to IL16 expression. L Differences in survival in the GSE31210 cohort stratified according to IL16 expression. M Differences in survival in the GSE72094 cohort stratified according to IL16 expression
3.3 High expression of IL11RA indicates a better prognosis
Survival analysis revealed that, in LUAD patients across three different cohorts, patients with higher expression of IL11RA had better overall survival than did those with lower expression of IL11RA. (Fig. 2E–G) In the GSE31210 and GSE72094 cohorts, patients with higher IL18R1 expression and higher IL16 expression exhibited improved prognosis, while there was no difference in survival among patients in the TCGA-LUAD cohort. (Fig. 2H–M). Then stratified analyses were employed to take into account the effects of other factors on overall survival of patients with lung adenocarcinoma, the results showed a robust association between IL11RA and overall survival, whereas the association between IL16/ IL18R1 and overall survival in patients with lung adenocarcinoma was disrupted by multiple factors. (Figures S2, S3, and S4).
3.4 Western blotting
According to the above results, only the expression pattern of IL11RA and its relationship with prognosis were validated in multiple cohorts, so we further explored the role of IL11RA in lung adenocarcinoma.
To validate the downregulation of IL11RA not only at the RNA level but also at the protein level in tumor tissues, Western blotting (WB) was employed to assess the protein expression of IL11RA in LUAD cell lines and HBEC cell lines. The results revealed that, in three lung adenocarcinoma cell lines (A549, H1299, and H441), the protein levels of IL11RA were lower than those in HBE cells. However, compared with HBE cells, the PC-9 cell line showed no significant difference in IL11RA expression. (Fig. 3A). The full blot images are shown in Figure S5.

Differential analyses and survival analyses of driver gene negative (DG-) patients. A Strip diagram and bar diagram of semi-quantitative results of the western blot illustrating the differences in the protein expression of IL11RA in HBEC cells and four different lung adenocarcinoma cell lines (H1299, H441, A549, PC9). B Differential expression of IL11RA in the GSE31210 DG- cohort. C Differential expression of IL11RA in the FAH cohort. D Survival differences in the GSE31210 DG- cohort based on IL11RA expression. E Differences in survival in the GSE72094 DG- cohort stratified according to IL11RA expression. F Differences in survival in the FAH cohort stratified by IL11RA expression
3.5 The relationship between IL11RA and prognosis was conserved in driver gene-negative patients
We compared the expression of negative driver genes between tumor tissue and normal tissue, and the results showed that IL11RA was still significantly downregulated in driver gene-negative patients. (Fig. 3B and C). The difference in survival between the high-low IL11RA expression group was compared in driver gene-negative patients: EGFR/KRAS-negative patients from the GSE72094 cohort, EGFR/KRAS/ALK-negative patients from the GSE31210 cohort, and pan-driver gene-negative patients from the FAH cohort. Remarkably, in all three cohorts, patients with high IL11RA expression continued to exhibit significantly better overall survival. (Fig. 3D–F).
3.6 Patients with more malignant LUAD tended to have lower expression of IL11RA
Differential analyses were also conducted between patients with different disease stages from both the TCGA-LUAD and FAH cohorts. In the TCGA-LUAD cohort, IL11RA expression was significantly downregulated in stage III patients compared to stage I and II patients. (Fig. 4A). Moreover, T2-stage patients exhibited lower IL11RA expression than T1-stage patients, and T3-stage patients had even lower IL11RA expression than T2-stage patients. (Fig. 4B). N2-stage patients had lower IL11RA expression compared to N1- and N0-stage patients. (Fig. 4C). However, no significant differences in IL11RA expression were observed between M0-stage patients and M1-stage patients. (Fig. 4D).

Differential expression of IL11RA among patients at different stages. A–D Differential expression of IL11RA among patients with different clinical stages/T stages/N stages/M stages in the TCGA-LUAD cohort. E–H Differential expression of IL11RA among patients with different clinical stages/T stages/N stages/M stages in the FAH cohort
According to the FAH cohort from our center, stage IV patients exhibited significantly lower IL11RA expression than stage I patients. (Fig. 4E). Additionally, T4-stage patients exhibited significantly lower IL11RA expression compared to T3-, T2-, and T1-stage patients. (Fig. 4F). Similarly, N3-stage patients had significantly downregulated IL11RA expression compared to N2- and N0-stage patients. (Fig. 4G). Although there were no significant differences in IL11RA expression between M0-stage patients and M1-stage patients, a significant negative correlation was observed between IL11RA expression and the number of circulating tumor cells in patients. Therefore, further assessment of its correlation with metastatic risk is warranted. (Figs. 4H and 5A).

Cell infiltration analysis results. A The correlation between IL11RA expression and the number of circulating tumor cells in the FAH cohort. B Correlations between IL11RA expression and stromal score, immune score, and ESTIMATE score in the TCGA-LUAD/GSE31210/GSE72094/FAH cohorts. C The correlation between IL11RA expression and 22 immune cell infiltration scores in the four cohorts. D The correlation between IL11RA expression and 64 immune cell and stromal cell infiltration scores in the four cohorts
We then performed Chi-square tests/Fisher’s exact test to compare the staging of patients with high or low IL11RA expression. There was no significant difference in the distribution of patients with high or low expression of IL11RA at the M0 and M1 stages, (p-value = 0.17). The same results were obtained in the FAH cohort (p-value = 0.29). (Table S6). However, it was observed that the T-stage distribution of patients with high or low expression of IL11RA was significantly different, whether in TCGA-LUAD (p = 4.3E-03) or FAH cohort (5.9E-03) (Table S6), which implied that higher IL11RA expression was associated with lower T staging.
3.7 Cell infiltration analyses
To explore how IL11RA impacts LUAD development, we computed immune scores and various cell infiltration scores for tumor patients from the TCGA-LUAD, GSE31210, GSE72094, and FAH cohorts. The estimated scores revealed a positive correlation between IL11RA expression and both stromal and immune scores across all four cohorts, resulting in decreased tumor purity (Fig. 5B).
The CIBERSORT results revealed significant heterogeneity in immune cell infiltration across the different cohorts. Notably, CD4 +memory-activated cells, M0 macrophages, and neutrophils were consistently negatively correlated with IL11RA expression (Fig. 5C).
The Xcell results indicated a consistent positive correlation between IL11RA expression and CD4 +T cell, fibroblast, mast cell, and iDC infiltration levels but a consistent negative correlation with epithelial cell and keratinocyte infiltration levels. These findings shed light on how IL11RA may influence the immune microenvironment in LUAD (Fig. 5D).
3.8 Co-expression and enrichment analyses
With respect to the four LUAD cohorts, we computed Pearson correlation coefficients between the expression levels of each gene and the expression level of IL11RA. We identified 538 genes that showed a positive correlation with IL11RA expression across all four cohorts. Further functional analyses through GO and KEGG pathway enrichment revealed that genes positively correlated with IL11RA expression were enriched in pathways related to GTPase-mediated signal transduction, such as the regulation of the RAS signaling pathway and cell–matrix adhesion. (Fig. 6A–C).

IL11RA co-expression gene and enrichment analyses. A The intersection of genes positively correlated with IL11RA expression in the four cohorts. B GO enrichment results for genes positively correlated with IL11RA expression. C KEGG enrichment results for genes positively correlated with IL11RA expression. D The intersection of genes negatively correlated with IL11RA expression in all four cohorts. E GO enrichment results for genes negatively correlated with IL11RA expression. F KEGG enrichment results for genes negatively correlated with IL11RA expression
Conversely, 263 genes exhibited a negative correlation with IL11RA expression. These genes were enriched primarily in processes related to glycosylation and the HIF-1 signaling pathway. These findings provide insights into the potential biological mechanisms and pathways associated with IL11RA in LUAD. (Fig. 6D–F).
3.9 Single-cell sequencing analyses
To further explore the expression profile and function of IL11RA, we merged single-cell data from the GSE131907, GSE171145, and GSE189357 cohorts. After performing PCA dimension reduction and clustering, these cells were categorized into 29 clusters. (Fig. 7A). Clustering analyses revealed significantly greater IL11RA expression in normal tissues than in tumor tissues. (Fig. 7B and C).

Results of single-cell RNA sequencing data clustering. A Uniform manifold approximation and projection (UMAP) plot of a total of 154,476 cells colored according to different clusters. B UMAP plot colored according to tissue type. C The expression of IL11RA in different tissues. D UMAP plot colored according to different cell types. E The expression of IL11RA in different cell types. F UMAP plot of 5348 fibroblasts classified by tissue type. G IL11RA + fibroblasts in tumor and normal tissues
After annotation, these cells were classified into 10 major cell categories. (Fig. 7D). IL11RA was expressed at higher levels in lung alveolar cells than in cancer cells, which is consistent with the findings obtained through Western blotting analyses. Furthermore, fibroblasts were the primary cell population expressing IL11RA (Fig. 7E). Given the known association between IL11RA and fibrosis, we performed subgroup analyses on 5348 fibroblasts extracted from the dataset. The results showed that there was a greater proportion of IL11RA+fibroblasts in normal tissues (30%) than in tumor tissues (13.5%) (Fig. 7F and G).
Trajectory analyses divided the fibroblasts into 3 states. (Fig. 8A). The differentiation proceeded from state 1 to state 2 and state 3. (Fig. 8B). IL11RA+fibroblasts were primarily located in state 1, representing an initial fibroblast state. (Fig. 8C and D). The proportion of IL11RA-expressing fibroblasts significantly decreased upon differentiation toward both ends. Thus, IL11RA+fibroblasts may represent a unique subgroup of fibroblasts and potentially serve as a protective factor for patients.

Trajectory analyses of fibroblasts. A The trajectory analyses categorize fibroblasts into three states. B The evolutionary process of fibroblasts in different states. C IL11RA + and IL11RA- fibroblasts in each state. D The differential expression of IL11RA in fibroblasts across different states. E Results of GO enrichment analyses of genes differentially expressed between IL11RA + and IL11RA- fibroblasts. F KEGG enrichment analysis results for genes differentially expressed between IL11RA + and IL11RA- fibroblasts
Subsequently, we conducted differential analyses between IL11RA+and IL11RA- fibroblasts, which revealed 227 differentially expressed genes. Functional enrichment analyses revealed that these DEGs were enriched primarily in processes related to epithelial cell differentiation and extracellular matrix construction. (Fig. 8E). KEGG analyses revealed enrichment in several classical pathways. These findings provide insights into the potential role of IL11RA+fibroblasts in lung adenocarcinoma and their association with protective mechanisms and relevant biological processes (Fig. 8F).
4 Discussion
Identifying effective therapeutic targets for the treatment of lung adenocarcinoma is urgently needed. Interleukin factor is believed to be closely related to the occurrence and development of cancer, but no studies have systematically investigated the effect of interleukin factor on lung adenocarcinoma. Our study fills this gap and provides an updated understanding of the role of interleukin factors in lung adenocarcinoma.
This study was the first study using MR and RNA-sequencing to investigate the relationship between the interleukin factors and LUAD. The results revealed causal associations between six interleukin factors (IL6RB, IL27RA, IL22RA1, IL16, IL18R1, and IL11RA) and LUAD. Among these factors, IL6RB, IL27RA, IL22RA1, and IL16 were identified as risk factors for the development of LUAD, while IL18R1 and IL11RA were found to be protective factors. Subsequent analyses at the transcriptomic level utilizing multiple LUAD patient cohorts confirmed that low expression of IL11RA is associated with an increased risk of LUAD development. This relationship remains conservative in LUAD driver gene-negative patients.
Our current research strongly suggests that IL11RA has the potential to serve as a valuable therapeutic target. Moreover, IL11RA may offer hope to LUAD patients, especially those with driver gene-negative patients. However, since IL11RA acts as a protective factor in LUAD patients, drug targeting is challenging. To distinguish this process from blocking risky targets, strategies for activating the upstream or downstream pathways of IL11RA may need to be explored to accurately determine its beneficial effects. This finding underscores the importance of further elucidating the physiological mechanisms of IL11RA in LUAD patients.
The estimated scores consistently indicated a positive correlation between IL11RA expression and immune scores as well as stromal scores across multiple datasets from different centers. Furthermore, IL11RA expression was negatively correlated with tumor purity, which aligns with its role as a protective factor in LUAD patients. Cibersort analyses suggested negative correlations between IL11RA and neutrophils, M0 macrophages, and CD4 memory-activated T cells. Studies have indicated that increased infiltration of neutrophils is associated with an elevated risk of brain metastasis in NSCLC patients [38]. Indeed, a higher level of neutrophil infiltration has been linked to poorer overall survival in LUAD patients [39]. DONG B and colleagues reported higher levels of M0 macrophage infiltration in metastatic LUAD patients [40]. The data from the FAH cohort further revealed the negative relationship between IL11RA expression and the circulating tumor cell count, although differential IL11RA expression between M0-stage and M1-stage LUAD patients was not observed. Furthermore, the decreased infiltration of CD4 memory-activated T cells may be attributed predominantly to the degree of cancer cell dormancy. The results from Xcell analyses indicated a positive correlation between IL11RA expression and fibroblast infiltration. Numerous studies have postulated that fibroblasts play a role in promoting immune evasion and driving the development of cancer [41,42,43]. However, fibroblasts can be classified into various subtypes, and their roles in the initiation and progression of tumors may vary significantly.
IL11 is regarded as a pivotal factor in promoting the progression of NSCLC. According to the TCGA-LUAD dataset, IL11 expression is significantly upregulated in LUAD patients, and this increase in IL11 expression demonstrates an adverse correlation with the prognosis of LUAD patients [44]. However, IL11RA, a receptor for IL11, paradoxically emerges as a protective factor in LUAD patients. Our current investigation revealed that the expression of IL11RA is decreased within the tumor microenvironment (TME) and that increased IL11RA expression is correlated with improved overall survival. Moreover, this association persisted even among LUAD driver gene-negative patients. Notably, Lei Gao et al. reported that patients exhibiting elevated IL11RA expression had superior overall survival [45]. This finding suggested that IL11RA might not only operate within the limits of the IL11 signaling pathway but also potentially engage with other factors to exert its anticancer effects.
Currently, there is limited research on the functional role of IL11RA in lung adenocarcinoma. We employed correlation analyses to identify a set of genes that exhibited a robust correlation with IL11RA expression in cancer patients. Subsequent enrichment analyses of these genes revealed that those genes that were positively correlated with IL11RA expression were enriched in the regulation of GTPase processes, with the well-known RAS signaling pathway being a prominent component. Hence, the upregulation of IL11RA may coincide with the modulation of the RAS signaling pathway. Conversely, genes that displayed a negative correlation with IL11RA expression were predominantly associated with glycosylation processes and cell cycle-related pathways. Aberrant glycosylation is widely recognized as a significant contributor to tumorigenesis [46]. The negative regulation of glycosylation processes to suppress cancer cell proliferation could represent one of the functional roles of IL11RA. Single-cell analyses reaffirmed that IL11RA is expressed at higher levels in normal tissue than in tumor tissue. IL11RA was predominantly expressed in fibroblasts, with a notably greater presence of IL11RA+fibroblasts in normal tissue than in tumor tissue. Consequently, we hypothesize that the anticancer effect of IL11RA could be partially mediated through fibroblasts. Indeed, previous research has highlighted the crucial role of IL11-IL11RA in cardiovascular fibrosis [47]. Anissa A. Widjaja's research has shown that inhibiting IL11/IL11RA can reduce the risk of liver fibrosis in mice [48]. These studies highlight the pivotal role of IL11RA in the process of fibrosis. However, the diverse classifications of fibroblast subpopulations and their varying impacts on tumors have prompted further investigation. Consequently, we isolated the fibroblast population for subclustering and conducted trajectory analyses. The results indicate that IL11RA+fibroblasts predominantly reside within a more immature fibroblast cluster, and their proportion significantly decreases with further differentiation. These findings suggested that IL11RA+fibroblasts may serve as inhibitory factors in the progression of LUAD. Christopher J. Hanley et al. classified tumor-associated fibroblasts into three groups: alveolar fibroblasts, adventitial fibroblasts, and myofibroblasts. Alveolar and adventitial fibroblasts can differentiate into myofibroblasts. Notably, myofibroblasts are associated with an unfavorable prognosis in lung adenocarcinoma patients, while alveolar fibroblasts play the opposite role [49]. The IL11RA+fibroblasts, as defined in our study, appear to overlap with alveolar fibroblasts and adventitial fibroblasts. We observed that IL11RA+fibroblasts constituted only 10–30% of the total fibroblasts. Therefore, further research is warranted to elucidate the role of IL11RA+fibroblasts in lung adenocarcinoma. Enrichment analyses of genes differentially expressed between IL11RA+and IL11RA- fibroblasts revealed significant enrichment in functions related to epithelial cell proliferation, extracellular matrix remodeling, and inflammatory responses. KEGG analyses also highlighted the enrichment of several classical pathways, such as the PI3K-AKT pathway, which may be important for IL11RA+fibroblasts to exert anticancer effects.
Notably, the expression patterns of IL16, IL6RB, IL27RA, IL22RA1, and IL18R1 in LUAD tissues are not completely consistent with the causal effect of plasma protein abundance on the occurrence of LUAD. This may be due to differences in gene expression patterns between blood and lung tissue, and transcriptomic analysis is also subject to reverse causality and confounding effects; therefore, further exploration of the role of these genes in lung adenocarcinoma is still necessary.
This study has several limitations. Although we have aggregated a substantial amount of GWAS data and conducted Mendelian randomization analyses, it is important to acknowledge that the results of Mendelian randomization may evolve with the continuous improvement of GWASs in the future. The GWAS datasets utilized in our study were derived from European populations, potentially limiting the generalizability of our findings to other ethnic groups. However, validation through transcriptomic data analyses conducted in our center’s Asian population cohort could improve the reliability of the findings. In this study, comprehensive correlation analyses were conducted to explore the potential mechanisms underlying the anticancer effects of IL11RA. Future experimental research will contribute to a deeper understanding of the mechanistic impact of IL11RA on LUAD.
5 Conclusions
Our study revealed that six interleukin factors are associated with the onset of LUAD. According to transcriptomic analyses, elevated expression of IL11RA has emerged as a prognostic indicator for improved overall survival in LUAD patients. Notably, as the malignancy of LUAD increases, the expression of IL11RA decreases. Importantly, this association was conserved even among driver gene-negative LUAD patients. Furthermore, the anticancer effect of IL11RA is likely mediated through its modulation of the Ras signaling pathway and glycosylation processes. IL11RA+fibroblasts are also speculated to play a pivotal role in this anticancer mechanism. Our study provides a deeper understanding of the role of Interleukin factors in LUAD.
Data availability
Summary statistics for the Mendelian randomization could be acquired from the IEU Open GWAS project [13]. The transcriptomic data for the LUAD cohort were publicly available through the TCGA [26] and GEO [27] websites. Survival information for the FAH cohort can be found in Table S5. Any additional information required to reanalyze the data reported in this paper is available from the corresponding author: leiyiyan@mail.sysu.edu.cn. Data is provided within the manuscript or supplementary information files.
Abbreviations
- CI:
-
Confidence interval
- DG-:
-
Driver gene negative
- FAH:
-
First Affiliated Hospital of Sun Yat-sen University
- GEO:
-
Gene Expression Omnibus
- GO:
-
Gene Ontology
- GWAS:
-
Genome-Wide Association Study
- HBEC:
-
Human bronchial epithelial cells
- IV:
-
Instrumental variable
- IVW:
-
Inverse-variance weighted
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LUAD:
-
Lung adenocarcinoma
- MR-PRESSO:
-
Mendelian Randomization Pleiotropy Residual Sum and Outlier
- OR:
-
Odds ratio
- OS:
-
Overall survival
- PCA:
-
Principal component analysis
- TCGA:
-
The Cancer Genome Atlas Program
- TKI:
-
Tyrosine kinase inhibitor
- UMAP:
-
Uniform manifold approximation and projection
References
Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49. https://doi.org/10.3322/caac.21660.
de Groot P, Munden RF. Lung cancer epidemiology, risk factors, and prevention. Radiol Clin North Am. 2012;50:863–76. https://doi.org/10.1016/j.rcl.2012.06.006.
Watanabe Y, Shimizu J, Oda M, et al. Results of surgical treatment in patients with stage IIIA non-small-cell lung cancer. Thorac Cardiovasc Surg. 1991;39:44–9. https://doi.org/10.1055/s-2007-1013929.
Myers DJ WJ. Lung Adenocarcinoma. In: StatPearls Treasure Island (FL): StatPearls Publishing; 2023. https://www.ncbi.nlm.nih.gov/books/NBK519578/. Accessed 12 Jun 2023.
Cui Y, Fang W, Li C, et al. Development and validation of a novel signature to predict overall survival in “driver gene-negative” lung adenocarcinoma (LUAD): results of a multicenter study. Clin Cancer Res. 2019;25:1546–56. https://doi.org/10.1158/1078-0432.Ccr-18-2545.
Seo JS, Ju YS, Lee WC, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22:2109–19. https://doi.org/10.1101/gr.145144.112.
Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020;20:662–80. https://doi.org/10.1038/s41568-020-0285-7.
Briukhovetska D, Dörr J, Endres S, et al. Interleukins in cancer: from biology to therapy. Nat Rev Cancer. 2021;21:481–99. https://doi.org/10.1038/s41568-021-00363-z.
Luo Z, He Z, Qin H, et al. Exercise-induced IL-15 acted as a positive prognostic implication and tumor-suppressed role in pan-cancer. Front Pharmacol. 2022;13:1053137. https://doi.org/10.3389/fphar.2022.1053137.
Tan Q, Duan L, Huang Q, et al. Interleukin -1β promotes lung adenocarcinoma growth and invasion through promoting glycolysis via p38 pathway. J Inflamm Res. 2021;14:6491–509. https://doi.org/10.2147/jir.S319433.
Sun G, Wang T, Shi M, et al. Low expression of IL6R predicts poor prognosis for lung adenocarcinoma. Ann Transl Med. 2021;9:1057. https://doi.org/10.21037/atm-21-36.
Sekula P, Del Greco MF, Pattaro C, et al. Mendelian randomization as an approach to assess causality using observational data. J Am Soc Nephrol. 2016;27:3253–65. https://doi.org/10.1681/asn.2016010098.
Bristol Uo. IEU open GWAS project. 2022. https://gwas.mrcieu.ac.uk/. Accessed 06 Nov 2022.
Ahola-Olli AV, Würtz P, Havulinna AS, et al. Genome-wide association study identifies 27 loci influencing concentrations of circulating cytokines and growth factors. Am J Hum Genet. 2017;100:40–50. https://doi.org/10.1016/j.ajhg.2016.11.007.
Suhre K, Arnold M, Bhagwat AM, et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat Commun. 2017;8:14357. https://doi.org/10.1038/ncomms14357.
Folkersen L, Fauman E, Sabater-Lleal M, et al. Mapping of 79 loci for 83 plasma protein biomarkers in cardiovascular disease. PLoS Genet. 2017;13: e1006706. https://doi.org/10.1371/journal.pgen.1006706.
Sun BB, Maranville JC, Peters JE, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558:73–9. https://doi.org/10.1038/s41586-018-0175-2.
Hillary RF, Trejo-Banos D, Kousathanas A, et al. Multi-method genome- and epigenome-wide studies of inflammatory protein levels in healthy older adults. Genom Med. 2020;12:60. https://doi.org/10.1186/s13073-020-00754-1.
Folkersen L, Gustafsson S, Wang Q, et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat Metab. 2020;2:1135–48. https://doi.org/10.1038/s42255-020-00287-2.
Gilly A, Park YC, Png G, et al. Whole-genome sequencing analysis of the cardiometabolic proteome. Nat Commun. 2020;11:6336. https://doi.org/10.1038/s41467-020-20079-2.
Fingene. Non-small cell lung cancer, adenocarcinoma, https://r9.risteys.finngen.fi/endpoints/C3_NSCLC_ADENO (2022, 2023).
Burgess S, Thompson SG. Bias in causal estimates from Mendelian randomization studies with weak instruments. Stat Med. 2011;30:1312–23. https://doi.org/10.1002/sim.4197.
Zheng J, Baird D, Borges MC, et al. Recent developments in mendelian randomization studies. Curr Epidemiol Rep. 2017;4:330–45. https://doi.org/10.1007/s40471-017-0128-6.
Verbanck M, Chen CY, Neale B, et al. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–8. https://doi.org/10.1038/s41588-018-0099-7.
Pereira TV, Patsopoulos NA, Salanti G, et al. Critical interpretation of Cochran’s Q test depends on power and prior assumptions about heterogeneity. Res Synth Methods. 2010;1:149–61. https://doi.org/10.1002/jrsm.13.
NIH. The Cancer Genome Atlas Program. 2006. https://portal.gdc.cancer.gov/
NCBI. Gene Expression Omnibus. 2000. https://www.ncbi.nlm.nih.gov/geo/. Accessed 06 Nov 2022.
Okayama H, Kohno T, Ishii Y, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Can Res. 2012;72:100–11. https://doi.org/10.1158/0008-5472.Can-11-1403.
Yamauchi M, Yamaguchi R, Nakata A, et al. Epidermal growth factor receptor tyrosine kinase defines critical prognostic genes of stage I lung adenocarcinoma. PLoS ONE. 2012;7: e43923. https://doi.org/10.1371/journal.pone.0043923.
Moreno Leon L, Gautier M, Allan R, et al. The nuclear hypoxia-regulated NLUCAT1 long non-coding RNA contributes to an aggressive phenotype in lung adenocarcinoma through regulation of oxidative stress. Oncogene. 2019;38:7146–65. https://doi.org/10.1038/s41388-019-0935-y.
Sivakumar S, Lucas FAS, McDowell TL, et al. Genomic landscape of atypical adenomatous hyperplasia reveals divergent modes to lung adenocarcinoma. Can Res. 2017;77:6119–30. https://doi.org/10.1158/0008-5472.Can-17-1605.
Schabath MB, Welsh EA, Fulp WJ, et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene. 2016;35:3209–16. https://doi.org/10.1038/onc.2015.375.
Kim N, Kim HK, Lee K, et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun. 2020;11:2285. https://doi.org/10.1038/s41467-020-16164-1.
Zhu J, Fan Y, Xiong Y, et al. Delineating the dynamic evolution from preneoplasia to invasive lung adenocarcinoma by integrating single-cell RNA sequencing and spatial transcriptomics. Exp Mol Med. 2022;54:2060–76. https://doi.org/10.1038/s12276-022-00896-9.
Yang L, He YT, Dong S, et al. Single-cell transcriptome analysis revealed a suppressive tumor immune microenvironment in EGFR mutant lung adenocarcinoma. J Immunother Cancer. 2022. https://doi.org/10.1136/jitc-2021-003534.
Lambrechts D, Wauters E, Boeckx B, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24:1277–89. https://doi.org/10.1038/s41591-018-0096-5.
Hu C, Li T, Xu Y, et al. Cell Marker 2.0: an updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. Nucl Acids Res. 2023;51:D870-d876. https://doi.org/10.1093/nar/gkac947.
Hu C, Wu J, Liu Y, et al. Relationship between neutrophil-to-lymphocyte ratio and brain metastasis in non-small cell lung cancer patients. Cancer Control. 2022;29:10732748221076804. https://doi.org/10.1177/10732748221076805.
Zuo Y, Leng G, Leng P. Identification and validation of molecular subtype and prognostic signature for lung adenocarcinoma based on neutrophil extracellular traps. Pathol Oncol Res POR. 2023;29:1610899. https://doi.org/10.3389/pore.2023.1610899.
Dong B, Wu C, Huang L, et al. Macrophage-related SPP1 as a potential biomarker for early lymph node metastasis in lung adenocarcinoma. Front Cell Dev Biol. 2021;9:739358. https://doi.org/10.3389/fcell.2021.739358.
Hanley CJ, Mellone M, Ford K, et al. Targeting the myofibroblastic cancer-associated fibroblast phenotype through inhibition of NOX4. J Natl Cancer Inst. 2018;110:109–20. https://doi.org/10.1093/jnci/djx121.
Papait A, Romoli J, Stefani FR, et al. Fight the cancer, hit the CAF! Cancers. 2022. https://doi.org/10.3390/cancers14153570.
Pereira BA, Vennin C, Papanicolaou M, et al. CAF subpopulations: a new reservoir of stromal targets in pancreatic cancer. Trends Cancer. 2019;5:724–41. https://doi.org/10.1016/j.trecan.2019.09.010.
Leung JH, Ng B, Lim WW. Interleukin-11: a potential biomarker and molecular therapeutic target in non-small cell lung cancer. Cells. 2022. https://doi.org/10.3390/cells11142257.
Gao L, Zhang L. Construction and comprehensive analysis of a ceRNA network to reveal potential prognostic biomarkers for lung adenocarcinoma. BMC Cancer. 2021;21:849. https://doi.org/10.1186/s12885-021-08462-8.
Lattová E, Skřičková J, Hausnerová J, et al. N-glycan profiling of lung adenocarcinoma in patients at different stages of disease. Mod Pathol. 2020;33:1146–56. https://doi.org/10.1038/s41379-019-0441-3.
Schafer S, Viswanathan S, Widjaja AA, et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature. 2017;552:110–5. https://doi.org/10.1038/nature24676.
Widjaja AA, Singh BK, Adami E, et al. Inhibiting interleukin 11 signaling reduces hepatocyte death and liver fibrosis, inflammation, and steatosis in mouse models of nonalcoholic steatohepatitis. Gastroenterology. 2019;157:777-792.e714. https://doi.org/10.1053/j.gastro.2019.05.002.
Hanley CJ, Waise S, Ellis MJ, et al. Single-cell analysis reveals prognostic fibroblast subpopulations linked to molecular and immunological subtypes of lung cancer. Nat Commun. 2023;14:387. https://doi.org/10.1038/s41467-023-35832-6.
Shen W, Song Z, Zhong X, et al. Sangerbox: a comprehensive, interaction-friendly clinical bioinformatics analysis platform. iMeta. 2022;1: e36. https://doi.org/10.1002/imt2.36.
Acknowledgements
We thank the authors for using their GWAS summary data and transcriptome datasets in this study. Some of the plots in our paper employed the Sanger box [50], and we appreciate that. Rongzhou He and Shuai Wang also assisted in this study, and we would like to express our gratitude for their contributions.
Funding
This work was supported by grants from the Development Center for Medical Science & Technology National Health Commission of the People’s Republic of China (No. WA2020RW10).
Author information
Authors and Affiliations
Contributions
Fei-Hang Zhi: Conceptualization, Data curation, Methodology, Software, Investigation, Visualization, Writing—original draft, Writing—review & Editing. Wei Liu: Writing—review & Editing. Hao-Shuai Yang: Methodology. Hong-He Luo: Supervision. Yan-Fen Feng and Yi-Yan Lei: Conceptualization, Writing—review & Editing, Supervision.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This project received approval from the Ethical Committee and Institutional Review Board of Sun Yat-sen University (No. 2021‐531), and written informed consent was obtained from all participants.
Consent for publication
All participants consented to the publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhi, FH., Liu, W., Yang, HS. et al. Exploring the relationship between the interleukin family and lung adenocarcinoma through Mendelian randomization and RNA sequencing analysis. Discov Onc 15, 436 (2024). https://doi.org/10.1007/s12672-024-01325-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-024-01325-1