
Abstract
Emerging evidence indicates that androgen receptor (AR) signaling plays a critical role in the pathogenesis of male-dominant urothelial cancer and its outgrowth. Meanwhile, latrophilins (LPHNs), a group of the G-protein-coupled receptors to which a spider venom latrotoxin (LTX) is known to bind, remain largely uncharacterized in neoplastic diseases. The present study aimed to determine the functional role of LPHN3 (encoded by the ADGRL3 gene), in association with AR signaling, in the progression of bladder cancer. In AR-positive bladder cancer lines, dihydrotestosterone considerably increased the expression levels of ADGRL3 and LPHN3, while chromatin immunoprecipitation assay revealed the binding of AR to the promoter region of ADGRL3. Treatment with LPHN3 ligands (e.g. α-LTX, FLRT3) resulted in the induction of ADGRL3 expression, as well as cell viability, in bladder cancer lines. By contrast, LPHN3 knockdown via shRNA virus infection significantly reduced the viability and migration of these cells. Immunohistochemistry in transurethral resection specimens further showed a strong correlation between LPHN3 and AR expression. Moreover, LPHN3 positivity in muscle-invasive bladder tumors, as an independent prognosticator, was associated with a significantly higher risk of disease progression and disease-specific mortality following radical cystectomy. These findings suggest that LPHN3 functions as a downstream effector of AR and promotes the growth of bladder cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Urinary bladder cancer has represented one of the most common malignancies, particularly among men, while the number of worldwide disease-specific deaths is considerably increasing [1, 2]. Patients with bladder cancer often present with non-fatal non-invasive disease but have a considerable risk of developing postoperative recurrence occasionally with invasive disease. Moreover, muscle-invasive bladder cancer is often associated with metastatic disease where overall oncologic outcomes remain poor (e.g. 5-year survival rate of 8.8% [3]). Although new drugs for targeted therapy have been clinically employed [4, 5], further identification of key molecules or signaling pathways responsible for the progression of urothelial cancer is thus required.
A growing body of evidence suggests the involvement of androgen receptor (AR), a member of the nuclear receptor superfamily, in the pathogenesis and progression of urothelial cancer. Specifically, AR activation results in the induction of tumor growth [6, 7], as well as resistance to conventional non-surgical therapy for bladder cancer such as intravesical BCG immunotherapy, cisplatin-based systemic chemotherapy, and radiotherapy [8]. Androgens and anti-androgens have thus been clearly demonstrated to promote and inhibit, respectively, the cell proliferation, migration, and invasion of various AR-positive bladder cancer lines [6, 7, 9]. However, precise mechanisms for how AR involves the outgrowth of urothelial cancer remain poorly understood.
Latrophilins (LPHNs; LPHN1/LPHN2/LPHN3), initially isolated as proteins to which a neurotoxin as a component of black widow spider venom, latrotoxin (LTX), binds [10, 11], are a group of the G-protein-coupled receptors [12]. Little is known about the biological functions of LPHNs, while LPHN3 is enriched particularly in the human brain [12,13,14]. Remarkably, alterations in their encoded genes (i.e. ADGRLs), particularly ADGRL3, have been implicated in susceptibility to attention-deficit/hyperactivity disorder [12, 15] whose risk is approximately 3 times higher in boys than in girls and is further increased by prenatal exposure to excess androgens [16, 17]. Meanwhile, the role of LPHNs in neoplastic diseases remains largely uncharacterized. In the present study, we aimed to determine whether LPHN3 could promote the growth of urothelial cancer via serving as a downstream effector of AR.
2 Methods
2.1 Cell lines
Human bladder urothelial carcinoma cell lines, UMUC3, 5637, 647 V, and TCCSUP, were originally obtained from the American Type Culture Collection and then authenticated by the institutional core facility. Sublines stably expressing AR-short hairpin RNA (shRNA) (i.e. UMUC3-AR-shRNA [18]) or human full-length wild-type AR (i.e. 5637-AR [18], 647 V-AR [19]) were established in our previous studies. Similarly, LPHN3-shRNA lentiviral particles (sc-60921-V, Santa Cruz Biotechnology) were infected and stably expressed in UMUC3, 5637, or 5637-AR cells. These parental cell lines and their stable sublines were maintained in DMEM (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin (both 100 units/mL) and then cultured in phenol red-free medium supplemented with 5% charcoal-stripped FBS for the experimental treatment with androgen/anti-androgen or 5% FBS for other experiments.
2.2 Chemicals and antibodies
We obtained dihydrotestosterone and hydroxyflutamide (HF) from Sigma-Aldrich, bicalutamide (BC) from Santa Cruz Biotechnology, α-LTX from Alomone Labs, and recombinant human FLRT3 protein from R&D Systems. Primary antibodies purchased were: LPHN3 (clone B-6, dilution for western blotting 1:100, Santa Cruz Biotechnology; or #PA5-145295, 1:100, Invitrogen); and GAPDH (clone 6C5, 1:5000; Santa Cruz Biotechnology).
2.3 Reverse transcription (RT) and polymerase chain reaction (PCR)
Total RNA isolated from cultured cells by TRIzol (Invitrogen) was reverse transcribed using oligo-dT primers and Omniscript reverse transcriptase (Qiagen). cDNAs were then amplified by PCR, as we described previously [6, 20], using primer sets specific for the ADGRL3 (forward, 5′-GCTCTTGCAGAGCCTATGTCCA-3′; reverse, 5′-CACTCGTAGTCAGGTCTCTCCA-3′) or GAPDH (forward, 5′-AAGGTGAAGGTCGGAGTCAAC-3′; reverse, 5′-GGGGTCATTGATGGCAACAATA-3′) gene. Real-time PCR was also performed using iQ™ SYBR® Green Supermix (Bio-Rad), as we described previously [6, 21].
2.4 Western blotting
Western blotting analysis was performed, as we recently described [22, 23]. Total proteins were extracted from the cells collected and washed twice with ice-cold 1× PBS with RIPA buffer supplemented with a protease and phosphatase inhibitor cocktail (Halt™; Thermo Fisher Scientific), and the DC Protein Assay kit (Bio-Rad) was used for the determination of protein concentration. Equal amounts of proteins (30 µg) obtained from the cell extracts were separated in 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membrane electronically, blocked, and incubated with a specific antibody and a secondary antibody (anti-mouse or anti-rabbit IgG HRP-linked antibody; Cell Signaling Technology), followed by scanning with an imaging system (ChemiDoc™ MP, Bio-Rad).
2.5 Chromatin immunoprecipitation (ChIP) assay
We first performed a bioinformatic search (LASAGNA-Search 2.0 available online at https://biogrid-lasagna.engr.uconn.edu/lasagna_search/) for identifying potential AR binding sites in the promoter of ADGRL3 and found a target site (see Fig. 1F). A ChIP assay was then performed, using the Magna ChIP kit (Sigma-Aldrich) according to the manufacturer’s recommended protocol with minor modifications, as we recently described [22,23,24]. Cells were cross-linked with 1% formaldehyde for 10 minutes at room temperature, and the lysates were sonicated in nuclear buffer (four 30-s pulses, output 3.0, duty cycle 30% in ice with 120-s rest between pulses; Branson Sonifier 450). Soluble chromatin was immunoprecipitated with an anti-AR antibody or normal mouse IgG (sc-2025, Santa Cruz Biochemistry) directly conjugated with protein A magnetic beads (Thermo Fisher Scientific). Immunoprecipitated DNA was eluted and reverse cross-linked, and DNA was extracted and purified using a spin filter column (Thermo Fisher Scientific). DNA samples were eventually analyzed by PCR, using the following primer set: forward, 5′-AAAGAACCGAAGAGACAGCG-3′; reverse, 5′-GAGCCACACAAACTCCTTCC-3′. The PCR products electrophoresed on 1% agarose gel and stained with ethidium bromide were visualized using Gel Doc XR+ (Bio-Rad).

Associations between AR signaling and LPHN3 expression in bladder cancer cells. A RT-PCR of ADGRL3 after 35 cycles of amplification in 4 cell lines. Real-time RT-PCR of ADGRL3 in UMUC3 cultured for 24 h with ethanol (mock) or 10 nM dihydrotestosterone (DHT) in AR-positive UMUC3/5637-AR (B) and AR-knockdown UMUC3-AR-shRNA (C). The expression of ADGRL3 normalized to that of GAPDH and representing the mean (± SD) of triplicate determinants is presented relative to that of mock treatment. *P < 0.05 (vs. mock treatment). Western blotting of LPHN3 in AR-positive (D) or AR-knockdown/negative (E) cells cultured for 48 h with ethanol (mock) or 10 nM DHT, as well as in UMUC3/647V-AR cells cultured for 24 h with ethanol (mock), 1 nM DHT, 5 µM BC, and/or 5 µM HF (F). GAPDH served as a loading control. G The ChIP assay, using UMUC3 and 647V-AR cell lysates immunoprecipitated with an anti-AR (or IgG as a negative control). The DNA fragments were PCR amplified with a set of primers specific for the promoter of ADGRL3, and the PCR products were electrophoresed on 1% agarose gel. Fractions of the mixture of protein-DNA complex (i.e. 1% of total cross-linked, reserved chromatin prior to immunoprecipitation) were used as “input” DNAs
2.6 MTT assay
The MTT assay was performed to assess the cell viability, as we recently described [22, 23]. Cells (3–8 × 103/well) seeded in 96-well tissue-culture plates were cultured for 96 h and then incubated with 0.5 mg/mL MTT (Sigma-Aldrich) in 100 µL medium for 4 h. MTT was dissolved by DMSO, and the absorbance was measured at a wavelength of 570 nm with background subtraction at 630 nm.
2.7 Wound-healing assay
The scratch wound-healing assay was performed to evaluate the ability of cell migration, as we recently described [22, 23]. Cells at a density of ≥ 90% confluence in 6-well tissue-culture plates were scratched with a 200 µL pipette tip. The wounded monolayers of the cells were allowed to heal in serum-free medium for 24 h. The normalized cell-free area in photographed pictures (24 h/0 h) was then quantitated, using the ImageJ (National Institute of Health).
2.8 Immunohistochemistry
A set of bladder tissue microarray (TMA) consisting of transurethral resection specimens was previously constructed upon appropriate approval by the Institutional Review Boards [25]. All the procedures were performed in accordance with the guidelines of the Declaration of Helsinki. Immunostaining was performed on 5 μm sections, using a primary antibody to LPHN3 (dilution 1:100), as we described previously [20,21,22,23, 25]. All stains were assessed by a board-certified pathologist (H.M.) who was blinded to sample identify. The immunoreactive scores (range: 0–12) calculated by multiplying the percentage of immunoreactive cells (0% = 0; 1–10% = 1; 11–50% = 2; 51–80% = 3; 81–100% = 4) by staining intensity (negative = 0; weak = 1; moderate = 2; strong = 3) were considered negative (0; 0–1), weakly positive (1+; 2–4), moderately positive (2+; 6–8), and strongly positive (3+; 9–12).
2.9 Statistical analysis
Chi-square test and Student’s t-test were used to evaluate categorized and numerical data, respectively. Time-to-event estimates of progression-free survival or cancer-specific survival in patients with muscle-invasive disease were calculated by the Kaplan-Meier method and compared by the log-rank test. The Cox proportional hazards model was also used to determine the significance of prognostic factors in a multivariable setting. All statistical analyses were performed, using EZR software [26] (R version 4.0.2; The R Foundation for Statistical Computing) or Prism version 10.2.3 (GraphPad Software). A P value of less than 0.05 was considered to be statistically significant.
3 Results
3.1 Impact of androgen/AR on LPHN3
Our DNA microarray analysis performed previously in control AR-positive bladder cancer UMUC3 cell line versus UMUC3-AR-shRNA [20] revealed that ADGRL3 was one of genes whose expression was considerably down-regulated in the AR-knockdown cells. We therefore anticipated that ADGRL3/LPHN3 could represent a downstream target of AR.
Semi-quantitative RT-PCR in 4 human bladder cancer lines showed that ADGRL3 expression was the highest in UMUC3 and weaker in others (Fig. 1A). We then assessed the effects of androgen (i.e. dihydrotestosterone) on the expression of ADGRL3/LPHN3. Dihydrotestosterone treatment significantly induced the levels of ADGRL3 in 2 AR-positive lines (Fig. 1B), but not in an AR-knockdown subline (Fig. 1C). Similarly, dihydrotestosterone considerably increased LPHN3 expression in AR-positive lines (Fig. 1D), but not in AR-knockdown or AR-negative cells (Fig. 1E). In addition, anti-androgens (i.e. BC, HF) restored androgen-induced LPHN3 expression (Fig. 1F).
A bioinformatics-driven search identified a putative AR binding site in the promoter region of ADGRL3, and ChIP assay was then performed (Fig. 1G). DNA fragments from UMUC3 or 647 V-AR cells immunoprecipitated with an anti-AR antibody were amplified by PCR with a set of primers specific for the ADGRL3 promoter. The PCR products for the potential binding site could be visualized from those precipitated by the AR antibody, but not control precipitants.
3.2 Impact of LPHN3 on cell growth
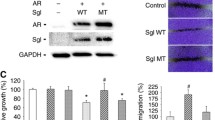
We next assessed the effects of LPHN3, via ligand (i.e. α-LTX, FLRT3) treatment and knockdown, on the growth of bladder cancer cells. As expected, α-LTX and FLRT3 increased ADGRL3 expression (Fig. 2A). α-LTX/FLRT3 treatment also induced the cell viability (via MTT assay; Fig. 2B). By contrast, knockdown of LPHN3 via its shRNA infection (Fig. 2C) resulted in the significant reduction in the cell viability (Fig. 2D) and migration (via a scratch wound-healing assay; Fig. 2E).

Effects of LPHN3 ligand treatment or knockdown on the growth of bladder cancer cells. A Real-time RT-PCR of ADGRL3 in UMUC3 cultured for 24 h with ethanol (mock), 0.1–1 nM α-LTX, or 0.175–1.75 nM FLRT3. The expression of ADGRL3 normalized to that of GAPDH and representing the mean (± SD) of triplicate determinants is presented relative to that of mock treatment. *P < 0.05 (vs. mock treatment). B MTT assay in UMUC3 and 647 V-AR cultured for 96 h with ethanol (mock), 1–10 nM α-LTX, or 1.75–17.5 nM FLRT3. Cell viability presented relative to that of mock-treated cells represents the mean (± SD) from three independent experiments. *P < 0.05 (vs. mock treatment). C Western blotting of LPHN3 in stable sublines expressing control-shRNA vs. LPHN3-shRNA. GAPDH served as a loading control. D MTT assay in control vs. LPHN3-knockdown sublines cultured for 96 h. Cell viability presented relative to that of control cells represents the mean (± SD) from three independent experiments. *P < 0.05 (vs. control-shRNA subline). E Wound-healing assay in control vs. LPHN3-knockdown sublines cultured for 24 h after scratching. Cell migration as the width of the wound area presented relative to that of control cells represents the mean (± SD) from three independent experiments. *P < 0.05 (vs. control-shRNA subline)
3.3 Expression of LPHN3 in surgical specimens
We stained immunohistochemically for LPHN3 in a total of 145 bladder tumors including 83 cases with non-muscle-invasive disease where LPHN3 had previously been stained [22]. Additionally, in all 145 cases, AR immunoreactivity had previously been determined [25]. Positive signals for LPHN3 were detected predominantly in the cytoplasm of bladder tumor cells (Fig. 3A). None of these tumors showed 3 + expression of LPHN3.

Immunohistochemistry of LPHN3 in bladder TMA. A A representative image of LPHN3 expression in bladder cancer. Kaplan-Meier curves for progression-free survival (B) and cancer-specific survival (C) in patients with LPHN3-negative vs. LPHN3-positive muscle-invasive tumor. The comparison between the two groups was made by the log-rank test
Associations between the levels (e.g. 0 vs. 1+/2+, 0/1 + vs. 2+) of LPHN3 expression and clinicopathologic features were assessed (Table 1). However, LPHN3 expression in 145 tumors was not strongly associated with patients’ sex, tumor grade, or pT or pN stage. Interestingly, positivity of LPHN3 and AR in tumors was strongly correlated (P = 0.048).
We then performed univariate survival analysis to assess possible associations of LPHN3 expression in 62 muscle-invasive tumors with oncologic outcomes after radical cystectomy. Patients with LPHN3-positive tumor had a significantly higher risk of disease progression (P = 0.011; Fig. 3B) or cancer-specific mortality (P = 0.007; Fig. 3C) than those with LPHN3-negative tumor.
To further determine if LPHN3 expression was an independent predictor of poorer prognosis, multivariable analysis of clinicopathologic factors was performed (Table 2). In the Cox regression model, LPHN3 positivity was associated with significantly worse progression-free survival (hazard ratio 2.320, 95% confidence interval 1.123–4.790, P = 0.023). AR positivity also showed a trend toward a higher progression rate (hazard ratio 2.202, 95% confidence interval 0.991–4.895, P = 0.053).
4 Discussion
G-protein-coupled receptors, as a large group of evolutionarily related proteins, are well known to mediate a variety of physiological and pathologic processes [12, 27, 28]. By contrast, only limited data have suggested the involvement of LPHNs, a group of G-protein-coupled receptors, in neoplastic diseases [12, 29,30,31,32]. In the present study, we have investigated the functional role of LPHN3 in the progression of urothelial cancer, which is a distinct step or event from urothelial tumorigenesis (i.e. tumor initiation, neoplastic transformation, tumor development), in relation to AR signaling, primarily via their activation (e.g. ligand treatment) and inactivation (e.g. knockdown) in cell line models for bladder cancer.
As aforementioned, we had originally identified LPHN3 as a potential AR target from our DNA microarray analysis in a control AR-positive UMUC3 cell line versus its AR-knockdown subline [20]. In not only UMUC3 cells but also other human bladder cancer sublines stably expressing a wild-type AR, we confirmed up-regulation of the expression of ADGRL3 gene and LPHN3 protein by androgen treatment, the latter of which was blocked by AR antagonists. Using ChIP assay in bladder cancer lines, we further demonstrated the interactions of AR with ADGRL3 at its promoter region, indicating the direct regulation of LPHN3 expression by AR. These results indicate that LPHN3 represents a downstream target of AR signaling in bladder cancer cells.
Again, it is known that LTX, a neurotoxin naturally found in the venom of widow spiders, binds and activates all 3 LPHNs [10,11,12]. In addition, it has been documented that FLRT3, a member of the fibronectin leucine rich transmembrane protein family, is an endogenous ligand for LPHN3 (and LPHN1) [12, 14, 33]. In UMUC3 cells, we demonstrated that α-LTX and FLRT3 induced the expression of ADGRL3.
The involvement of ADGRL3/LPHN3 in the progression, as well as therapeutic resistance, has been suggested in several types of malignancy. The observations in these studies include: (1) an association of reduced ADGRL3 expression in tumor tissues with shorter overall survival in patients with ependymoma [32]; and (2) elevated ADGRL3 expression in acute myeloid leukemia cell lines possessing P-glycoprotein variants that have been implicated in chemoresistance [30]. Recently, we have additionally demonstrated that LPHN3 promotes: (1) the chemical carcinogen-mediated neoplastic transformation of urothelial cells [22]; and (2) the cell proliferation and migration of both AR-positive and AR-negative prostate cancer lines [23]. Nevertheless, loss-of-function mutations within the ADGRL3 gene have been identified in a few malignancies, including bladder cancer [12, 34], implying its role as a tumor suppressor. We herein found that α-LTX and FLRT3 could induce the proliferation of bladder cancer cells derived primarily from invasive urothelial carcinoma. Correspondingly, LPHN3 knockdown resulted in the reduction of their growth. These findings suggest that ADGRL3/LPHN3 functions as a promoter of bladder cancer progression. However, signal transduction pathways downstream of LPHN3 in tumor cells, as well as the roles of other LPHNs in urothelial cancer, need to be further elucidated. Moreover, it is interesting to determine if LPHN3 also induces the growth of low-grade and/or non-invasive urothelial cancer cells.
The expression status of ADGRL genes and LPHN proteins in the bladder or urothelial tumor remained uncertain. We immunohistochemically examined LPHN3 expression in transurethral resection specimens and its prognostic significance in patients with muscle-invasive disease. There were no significant associations of LPHN3 expression with the histopathology including tumor grade and stage. Nonetheless, LPHN3 positivity in muscle-invasive bladder tumors, as an independent prognosticator, was associated with a significantly higher risk of postoperative disease progression and resultant disease-specific mortality. Meanwhile, positivity of LPHN3 vs. AR in bladder tumors was strongly correlated. All these immunohistochemical findings in surgical specimens strongly support our in vitro data indicating that LPHN3, presumably as a consequence of AR activation, induces the progression of urothelial tumor. In addition, it would be possible that, as documented in not only AR itself [35] but also some effectors as its direct downstream targets, such as FOXO1 [36], GABBR2 [24], and GULP1 [37], LPHN3 might contribute to reducing cisplatin sensitivity, and LPHN3 overexpression was thereby associated with a poorer prognosis particularly in patients with muscle-invasive bladder cancer who received cisplatin-based chemotherapy.
There have been sex-related differences in the incidence and prognosis of bladder cancer [1,2,3], while sex hormone receptors, including AR and estrogen receptors (ERs), have been implicated in the development and progression of urothelial cancer [7, 25, 38]. Although the expression of AR was found to be positively correlated with that of LPHN3, as a direct downstream, in bladder cancer cells, there was no significant difference in LPHN3 expression between male vs. female tumors in our cohort. Remarkably, the majority of the previous studies, where the expression levels of AR, ERα, and/or ERβ were compared between male and female bladder tissues, failed to show significant differences [7, 25, 38, 39], although their activities in cells might vary depending on the levels of sex hormones. As aforementioned, LPHN3 could similarly induce the growth of AR-positive and AR-negative prostate cancer cells [23]. LPHN3 might thus simply represent a direct target of AR. Otherwise, as demonstrated in other AR targets including FOXO1 [40] and GULP1 [41], ERβ may also possibly regulate the expression of LPHN3 in bladder cancer cells, particularly leading to the promotion of the proliferation of female tumor cells. In any case, although it remains to be determined, LPHN3 may not mediate distinct further downstream pathways to promote the growth of bladder cancer cells in males vs. females.
In conclusion, LPHN3 was found to be a downstream effector of AR in bladder cancer cells and promote their growth. Accordingly, although no specific inhibitors are currently available, LPHN3 represents a potential therapeutic target for advanced bladder cancer. Moreover, LPHN3 overexpression may serve as a useful prognosticator especially in patients with muscle-invasive bladder cancer. Meanwhile, further studies are warranted to not only validate our results but also elucidate the molecular mechanisms underlying LPHN-mediated urothelial cancer progression.
Data availability
Data are provided within the manuscript, while raw data underlying this study are available upon the request to the corresponding author.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108.
Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229–63.
SEER Cancer Stat Facts: Bladder Cancer. National Cancer Institute, Bethesda MD. 2024. http://seer.cancer.gov/statfacts/html/urinb.html. Accessed 8 Jul 2024.
Goto T, Miyamoto H. Why has the prognosis for muscle-invasive bladder cancer not significantly improved after decades of therapeutic advancements? Expert Opin Anticancer Ther. 2020;20:229–31.
Esteban-Villarrubia J, Torres-Jiménez J, Bueno-Bravo C, Garcia-Mondaray R, Subiela JD, Gajate P. Current and future landscape of perioperative treatment for muscle-invasive bladder cancer. Cancers. 2023;15:566.
Miyamoto H, Yang Z, Chen YT, et al. Promotion of bladder cancer development and progression by androgen receptor signals. J Natl Cancer Inst. 2007;99:558–68.
Inoue S, Mizushima T, Miyamoto H. Role of the androgen receptor in urothelial cancer. Mol Cell Endocrinol. 2018;465:73–81.
Ide H, Miyamoto H. Steroid hormone receptor signaling in bladder cancer: a potential target for enhancing the efficacy of conventional non-surgical therapy. Cells. 2021;10:1169.
Kawahara T, Ide H, Kashiwagi E, et al. Enzalutamide inhibits androgen receptor-positive bladder cancer cell growth. Urol Oncol. 2016;34:e43215–23.
Davletov BA, Shamotienko OG, Lelianova VG, Grishin EV, Ushkaryov YA. Isolation and biochemical characterization of a Ca2+-independent α-latrotoxin-binding protein. J Biol Chem. 1996;271:23239–45.
Krasnoperov VG, Beavis R, Chepurny OG, Little AR, Plotnikov AN, Petrenko AG. The calcium-independent receptor of α-latrotoxin is not a neurexin. Biochem Biophys Res Commun. 1996;227:868–75.
Meza-Aguilar DG, Boucard AA. Latrophilins updated. Biomol Concepts. 2014;5:457–8.
Sugita S, Ichtchenko K, Khvotchev M, Südhof TC. α-Latrotoxin receptor CIRL/latrophilin 1 (CL1) defined an unusual family of ubiquitous G-protein-linked receptors: G-protein coupling not required for triggering exocytosis. J Biol Chem. 1998;273:32715–24.
Boucard AA, Maxeiner S, Südhol TC. Latrophilins function as heterophilic cell-adhesion molecules by binding to teneurins. J Biol Chem. 2014;289:387–402.
Regan SL, Williams MT, Vorhees CV. Review of rodent models of attention deficit hyperactivity disorder. Neurosci Biobehav Rev. 2022;132:621–37.
Lombardo MV, Ashwin E, Auyeung B, et al. Fetal programming effects of testosterone on the reward system and behavioral approach tendencies in humans. Biol Psychiatry. 2012;72:839–47.
Kosidou K, Dalman C, Widman L, et al. Maternal polycystic ovary syndrome and risk for attention-deficit/hyperactivity disorder in the offspring. Biol Psychiatry. 2017;82:651–9.
Zheng Y, Izumi K, Yao JL, Miyamoto H. Dihydritestosterone upregulates the expression of epidermal growth factor receptor and ERBB2 in androgen receptor-positive bladder cancer cells. Endocr Relat Cancer. 2011;18:451–64.
Li Y, Ishiguro H, Kawahara T, Miyamoto Y, Izumi K, Miyamoto H. GATA3 in the urinary bladder: suppression of neoplastic transformation and down-regulation by androgens. Am J Cancer Res. 2014;4:461–73.
Mizushima T, Jiang G, Kawahara T, et al. Androgen receptor signaling reduces the efficacy of bacillus Calmette-Guérin therapy for bladder cancer via modulating Rab27-induced exocytosis. Mol Cancer Ther. 2020;19:1930–42.
Izumi K, Zheng Y, Hsu JW, Chang C, Miyamoto H. Androgen receptor signals regulate UDP-glucuronosyltransferases in the urinary bladder: a potential mechanism of androgen-induced bladder carcinogenesis. Mol Carcinogen. 2013;52:94–102.
Goto T, Yasui M, Teramoto Y, Nagata Y, Mizushima T, Miyamoto H. Latrophilin-3 as a downstream effector of the androgen receptor induces urothelial tumorigenesis. Mol Carcinog. 2024;63:1847–54.
Teramoto Y, Elahi Najafi MA, Matsukawa T, Sharma A, Goto T, Miyamoto H. Latrophilins as downstream effectors of androgen receptors including a splice variant, AR-V7, induce prostate cancer progression. Int J Mol Sci. 2024;25:7289.
Elahi Najafi MA, Yasui M, Teramoto Y, Tatenuma T, Jiang G, Miyamoto H. GABBR2 as a downstream effector of the androgen receptor induces cisplatin resistance in bladder cancer. Int J Mol Sci. 2023;24:13733.
Miyamoto H, Yao JL, Chaux A, et al. Expression of androgen and oestrogen receptors and its prognostic significance in urothelial neoplasm of the urinary bladder. BJU Int. 2012;109:1716–26.
Kanda Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transpl. 2013;48:452–8.
Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7:79–94.
Qin K, Dong C, Wu G, Lambert NA. Inactive-state preassembly of Gq-coupled receptors and Gq heterodimers. Nat Chem Biol. 2011;7:740–7.
Jeon MS, Song SH, Yun J, et al. Aberrant epigenetic modification of LPHN2 function as a potential cisplatin-specific biomarker for human gastrointestinal cancer. Cancer Res Treat. 2016;48:676–86.
Kocibalova Z, Guzyova M, Imrichova D, Sulova Z, Breier A. Overexpression of the ABCB1 drug transporter in acute myeloid leukemia cells is associated with downregulation of latrophilin-1. Gen Physiol Biophys. 2018;37:353–7.
Yasinska IM, Sakhnevych SS, Pavlova L, et al. The Tim-3-galectin-9 pathway and its regulatory mechanisms in human breast cancer. Front Immunol. 2019;10:1594.
Wang J, Xi S, Zhao Q, et al. Driver mutations in ADGRL3 are involved in the evolution of ependymoma. Lab Invest. 2022;102:702–10.
O’Sullivan ML, de Wit J, Savas JN, et al. Postsynaptic FLRT proteins are endogenous ligands for the black widow spider venom receptor latrophilin and regulate excitatory synapse development. Neuron. 2012;73:903–10.
Kan Z, Jaiswal BS, Stinson J, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–73.
Kashiwagi E, Ide H, Inoue S, et al. Androgen receptor activity modulates responses to cisplatin treatment in bladder cancer. Oncotarget. 2016;7:49169–79.
Ide H, Goto T, Teramoto Y, et al. FOXO1 inactivation induces cisplatin resistance in bladder cancer. Cancer Sci. 2020;111:3397–400.
Teramoto Y, Jiang G, Goto T, et al. Androgen receptor signaling induces cisplatin resistance via down-regulating GULP1 expression in bladder cancer. Int J Mol Sci. 2021;22:10030.
Goto T, Miyamoto H. The role of estrogen receptors in urothelial cancer. Front Endocrinol. 2021;12:643870.
Nagata Y, Miyamoto H. The prognostic role of steroid hormone receptor signaling pathways in urothelial carcinoma. Transl Cancer Res. 2020;9:6596–608.
Ide H, Mizushima T, Jiang G, et al. FOXO1 as a tumor suppressor inactivated via AR/ERβ signals in urothelial cells. Endocr Relat Cancer. 2020;27:231–44.
Tatenuma T, Matsukawa T, Goto T, et al. GULP1 as a downstream effector of the estrogen receptor-β modulates cisplatin sensitivity in bladder cancer. Cancer Genom Proteom. 2024 (in press).
Acknowledgements
Not applicable.
Funding
This study received no external funding.
Author information
Authors and Affiliations
Contributions
T.G. collected and elaborated data and wrote the original manuscript. Y.T. and Y.N. collected data. H.M. conceived, designed, and supervised the study. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and informed consent
Appropriate approval from the Institutional Review Boards at the University of Rochester Medical Center and The Johns Hopkins Hospital, including the request to waive the documentation of informed consent from the patients, was obtained before construction and use of the set of bladder TMA.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Goto, T., Teramoto, Y., Nagata, Y. et al. Latrophilin-3 as a downstream effector of the androgen receptor induces bladder cancer progression. Discov Onc 15, 440 (2024). https://doi.org/10.1007/s12672-024-01324-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-024-01324-2