
Abstract
Hepatocellular carcinoma (HCC) is a highly heterogeneous disease, making the prognostic prediction challenging. Ferroptosis, an iron-dependent form of cell death, is a key regulator in the initiation, progression, and metastasis of HCC. However, the expression and function of ferroptosis-related genes (FRGs) in HCC remained largely unclear. In this study, we analyzed TCGA datasets and identified 58 survival-related deferentially expressed FRGs (DE-FRGs). Then, based on the results of LASSO analysis, we developed a novel prognostic model based on 12 survival-related DE-FRGs. Survival assays indicated a strong prognostic ability of this new model in predicting clinical prognosis of HCC patients. In addition, we conducted an exploration of molecular subtypes related to HCC and delved into the associated immune characteristics and gene expression patterns. Among the 12 survival-related DE-FRGs, our attention focused on ABHD12 (abhydrolase domain containing 12) which was highly expressed in HCC and associated with advanced clinical stages. Multivariate assays confirmed that ABHD12 was a significant prognostic factor for HCC patients. Immune analysis revealed that ABHD12 may play an important role in tumor microenvironment. Finally, we performed RT-PCR and confirmed that ABHD12 was highly expressed in HCC cells. Functional experiments revealed that ABHD12 knockdown may suppress the proliferation and migration of HCC cells. These findings emphasized the significance of ABHD12 as a potential prognostic marker for HCC and its crucial role in the field of tumor biology. Additionally, the study introduces a novel survival model that holds promise for enhancing prognostic predictions in HCC patients. Overall, this research provided valuable insights for a deeper comprehension of the complexity of HCC and the development of personalized treatment strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Liver cancer is the sixth most prevalent malignant digestive system cancer and the third leading cause of cancer-related death around the world [1]. Hepatocellular carcinoma (HCC) is the most common form of liver cancer, accounting for 75–95% of all cases [2]. The incidence of HCC varies significantly across the world. In Asian regions, particularly in China, Taiwan, South Korea, and Vietnam, the incidence of HCC is relatively high [3]. This is partly attributed to the high prevalence of hepatitis B virus (HBV) and hepatitis C virus (HCV) infections, as well as certain dietary and lifestyle habits in some areas. There is a certain association between non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) with HCC (Hepatocellular carcinoma). The prevalence of these diseases is on the rise globally, which could potentially lead to an increase in the incidence of HCC. The higher risk of HCC in males compared to females may be associated with differences in sex hormones and the reproductive system [4, 5]. Although surgery, radiation, chemotherapy, and immunotherapy have all made significant strides in HCC treatment, the prognosis of HCC remains poor, with an estimated 5-year overall survival of fewer than 20%. There is currently no approved medication that slows disease development or stops recurrence. Thus, there is an immediate need for methodologies that can predict patient prognosis, so that targeted therapy and management strategies for subgroups of HCC patients can be developed.
Ferroptosis is a relatively new and distinct form of cell death, different from other known types of cell death such as apoptosis, necrosis, and autophagy [6]. It occurs when there is an accumulation of iron ions within the cell, under conditions of oxidative stress, typically accompanied by lipid peroxidation, leading to the rupture of cell membranes and cell death [7, 8]. The occurrence of ferroptosis requires the accumulation of free iron ions within the cell, which is typically associated with disruptions in iron metabolism. Iron plays a crucial role in various biochemical processes within the cell, including maintaining cell survival and responding to oxidative stress. Damage to cell membranes resulting from oxidative processes within lipid molecules is a hallmark of ferroptosis, known as lipid peroxidation. In ferroptosis, this procedure is essential. The occurrence of ferroptosis is associated with intracellular oxidative stress, where an excess of reactive oxygen species (ROS) is present, leading to oxidative damage [9]. Cysteine inhibitors can reduce these oxidative stress reactions, aiding cells in maintaining oxidative balance. Some studies suggest that ferroptosis may play a role in inhibiting tumor growth. Cancer cells often have a higher demand for iron to support their rapid growth and division [10]. By interfering with iron metabolism and inducing ferroptosis, it is possible to suppress the proliferation of tumor cells. The potential of ferroptosis as a cancer treatment has captured the attention of scientists. Various medicines and treatment strategies have been devised to promote ferroptosis in tumor cells [11, 12]. The liver serves as a major reservoir and metabolic hub for iron in the body, and HCC often accompanies abnormal iron accumulation. The elevated iron requirements of HCC cells may be related to their prodigious growth and proliferation. Because of their ability to disrupt cellular structure and function, oxidative stress and lipid peroxidation, two biological processes strongly linked to ferroptosis, also play crucial roles in HCC [13, 14]. Interventions targeting iron metabolism are therefore being investigated as a possible therapy option for HCC. The goal is to trigger ferroptosis and so reduce tumor cell growth. Although ferroptosis is a promising new approach to cancer treatment, more studies are needed to determine its precise roles and efficacy in HCC therapy. HCC is a multifaceted disease, and its treatment typically requires consideration of multiple factors. While ferroptosis represents a promising new direction for future HCC treatment, it is currently in the early stages of research and clinical trials.
In this study, we analyzed TCGA datasets and explored the expressing pattern and prognostic values of ferroptosis-related genes in HCC. Then, we further explored the association between ferroptosis-related genes and immune microenvironment. Finally, we identified a critical ferroptosis-related gene ABHD12 and further performed in vitro experiments to explore its potential function in HCC. This study not only enhances our understanding of the molecular mechanisms and prognostic factors in HCC but also offers critical insights for immunotherapy and potential therapeutic target research. It holds profound clinical and scientific significance.
2 Materials and methods
2.1 Cell lines and cell culture
Various HCC cancer cell lines, including Huh-7, JHH-4, JHH-2, SNU-398, Hep3B, SNU-182 and HepG2 cells were obtained from Shanghai Cell Bank (Xuhui, Shanghai, China). A human normal liver cell line, LO2 cell line, was purchased from HongShun Biotechnology (Minhang, Shanghai, China). All the cells were cultured in RPMI-1640 media containing 10% fetal bovine serum (FBS, Gibco, Carlsbad, CA, USA), penicillin (100 U/ml) and streptomycin (100 μg/ml). The penicillin and streptomycin were obtained from Beyotime Biotechnology (Haimen, Jiangsu, China). All the cells were maintained in a humidified 37 °C incubator with 5% CO2 with changing the refresh media every 2–3 days.
2.2 siRNA synthesis and cell transfection
The siRNAs targeting ABHD12 (siRNA-1 and siRNA-2) were synthesized by Jushengwu Biotechnology (Wuhan, Hubei, China). Lipofectamine™ 3000 reagent kits (Invitrogen, Carlsbad, CA, USA) were employed for siRNAs transfection. In brief, Huh-7 or Hep3B cells were cultured in proper plates, and when the cell confluence reached about 70%, the diluted siRNAs were mixed with Lipofectamine™ 3000 reagents and the resulting siRNA-Lipofectamine™ 3000 mixture was added into the HCC cells. After being incubated for 5 h in incubator, the media were changed, replaced with fresh media and incubated for an additional 48 h for the following experiments.
2.3 Real-time PCR assays
The mRNAs of ABHD12 in the present study were examined by qRT-PCR assays. In brief, total RNA isolation from Huh-7 or Hep3B cells after siRNAs transfection was performed using the RNeasy Mini kits (Qiagen, Germantown, MD, USA) according to the manufacturer’s protocol. The QuantiFast™ SYBR Green RT-PCR kits (Qiagen, Germantown, MD, USA) were applied for synthesizing the first strand and qRT-PCR amplification in a one-step, single-tube format. The PCR condition was as following: 10 min at 55 °C, 5 min at 95 °C, then 40 cycles of 5 s at 95 °C for denaturation, and 10 s at 60 °C for annealing and elongation (one-step). As an internal standard, we used GAPDH. The relative expression of ABHD12 was calculated using 2−∆∆Ct methods. The primers were synthesized by Generay Technologies (Pudong, Shanghai, China), and the primer sequences were: F-ABHD12: 5ʹ-GTCCTGGAATACAGGCCAAAC-3ʹ; R-ABHD12: 5ʹ-CAATGGTCACGTCTTCCTCTG-3ʹ F-GAPDH: 5ʹ-CCACATCGCTCAGACACCAT-3ʹ; R-GAPDH: 5ʹ-ACCAGGCGCCCAATACG-3ʹ.
2.4 Cell proliferation evaluation
The cell proliferation was assessed by using CCK-8 assays and 5-ethynyl-2ʹ-deoxyuridine (EdU) incorporation assays. For CCK-8 assays, the Huh-7 or Hep3B cells after siRNAs transfection were seeded in 96-well plates at a density of 3000 cells/well. After attachment, the HCC cells were incubated with fresh media containing CCK-8 reagents (10% concentration; 100 µl per well; Beyotime, Haimen, Jiangsu, China) for 2 h at 37℃. Afterwards, the absorbance values of each wells at 450 nm were detected using a BioTek microplate reader (Winooski, VT, USA). The cellular proliferation was also determined by using EdU incorporation assays using Click-iT EdU kits from Invitrogen (Grand Island, NY, USA). In brief, the Huh-7 or Hep3B cells after siRNAs transfection were seeded in 96-well plates at a density of 5 × 103 cells/well. Twenty minutes after being cultured with 10 μM EdU for 24 h, the cells were fixed with 4% paraformaldehyde and 0.5% Triton X-100. The cell nuclei were stained by DAPI solution. The fluorescence was observed by using an inverted fluorescence microscope.
2.5 Cell migration ability detection
After transfection with siRNAs, the migratory capacity of HCC cells was evaluated using a transwell migration assay in transwell chambers (Corning, NY, USA). Briefly, on the top transwell chamber, siRNA-transfected Huh-7 or Hep3B cells (3 × 103 cells per chamber) were planted while suspended in serum-free media. After that, 600 μl of full media was poured into the bottom chamber to serve as an enticement. Following incubation for 24 h, migrating cells were stained with 0.1% crystal violet (Beyotime biotech, Haimen, Jiangsu, China) for 15 min, washed, then analyzed by counting and photographing under an optical microscope (Olympus IX73, Tokyo, Japan).
2.6 Malondialdehyde (MDA) assays
The relative MDA concentration was evaluated by using lipid peroxidation kits (#ab118970; Abcam, Pudong, Shanghai, China). In brief, HCC cells in the 6-well plates were transfected with siRNAs. After 48 h, they were washed PBS and placed on ice in 300 μl of the MDA lysis buffer, followed by being centrifuged (12,000 × g, 5–10 min), and the supernatants were collected. Then, 20 μl of the supernatants and 60 μl of the thiobarbituric acid (TBA) solution were mixed in a 96-well plate, followed by incubation at 95 ℃ for 50 min. Subsequently, the absorbance at a wavelength of 532 nm was measured using a BioTek microplate reader (Winooski, VT, USA).
2.7 Acquisition of public data
The 564 ferroptosis-related genes (FRGs) were obtained from the FerrDb database, which is the first database that dedicates to ferroptosis regulators including driver, suppressor, marker, and unclassified regulator. The RNA-sequencing expression (level 3) and corresponding clinical information of liver hepatocellular carcinoma (LIHC) including 371 HCC samples and 50 paired normal samples were downloaded from database. In addition, the cell line mRNA expression matrix of tumors was obtained from the CCLE dataset.
2.8 Identification of deferentially expressed ferroptosis-related genes (DE-FRGs) and prognostic signature model construction
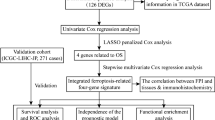
Using the "limma" tool in R (Version 4.0.3), we analyzed TCGA data to determine the DEGs of HCC. Both the logFC and the adjusted p value (adj.P) were set to > 1.5 and 0.05, respectively. The volcano plot of HCC DEGs was displayed using R software. Then, the genes with significant overall survivals in HCC were obtained by using UALCAN database (https://ualcan.path.uab.edu/index.html) and there were 5759 genes (Significant-OS). The overlap genes among DEGs, FRGs and Significant-OS genes were obtained by using Venny 2.1 website (https://bioinfogp.cnb.csic.es/tools/venny/), and there were a total of 58 deferentially expressed ferroptosis-related genes (DE-FRGs) including 56 up-regulated and 2 down-regulated DE-FRGs. In addition, using the LASSO Cox regression, a prognostic model including the 56 up-regulated DE-FRGs was built using the R software “survival” and “glmnet” packages. In the prognostic signature, the risk score formula is: risk score = (coefficient mRNA1 * expression of mRNA1) + (coefficient mRNA2 * expression of mRNA2) + … + (coefficient mRNAn * expression mRNAn). Afterwards, all the HCC samples were separated into high- and low-risk sub-groups based on the median of the risk scores.
2.9 Immune infiltration analysis
CIBERSORT is a computational tool and method used for analyzing the relative abundance of different cell types within complex mixed cell populations [15, 16]. It is a computational biology tool based on gene expression data and is widely applied in biomedical research and the field of bioinformatics. The "CIBERSORT" package in R software was utilized for immune cell infiltration analysis, encompassing immune scores of various immune cells such as T cell follicular helper, Tregs, Macrophage M0, Monocyte, B cell naive, and B cell memory. This analysis also examined the distribution of immune cells across the four molecular subtype-groups of HCC. Besides, the relationships of the expression, copy number variants (CNA), methylation of gene and tumor infiltrating lymphocytes (TILs), as well as other immune factors including immunoinhibitors, immunostimulators, MHC molecules, chemokines and receptors were analyzed by using TISIDB database (http://cis.hku.hk/TISIDB/). Moreover, the immune interacting networks of each four HCC molecular subtype-groups or HCC grades and the DE-FRGs were constructed by using R software package “immuneeconv” based on EPIC algorithm, and the visualization were performed by the R software “ggClusterNet” package.
2.10 Stemness indices evaluation
The MRNAsi score is a transcriptomic-based OCLR stemness indicator [17]. One-class logistic regression is a variant of logistic regression that is primarily used for anomaly detection and classification problems where one class is significantly underrepresented compared to the other class (class imbalance) [18]. In a traditional binary logistic regression, the goal is to model the relationship between a binary dependent variable and one or more independent variables. However, in one-class logistic regression, the focus is on modeling the characteristics of the majority class while ignoring the minority class. In the current investigation, a one-class logistic regression (OCLR) machine-learning technique was employed to construct stemness indices (mRNAsi scores). Stronger stem cell characteristics were indicated by mRNAsi scores closer to 1, within a range from 0 to 1. Samples without detectable mRNAsi were excluded. To assess associations between clinical factors such as age, pN-stage, pM-stage, grade, or radiation, and low or high stemness scores in HCC samples, mRNAsi values were ordered from low to high stemness index.
2.11 Online bioinformatics analysis websites
The Gene Set Cancer Analysis (GSCA) database (http://bioinfo.life.hust.edu.cn/GSCA/#/) was used for analyzing gene expression, survivals (including OS, PFS, DSS), SNV, CNV and methylation in pan-cancers based on TCGA data. The expression of ABHD12 in pan-cancers and human tissues were analyzed respectively using TIMER 2.0 database (http://timer.cistrome.org/) and the Human protein atlas (HPA, https://www.proteinatlas.org/). The STRING database (https://cn.string-db.org/) was utilized for generating PPI network of 12 DE-FRGs. In addition, to identify other potential interacting proteins of each 12 DE-FRGs, functional protein networks were built by GeneMANIA database (http://genemania.org/search/homo-sapiens/). The KEGG pathway enrichment analysis was further conducted using R software “ClusterProfiler”, and the top 20 signaling pathways were plotted by using Chiplot website (https://www.chiplot.online/#).
2.12 Statistical analysis
R (version 4.2.0; The R Foundation for Statistical Computing, Vienna, Austria) and GraphPad Prism (version 8.02; GraphPad Software, San Diego, CA, USA) were used for all statistical analysis and graphical representations. Student's t-test or Wilcoxon signed-rank test was used to compare the two groups. Two-tailed p < 0.05 was considered statistically significant. p values are indicated by asterisks as follows: ∗ p < 0.05, ∗ ∗ p < 0.01, ∗ ∗ ∗ p < 0.001.
3 Results
3.1 DEGs identification and prognostic signature model construction based on deferentially expressed FRGs
Firstly, we analyzed the DEGs in HCC tumor tissues when compared with the corresponding normal tissue samples, and consequently 2451 up-regulated and 446 down-regulated genes were obtained. The DEGs were presented in Fig. 1A by using volcano plot. Afterwards, we attempted to narrow the range of these DEGs, and thereby the overlap genes among DEGs, FRGs and genes with significant overall survivals from UALCAN database were obtained, and there were a total of 58 deferentially expressed ferroptosis-related genes (DE-FRGs) (Fig. 1B). Thereafter, the LASSO regression was performed to develop the prognostic signature model in HCC using the 58 DE-FRGs, and their coefficient was shown in Fig. 1C. Following cross-validation, 12 genes achieved the minimum partial likelihood deviance (Fig. 1D). The formula used for risk score computation was as follows: riskscore = (0.0658)*G6PD + (0.1677)*TIMM9 + (0.0901)*SLC7A11 + (0.1357)*CFL1 + (-0.1116)*FTH1 + (0.0814)*SQSTM1 + (0.006)*NEDD4L + (0.008)*ABHD12 + (0.1561)*KIF20A + (0.0165)*ETV4 + (0.0263)*EZH2 + (0.0369)*TXNRD1. Then, we divided the training cohort into high-risk and low-risk groups, and the data suggested that poor survival rates were more common in the high-risk group than in the low-risk group, and a heatmap of the expression of 12 genes in HCC was also compiled. (Fig. 1E). Besides, the Kaplan–Meier analysis demonstrated that the patients in the high-risk group had a significantly poor overall survivals than that in the low-risk patients (Fig. 1F). Additionally, to determine the predictive accuracy of this prognostic model, we performed a ROC curve analysis based on TCGA-LIHC cohort, which demonstrated that the area under the curve (AUC) for 1-, 3-, and 5-year was 0.804, 0.733, and 0.7, respectively (Fig. 1G).

DE-FRGs identification and the prognostic signature associated with DE-FRGs. A Volcano map of DEGs in HCC. B Venn plots displaying the DE-FRGs. Significant-OS: the genes with significant overall survivals in HCC were obtained by using UALCAN database. C and D By applying LASSO Cox regression analysis, we have successfully established a prognostic feature signature with predictive potential. This signature can be used to assess the individual's survival status and event risk. E The distribution of risk scores, the survival status of patients, and the expression profiles of 12 DE-FRGs were calculated based on the prognostic signature. F Survival curves depicting the outcomes of HCC patients categorized into low- and high-risk groups. G ROC curves illustrating the performance of the prognostic signature in predicting outcomes at 1-year, 3-years, and 5-years time points
3.2 The expression and prognostic value of deferentially expressed ferroptosis-related genes
Since we obtained 12 deferentially expressed ferroptosis-related genes (DE-FRGs) which were included in our prognostic model in HCC, we next attempted to investigate their expression and prognostic value in pan-cancers. Differential expression analysis using GSCA database between tumor-normal paired samples from kinds of TCGA cancer types indicated that the expression levels of most 12 DE-FRGs were highly expressed in pan-cancers (Fig. 2A). Moreover, the associations between subtypes of various cancer types and 12 DE-FRGs expression were also explored, and the data suggested that almost all the 12 DE-FRGs displayed a subtype-specific expression pattern in BRCA, KIRC, LUAD, LUSC, and STAD (Fig. 2B). In addition, the trend of mRNA expression of 12 DE-FRGs from early pathologic stage to late pathologic stage was also displayed by trend plot, and the data revealed that most 12 DE-FRGs were up-regulated in late pathologic stage in many tumor types (Fig. 2C). Besides, overall survival analysis revealed that the expression of 12 DE-FRGs impacted the prognosis of various tumors in varying ways, and high expression of most 12 DE-FRGs was positively correlated with poor overall survivals especially in KIRC, LIHC,KIRP, ACC, and LGG (Fig. 2D).

The expression and survival analysis of 12 DE-FRGs. A The expression of 12 DE-FRGs in pan-cancers. B The associations between cancer subtypes and 12 DE-FRGs expression. C The trend of 12 DE-FRGs mRNA expression from early pathologic stage to late pathologic stage in pan-cancers. D The overall survival analysis of 12 DE-FRGs in pan-cancers
3.3 Mutational landscape of DE-FRGs in pan-cancers
Somatic mutations are closely related to the occurrence and development of tumors. Therefore, we next explored the SNP data of 12 DE-FRGs in pan-cancers. The single nucleotide variation (SNV) percentage heatmap proved that many of the 12 DE-FRGs had high mutation frequency in UCEC, SKCM, COAD, STAD, BLCA, CESC and UCS, and NEDD4L as well as EZH2 had the most mutation frequency in cancers especially in UCEC (Fig. 3A). In addition, the majority of the variation classification types were missense mutations, as shown by an SNV summary graphic, and the most common SNV class was C > T (Fig. 3B and C). A waterfall plot showed that the mutation frequencies of NEDD4L, EZH2, TXNRD1, KIF20A, G6PD, SLC7A11, SQSTM1, ETV4, ABHD12, and FTH1 was 23%, 20%, 16%, 15%, 14%, 13%, 11%, 9%, 8% and 5%, respectively (Fig. 3D). Survival analysis found that there was no significant survival difference (OS and PFS) between mutant and wild-type of most 12 DE-FRGs in pan-cancers (Fig. 3E).

Mutation analysis of 12 DE-FRGs in pan-cancers. A The single nucleotide variation (SNV) percentage heatmap. B The variant classification type. C The SNV class. D A waterfall plot visualizing the mutation patterns of the top 10 most frequently mutated genes, arranged in descending order based on their mutation frequency. E Survival difference between mutant and wild-type of 12 DE-FRGs
3.4 Copy number variation (CNV) landscape of DE-FRGs in pan-cancers
CNV is an important hallmark of cancer development and progression. For this reason, we next sought to study the CNV changes of 12 DE-FRGs in pan-cancers. The data from bubble plots certified that all the 12 DE-FRGs had many heterozygous copy number amplifications and deletions in most cancer types, and homozygous CNV amplifications and deletions of these genes were observed in more than half of the cancer types (Fig. 4A and B). The correlation analysis revealed that the mRNA expression levels of most 12 DE-FRGs were positively correlated with their copy number levels in most cancers, especially BRCA, LUSC, OV, HNSC, LUAD, COAD, STAD, BLCA and ESCA (Fig. 4C). In addition, the survival analysis including OS, PFS and DSS demonstrated that CNVs of most 12 DE-FRGs were significantly associated with prognosis in UCEC, LGG, KIRP, MESO, ACC and KIRC (Fig. 4D).

Copy number variation (CNV) analysis of 12 DE-FRGs in pan-cancers. A Heterozygous CNV of 12 DE-FRGs in each cancer. B Homozygous CNV of 12 DE-FRGs in each cancer. C The correlation of CNV with 12 DE-FRGs in pan-cancers. D The survival difference between CNV and wild-type
3.5 Methylation analysis of DE-FRGs in pan-cancers
Methylation accounts for tumor development and progression in kinds of cancers. Thus, we next sought to describe the methylation information of 12 DE-FRGs in pan-cancers. The methylation difference of the 12 DE-FRGs in different cancer types was assessed, and we found that more than half of the 12 DE-FRGs were low methylation in many cancer types including KIRC, LUAD, LIHC, THCA, LUSC and BRCA (Fig. 5A). In addition, the correlation between methylation and mRNA expression of 12 DE-FRGs in pan-cancers was also evaluated, and the data suggested that the methylation levels of 12 DE-FRGs were general negatively correlated with their corresponding mRNA levels in most cancer types (Fig. 5B). Besides, we also detected the correlation between methylation and 12 DE-FRGs mRNA expression levels in HCC, and the data indicated that all the 12 DE-FRGs mRNA expression levels in LIHC were obviously negatively correlated with their corresponding methylation levels (Fig. 5C). Moreover, we examined the survival difference (including OS, PFS and DSS) between high and low methylation in each cancer, and the results proved that most of the 12 DE-FRGs methylation difference did not affect the survivals in most cancer types except LGG (Fig. 5D).

Methylation landscape of 12 DE-FRGs in pan-cancers and HCC. A Methylation difference in each cancer. B The correlation between methylation and mRNA expression in pan-cancers. C The correlation between methylation and 12 DE-FRGs mRNA expression in HCC. D The survival difference between high and low methylation groups in pan-cancers
3.6 Protein–protein interacting (PPI) network construction
Constructing a protein–protein interaction (PPI) network of 12 DE-FRGs was conducted with STRING database. According to the data, FTH1, SQSTM1, G6PD, SLC7A11 and TXNRD1 formed a core interacting network, and the rest of 12 DE-FRGs had no direct interaction with each other except EZH2 and KIF20A (Fig. 6A). In addition, to identify other potential interacting proteins of each 12 DE-FRGs, functional protein networks were built by GeneMANIA database. According to the data, other proteins interacting with most 12 DE-FRGs were physical interactions, while the types of ABHD12 and SLC7A11 were mainly genetic interactions (Fig. 6B).

PPI-networks of 12 DE-FRGs or each alone. A A protein–protein interaction (PPI) network of 12 DE-FRGs was conducted with STRING database. B PPI-networks of each 12 DE-FRGs with other genes were generated by using GeneMANIA database
3.7 Interaction map and pathway activity analysis of DE-FRGs
We next wonder whether the 12 DE-FRGs were incorporated in some crucial signaling pathways which account for tumor development and progression. Hence, using the GSCA database's pathway activity module, we compared gene expressions in pan-cancers belonging to the pathway active and inhibit categories, as determined by pathway scores. An interaction map of 12 DE-FRGs and various pathways across pan-cancers was constructed. The data indicated that mRNA expression of these 12 DE-FRGs was closely associated with the activity of cancer-related pathways across different types of cancer. Interestingly, we observed that these DE-FRGs could exert diverse or even opposing regulatory effects on the same pathway in different cancer types (Fig. 7A). Next, we further evaluated the pathway activity of 12 DE-FRGs in LIHC. The heatmap presented the percentage of 12 DE-FRGs activated or inhibited in cancer-related pathways, and the data revealed that some of the 12 DE-FRGs such as EZH2 and KIF20 were able to activate cell cycle, and several other genes such as NEDD4L could inhibit cell cycle, which implied that each of the 12 DE-FRGs might exert different functions in different pathways in LIHC (Fig. 7B).

Gene interaction map and pathway activity analysis. A The interaction map of 12 DE-FRGs and kinds of pathways in pan-cancers. B The heatmap presented the percentage of 12 DE-FRGs activated or inhibited in cancer-related pathways
3.8 Identification of 12 DE-FRGs-related molecular subtypes in HCC
Tumor heterogeneity is a critical factor which accounts for tumorigenesis, tumor development and tumor drug resistance. Thus, we next attempted to clarify the molecular subtypes in HCC based on the 12 DE-FRGs. To achieve this, we conducted unsupervised consensus clustering analysis on 371 HCC samples, setting the k value within the range of 2–6. When k = 5, we identified five molecular subtypes of HCC. These subtypes, designated as Group 1 to Group 5, comprised 62, 114, 60, 87, and 48 HCC samples, respectively (Fig. 8A–C). The relative expression of the 12 DE-FRGs in each group (group 1 to group 5) was shown in Fig. 8D using heatmap plot. Subsequently, the overall survivals of each groups were analyzed and the data suggested that there was significant overall survival difference among group 1 to group 5 molecular subtypes of 371 HCC samples (Fig. 8E).

Identification of HCC molecular subtypes. A The cumulative distribution function (CDF) for consensus clustering. B A delta area curve representing the changes in the cumulative distribution function. C Heatmap of sample clustering when k = 5. D Heatmap of 12 DE-FRGs mRNA levels in five HCC molecular subtypes. E KM survival curve of five HCC molecular subtypes
3.9 Gene correlation analysis and clinical features distribution of 12 DE-FRGs in molecular subtypes of HCC
Next, we sought to investigate the expression correlation of 12 DE-FRGs in TCGA-LIHC cohort, ICGC (International Cancer Genome Consortium) database, and the above identified HCC molecular subtypes. Since the above identified group 5 molecular subtypes of HCC samples contained the least HCC samples (only 48 HCC samples), we thereby excluded the group 5 molecular subtypes in here or the following analysis. Therefore, we first analyzed the expression correlation of 12 DE-FRGs in group 1, group 2, group 3 and group 4 molecular subtypes of HCC samples. The data suggested that the expression of more than a half of the 12 DE-FRGs was positively correlated with each other in all the four molecular subtype-groups, while the rest genes such as KIF20A and ETV4, were negatively correlated with each other (Fig. 9A–D). Afterwards, the expression correlation of 12 DE-FRGs in TCGA-LIHC (371 HCC samples) were analyzed, and all the 12 DE-FRGs were positive correlation with each other, which was different with the results in four molecular subtype-groups, indicating that the molecular subtypes of HCC identified by the 12 DE-FRGs were more proper to be used for HCC study (Fig. 9E). Besides, we also assessed the expression correlation of 12 DE-FRGs in ICGC HCC cohort from France (160 HCC samples), and similar result with that of the molecular subtype-group was observed (Fig. 9F). Finally, we evaluated the clinical features among the four molecular subtypes of HCC including gender (Fig. 10A), race (Fig. 10B), pT stage (Fig. 10C), pN stage (Fig. 10D), pM stage (Fig. 10E), pTNM stage (Fig. 10F), grade (Fig. 10G), and primary & recurrence (Fig. 10H). The data from the clinical feature distribution analysis revealed that the gender, pT stage, pTNM stage and grade had significance among each molecular subtype-groups.

Gene correlation analysis of 12 DE-FRGs in HCC molecular subtypes. A–D Gene correlation analysis of 12 DE-FRGs in group 1 to group 4 HCC molecular subtypes. E Gene correlation analysis based on TCGA-LIHC cohort. F Gene correlation analysis of 12 DE-FRGs based on ICGC-HCC-France cohort

The clinical features distribution of 12 DE-FRGs in HCC molecular subtypes. A Gender. B Race. C pT stage. D pN stage. E pM stage. F pTNM stage. G Grade. H Primary and recurrence
3.10 Immune infiltration analysis and immune interacting network construction in molecular subtype-groups of HCC
Immune infiltration is a crucial factor involved in the tumor microenvironment, and understanding the constitution of immune cells in tumors is critical for expanding immunotherapy efficacy. Hence, we next first sought to carry out the immune infiltration analysis in the above identified four HCC molecular subtype-groups. According to the data analyzed by using CIBERSORT algorithm, the immune scores of many immune cells including T cell follicular helper, Tregs, Macrophage M0, Monocyte, B cell naive, B cell memory and Myeloid dendritic cell resting were significantly different in the above identified four HCC molecular subtype-groups (Fig. 11A). In addition, the distribution of the immune cells across the four HCC molecular subtype-groups was also assessed, and we could deduce that the most common immune cell fractions were B cell naive, Macrophage M0, T cell follicular helper and Tregs (Fig. 11B). Furthermore, we also evaluated the relative expression of immune checkpoints in the four HCC molecular subtype-groups. As the data presented in Supplementary Figure S1A, there were obviously different expression of CD274, CTLA4, HAVCR2, LAG3, PDCD1, TIGIT and SIGLEC15 in the subtype-groups of HCC. Besides, the immune interacting networks of each four HCC molecular subtype-groups and the 12 DE-FRGs were also separately constructed based on EPIC algorithm (Supplementary Figure S1B-E). According to the results, the 12 DE-FRGs seemed to be interact with different immune cells in different HCC molecular subtype-groups. For example, in HCC molecular subtype group 1 and group 3, most of the 12 DE-FRGs were positively correlated with Macrophage, Endothelial cell, T cell CD8 + , T cell CD4 + , and B cell, while negatively relevant with other uncharacterized cell. However, in HCC molecular subtype group 2 and group 4, most of 12 DE-FRGs were negatively correlated with Endothelial cell, T cell CD8 + , T cell CD4 + , and B cell.

Composition of immune cells in HCC molecular subtypes. A The heatmap of different immune cells in group 1 to group 4 of HCC molecular subtypes. B The distribution of the immune cells across the four HCC molecular subtype-groups
3.11 ABHD12 might be selected as a proper prognostic factor of HCC
Among the 12 deferentially expressed ferroptosis-related genes, ABHD12 attracted our attention because ABHD12 is belonged to the lysophosphatidylserine lipase family which regulates kinds of biological processes including immunological processes and tumor development, and ABHD12 has not been studied in HCC. Therefore, we first utilized the R software package “ggrisk” to analyze the ABHD12 with survival time and survival status. The data suggested that when ABHD12 expression was sorted from low expression group to high expression group, the corresponding middle scatter plot from left to right presented a trend of patients dying more with shorter time (Supplementary Figure S2A). The overall survival analysis revealed that the survivals of HCC patients with ABHD12 high expression were poor, and correspondingly the survivals of the ABHD12 low expression were high (Supplementary Figure S2B). Afterwards, the analysis from the receiver operating characteristic (ROC) curves demonstrated that the area under curve (AUC) values for 1-, 3-, and 5-year survival was respectively 0.7, 0.6, and 0.618, indicating that the model of ABHD12 as signature had good accuracy in HCC (Supplementary Figure S2C). Moreover, in univariate Cox proportional hazard regression, ABHD12, pT-stage and pTNM-stage were identified as significant prognostic factors for OS, while in multivariate Cox proportional hazards regression, ABHD12 and pT-stage were identified as significant prognostic factors for OS (Supplementary Figure S2D and E).
3.12 The expression, genetics and methylation landscapes of ABHD12 in pan-cancers
The expression of ABHD12 in pan-cancers was determined by using TIMER 2.0 database, and the results indicated that most cancer types expressed high levels of ABHD12 in tumor samples than that in the corresponding normal samples (Supplementary Figure S3A). Besides, the data from HPA database revealed that ABHD12 expression was relatively lower in most human tissues except nerve system and pancreas (Supplementary Figure S3B). The mutation analysis then proved that the ABHD12 mutation frequency in all TCGA cancers was 29%,and UCEC and SKCM had the highest ABHD12 mutation frequency (Supplementary Figure S3C). The ABHD12 CNV analysis in pan-cancers was also conducted and the data suggested that most cancers had ABHD12 heterozygous copy number amplifications (Supplementary Figure S3D). Finally, the ABHD12 methylation across TCGA cancers was investigated and the data suggested that ABHD12 was low methylation in more than half of these TCGA cancers (Supplementary Figure S3E).
3.13 The correlation between ABHD12 and immune profiles in pan-cancers
Next, we attempted to study the relationship of the expression, copy number variants (CNA), methylation of ABHD12 and tumor infiltrating lymphocytes (TILs), as well as other immune factors using TISIDB database. The data suggested that in most cancers, the ABHD12 was positive correlation with most TILs, while CNA of ABHD12 was negatively correlated with TILs abundance in most cancer types (Supplementary Figure S4A and B). The correlation of ABHD12 methylation was also positively relevant with TILs abundance in most cancer types (Supplementary Figure S4C). In addition, the correlations between ABHD12 expression and immunomodulators including immunoinhibitors, immunostimulators and MHC molecules were further explored. The data demonstrated that ABHD12 expression was positively correlated with many immunoinhibitors in more than half of the TCGA cancer types, while the correlation between ABHD12 expression and immunostimulators was very variant in different cancers (Supplementary Figure S4D and E). However, in most cancers, ABHD12 expression was positively correlated with most MHC molecules (Supplementary Figure S4F). Although the correlation between ABHD12 expression and chemokines was very variant in different cancers, the correlation between ABHD12 expression and receptors was negative in most cancer types (Supplementary Figure S4G and H).
3.14 The mRNA and protein expression of ABHD12 in different subgroups of HCC
We next sought to investigate the mRNA and protein expression of ABHD12 in HCC using UALCAN database. First, we analyzed the ABHD12 mRNA levels based on HCC patients’ sample type, gender, age, race and weight, and the results suggested that the ABHD12 mRNA level was high in all the subgroups of HCC when compared with the paired control samples (Supplementary Figure S5A-E). Then, the data from the study of ABHD12 mRNA level in other subgroups of HCC based on TP53 mutation status, individual cancer stages, tumor grade, nodal metastasis status, suggested that ABHD12 mRNA level was high expressed in all these HCC subgroups (Supplementary Figure S5F-I). Besides, the protein expression of ABHD12 in HCC subgroups was also investigated. The data indicated that ABHD12 protein expression levels in total HCC samples (165 samples), female (32 samples) or male (128 samples) HCC samples, different age HCC samples, were high that that of the corresponding normal samples (Supplementary Figure S5J-L).
3.15 Clinical features and survival analysis of ABHD12 in HCC
Next, we wonder whether the high or low expression of ABHD12 was correlated with the clinical features and survivals of HCC. To achive that, we employed R software package “ggalluvial” to build Sankey diagram which connected the clinical features including age, gender, race, pTNM stage, grade, radiation, pT stage, pN stage, pM stage, and ABHD12 high or low group as well as live or dead status, and the results were presented in Supplementary Figure S6A. The overall survival analysis using Kaplan–Meier Plotter database further revealed that the patients with ABHD12 high expression had poorer survivals than that with low ABHD12 expression (Supplementary Figure S6B). Besides, we also investigate the overall survival of ABHD12 high and low expression using another HCC cohort, ICGC HCC data from RIKEN. The result was similar with the above result based on TCGA data the HCC patients with ABHD12 high expression had poor survivals (Supplementary Figure S6C).
3.16 The ABHD12 mRNA expression based stemness scores evaluation in HCC
Given that our above analyses deeply uncovered the potential prognosis value of ABHD12, we next wonder whether ABHD12 is relevant with tumor stemness which serves as a critical role in regulating tumor development and progression. We used the OCLR algorithm to determine mRNAsi values, then ranked the HCC samples from lowest to highest mRNAsi value to see if any clinical feature correlated with low or high stemness scores; our findings indicated that high ABHD12 expression was significantly linked to high stemness scores (Supplementary Figure S7A). Afterwards, we carried out the stemness score analysis of ABHD12 high and low expression group in different HCC grades (grade 1 to grade 3), and the data indicated that the mRNAsi scores were significantly higher in the ABHD12 high expression group across HCC grade 1 to grade 3, suggesting that ABHD12 high expression was obviously relevant with tumor stemness (Supplementary Figure S7B). Subsequently, the correlation analysis of stemness scores and ABHD12 gene expression in different HCC grades (grade 1 to grade 4) was also conducted, and the results demonstrated that in all the four grades of HCC, ABHD12 gene expression was notably positively correlated with stemness scores (Supplementary Figure S7C). Then, the HCC samples were divided into groups with or without HBV, and the correlation analysis proved that in both HBV positive and negative HCC samples, ABHD12 gene expression was positively correlated with stemness scores (Supplementary Figure S7D). Besides, the correlation was also conducted between ABHD12 expression and stemness scores in the TP53 mutation and non-mutation HCC samples, and the data suggested that ABHD12 expression and stemness score was positive correlation only in TP53 wild-type HCC samples but not TP53 mutant samples (Supplementary Figure S7E).
3.17 The immune analysis in different HCC grades based on ABHD12 high and low expression and ABHD12 immune interacting network construction
First, the immune scores in different HCC grades (grade 1 to grade 4) and the correspondingly paired normal tissues were estimated, and the data suggested that the immune cells were obviously different among the different HCC grades samples and normal tissue samples (Supplementary Figure S8A). Then, the immune score analysis in different grades of ABHD12 high expression HCC samples was assessed, and the data indicated that only T cell CD4 + was significantly different among the four grades of ABHD12 high expression HCC samples (Supplementary Figure S8B). However, in the ABHD12 low expression HCC samples, there were marked differences of macrophage M0 and Tregs among the four grades of HCC samples (Supplementary Figure S8C). Afterwards, the immune checkpoints expressions in ABHD12 high and low expression HCC samples were evaluated, and the data revealed that the expressiosn of CTLA4, HAVCR2, LAG3, PDCD1 and TIGIT were significantly higher in the ABHD12 high expression HCC samples than that of the ABHD12 low expression HCC samples (Supplementary Figure S8D). In addition,using the TIDE score and the TIDE algorithm, we were able to predict how ABHD12 high and low expression groups will react to an immune checkpoint blockade (ICB). The data suggested that the TIDE score was obviously higher in ABHD12 high expression group than that in the ABHD12 low expression group, indicating that high ABHD12 expression HCC patients were more sensitive to ICB therapy (Supplementary Figure S8E). Thereafter, the interacting networks between ABHD12 and kinds of immune cells in different grades of HCC were built (Supplementary Figure S9A-D). The data suggested that in the low grades (grade 1 and grade 2), ABHD12 was negatively correlated with T cells, B cells, endothelial cells and NK cells, while in the high grades (grade 3 and grade 4), ABHD12 was positively correlated with these cells. And in all the four grades, ABHD12 was positively relevant with macrophage.
3.18 Real-time PCR detects ABHD12 expression in HCC cell lines and after siRNAs transfection
Since the above analyses indicated that ABHD12 might act as crucial roles in modulating HCC development and progression, we next attempted to carry out experiments to examine the functions of ABHD12 in HCC cells. Firstly, the ABHD12 mRNA expression data in more than 900 cancer cell lines were downloaded in CCLE database, and the relative ABHD12 mRNA levels in kinds of cancer types were presented in Supplementary Figure S10A. Then, we drew the lollipop chart to display the relative ABHD12 mRNA levels in 25 HCC cell lines, and it was shown that all the 25 HCC cell lines expressed ABHD12 (Supplementary Figure S10B). Thereafter, the qRT-PCR assays were applied for the determination of the ABHD12 mRNA levels in several HCC cell lines including Huh-7, JHH-4, JHH-2, SNU-398, Hep3B, SNU-182 and HepG2, and the data demonstrated that when compared with the normal control cell line LO2, other HCC cell line expressed high levels of ABHD12 (Supplementary Figure S10C). Since Huh-7 and Hep3B expressed the highest ABHD12 levels among these HCC cell lines, we next selected these two cell lines for further investigation. The siRNAs targeting ABHD12 were synthesized (siRNA-1 and siRNA-2) and respectively transfected into Huh-7 and Hep3B cells. Then, the qRT-PCR assays were utilized for detecting the ABHD12 levels, and the results displayed that the siRNAs were capable to dramatically silence the ABHD12 expression in both Huh-7 and Hep3B cells (Supplementary Figure S10D). These findings underscored the pivotal role of ABHD12 in HCC, suggesting its potential as a therapeutic target. The study lays the groundwork for further investigations aimed at understanding the molecular mechanisms by which ABHD12 influences HCC progression, ultimately contributing to the development of novel therapeutic strategies for treating hepatocellular carcinoma.
3.19 Silencing ABHD12 expression inhibits HCC cell growth and migration
Then, we sought to study the affection of ABHD12 knockdown on HCC cell proliferation and migration. We first employed CCK-8 assays to determine the cellular growth of HCC cells, and the data suggested that the HCC cell growth including Huh-7 and Hep3B cells was notably suppressed by siRNAs transfection (Fig. 12A and B). Afterwards, we carried out EdU incorporated assays to further evaluate the impact of the ABHD12 knockdown on Huh-7 and Hep3B cell proliferation. The result from the EdU incorporated assays indicated that ABHD12 depletion resulted in significantly decreased proliferate cells in both the Huh-7 and Hep3B cells (Fig. 12C and D). Besides, the affection of ABHD12 depletion on HCC cells migration was also assessed by using transwell migration assays. The results suggested that suppressing ABHD12 expression by transfecting siRNAs caused markedly decreased migration cell number of both Huh-7 and Hep3B cells (Fig. 12E). These findings underscored the critical role of ABHD12 in promoting HCC cell proliferation and migration, highlighting its potential as a therapeutic target for inhibiting tumor progression in hepatocellular carcinoma.

Cell proliferation and migration analysis. A and B CCK-8 assays determined the proliferation of Huh-7 and Hep3B cells treated with siRNAs targeting ABHD12. C and D EdU staining. The red represents the proliferate cells and the blue represents nuclei stained by DAPI dye. E Transwell migration assays assessed the migration ability of Huh-7 and Hep3B cells treated with siRNAs targeting ABHD12. ∗p < 0.05, ∗∗ p < 0.01, ∗∗∗p < 0.001
4 Discussion
Ferroptosis is closely related to HCC. Firstly, iron overload is a significant factor that can significantly increase the risk of developing HCC. Prolonged iron overload can damage the liver and potentially lead to chronic liver diseases, including cirrhosis, which is one of the primary risk factors for HCC. The danger of developing HCC is greatly amplified when liver damage has occurred [19, 20]. Second, ferroptosis is a method of cell death that occurs as a result of an excess of iron. Accumulation of iron within cells results from iron overload, which in turn causes oxidative stress reactions and releases numerous free radicals. The cell's nucleic acids, proteins, and lipids can all be damaged by these noxious chemicals, which in turn raises the likelihood of cellular mutations and fosters tumor growth [21]. Furthermore, studies have linked iron deficiency to the initiation and progression of several tumor types, including HCC [22, 23]. HCC is exacerbated by the immune system's failure to recognize and combat tumor cells, potentially exacerbated by the accumulation of iron within cells. In this study, we identified 58 survival-related DE-FRGs. Subsequently, LASSO regression analysis was performed using these 58 DE-FRGs, ultimately selecting 12 genes to construct a prognostic signature model. This model categorizes HCC patients into high-risk and low-risk groups, with the high-risk group showing poorer survival outcomes. Kaplan–Meier analysis confirmed significantly lower survival rates among patients in the high-risk group. In addition, pan-cancer assays confirmed that the 12 genes may exhibit an important role in the progression of several tumors. This study revealed the mutational, copy number variation, and methylation patterns of DE-FRGs in various cancer types. Some DE-FRGs exhibited high mutation frequencies and copy number variations, which may have implications for tumor development and prognosis. However, in most cases, differences in methylation of DE-FRGs did not appear to directly impact patient survival. These findings underscore the complexity of DE-FRGs in cancer biology and provide valuable insights for further research into their functions and clinical significance in specific cancer types. These research findings indicate the potential clinical utility of this prognostic model, serving as a valuable tool to guide the management and treatment decisions for HCC patients.
Tumor heterogeneity refers to the presence of various types of cells, subtypes, or genetic variations within a tumor, exhibiting differences in biological characteristics, molecular marker expression, genetic alterations, and more [24, 25]. This diversity can exist within different regions of the same tumor tissue or among tumors in different individuals. Tumor heterogeneity can lead to varying sensitivities to drugs within different parts of the tumor. Some tumor cells may exhibit resistance to a particular therapeutic drug, while others may be sensitive to the same drug [26, 27]. Understanding tumor heterogeneity is crucial for cancer treatment because it can assist doctors in selecting more precise treatment approaches, personalized treatment strategies, and the development of new targeted therapies. In this study, we conducted cluster analysis using 12 DE-FRGs on 371 HCC samples, identifying five distinct molecular subtypes of HCC. These subtypes exhibited significant differences in the gene expression levels of DE-FRGs, patient survival outcomes, and clinical characteristics. Further analysis revealed that the expression of DE-FRGs was correlated within these subtypes, a finding validated in other datasets. Additionally, the researchers observed significant variations in clinical features such as gender, staging, and grading among the different subtypes. Our findings provide a foundation for more precise HCC classification and personalized treatment, with the potential to enhance the management and treatment of HCC patients.
The Tumor Microenvironment (TME) refers to the biological and chemical environment within cancerous tissue, encompassing not only the tumor cells themselves but also the surrounding cells, blood vessels, extracellular matrix (ECM), immune cells, inflammatory factors, and various enzymes that interact with them [28, 29]. TME is critical to tumor initiation, development, and resistance to therapy. T cells, B cells, natural killer cells, antigen-presenting cells, and many others are all present in the tumor microenvironment. When the extracellular matrix (ECM) is present in high concentrations in the tumor microenvironment, it can impede the metastasis of tumor cells and the delivery of therapeutic medications. In this research, immune infiltration and immune interaction networks were analyzed and built for molecular subtypes of HCC [30, 31]. Immunological cell composition and the expression of immunological checkpoints were shown to vary significantly between HCC molecular subtype groups. The variation in immune cell composition between these HCC subtype groups influenced the immune characteristics of the tumor microenvironment. Furthermore, the study identified distinct interaction patterns between 12 DE-FRGs and immune cells in different subtype groups. These findings contribute to a better understanding of the immune characteristics of HCC, offering valuable insights for personalized immunotherapy and the modulation of the tumor microenvironment. In summary, the immune features of molecular subtypes in HCC hold significant relevance for cancer treatment and management.
Among the 12 DE-FRGs, our attention focused on ABHD12 (abhydrolase domain containing 12). ABHD12 is an enzyme encoded by the ABHD12 gene and belongs to the lipase family [32]. Its primary function in the human body is closely related to lipid metabolism and neurological functions. The main role of ABHD12 is to regulate lipid metabolism in the nervous system. It does so by hydrolyzing a specific lipid molecule called N-acylphosphatidylethanolamine (NAPE), which is crucial for maintaining the normal functioning of the nervous system [33,34,35]. Defects or mutations in the ABHD12 gene can lead to a rare genetic disorder known as N-acylphosphatidylethanolamine phospholipase D (NAPE-PLD) deficiency or neurodegeneration with brain iron accumulation 4 (NBIA4). In recent years, several studies have reported that ABHD12 was involved in tumor progression. For instance, Jun et al. reported that ABHD12 was highly expressed in breast cancer cells and its knockdown distinctly suppressed the proliferation, migration and invasion of breast cancer cells, indicating it as a tumor promotor [36]. It was also reported that blockade of the lysophosphatidylserine Lipase ABHD12 potentiated ferroptosis in cancer cells [37]. However, the expression and function of ABHD12 in HCC have not been investigated. The study first conducted an analysis of ABHD12's association with the survival of HCC patients using the R software package "ggrisk." The findings revealed that high expression of ABHD12 was correlated with poorer survival outcomes. Furthermore, the model utilizing ABHD12 as a prognostic marker demonstrated excellent accuracy in predicting outcomes in HCC patients. Across different cancer types, ABHD12 expression levels were typically higher in tumor tissues compared to normal tissues. In TCGA cancer datasets, the mutation frequency of ABHD12 was 29%, with the highest frequencies observed in UCEC and SKCM. ABHD12 methylation levels were generally low. ABHD12 exhibited a positive correlation with tumor-infiltrating lymphocytes (TILs) and various immune-related factors, indicating its potential role in immune responses. Finally, both mRNA and protein levels of ABHD12 were consistently elevated across different subgroups. These findings collectively support ABHD12 as a promising prognostic marker for HCC and suggest its significance in the context of HCC and other malignancies. Finally, we confirmed that ABHD12 was highly expressed in HCC cells and its knockdown distinctly suppressed the proliferation and migration of HCC cells. Despite identifying ABHD12 as a potential therapeutic target due to its high expression in HCC cells and its significant impact on cell proliferation and migration upon knockdown, our study acknowledges the need for further research to elucidate effective therapeutic strategies and their clinical efficacy.
The study is subject to certain limitations. Firstly, The analysis is based on TCGA database, where potential biases in sample selection and data quality may limit the generalizability and applicability of the study findings. Secondly, while the study emphasizes the importance of ABHD12 in HCC, its specific molecular mechanisms and exact role in the ferroptosis pathway remain incompletely elucidated. Thirdly, although ABHD12 has been identified as a significant prognostic factor for HCC, further clinical cohort studies are needed to assess its general applicability and clinical prospects across different populations.
5 Conclusion
We screened 58 survival-related DE-FRGs and developed a novel prognostic model based on 12 of these genes. Additionally, we identified differences among molecular subtypes of HCC, including variations in gene expression patterns, immune infiltration, and interactions. These findings offer crucial insights into the heterogeneity of HCC and the development of personalized treatment strategies. Furthermore, we provided evidence that ABHD12 serves as a novel prognostic biomarker for HCC patients, and its knockdown suppresses the proliferation and migration of HCC cells. Overall, these research findings not only enhance our understanding of the biological characteristics of HCC but also open new avenues for the diagnosis, treatment, and prognosis assessment of this disease.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33.
Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet (London, England). 2018;391(10127):1301–14.
Wen N, Cai Y, Li F, Ye H, Tang W, Song P, Cheng N. The clinical management of hepatocellular carcinoma worldwide: a concise review and comparison of current guidelines: 2022 update. Biosci Trends. 2022;16(1):20–30.
Alawyia B, Constantinou C. Hepatocellular carcinoma: a narrative review on current knowledge and future prospects. Curr Treat Options Oncol. 2023;24(7):711–24.
Grandhi MS, Kim AK, Ronnekleiv-Kelly SM, Kamel IR, Ghasebeh MA, Pawlik TM. Hepatocellular carcinoma: from diagnosis to treatment. Surg Oncol. 2016;25(2):74–85.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–82.
Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, Sun B, Wang G. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88.
Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–25.
Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22(7):381–96.
Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054–81.
Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. 2022;82(12):2215–27.
Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185(14):2401–21.
Wu J, Wang Y, Jiang R, Xue R, Yin X, Wu M, Meng Q. Ferroptosis in liver disease: new insights into disease mechanisms. Cell Death Discov. 2021;7(1):276.
Nie J, Lin B, Zhou M, Wu L, Zheng T. Role of ferroptosis in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2018;144(12):2329–37.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–7.
Gershon R, Polevikov A, Karepov Y, Shenkar A, Ben-Horin I, Alter Regev T, Dror-Levinsky M, Lipczyc K, Gasri-Plotnitsky L, Diamant G, et al. Frequencies of 4 tumor-infiltrating lymphocytes potently predict survival in glioblastoma, an immune desert. Neuro Oncol. 2024;26(3):473–87.
Wang C, Qin S, Pan W, Shi X, Gao H, Jin P, Xia X, Ma F. mRNAsi-related genes can effectively distinguish hepatocellular carcinoma into new molecular subtypes. Comput Struct Biotechnol J. 2022;20:2928–41.
Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN, Kamińska B, Huelsken J, Omberg L, Gevaert O, et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell. 2018;173(2):338-354.e315.
He Y, Wu Y, Song M, Yang Y, Yu Y, Xu S. Establishment and validation of a ferroptosis-related prognostic signature for hepatocellular carcinoma. Front Oncol. 2023;13:1149370.
Chen J, Li X, Ge C, Min J, Wang F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 2022;29(3):467–80.
Capelletti MM, Manceau H, Puy H, Peoc’h K. Ferroptosis in liver diseases: an overview. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21144908.
Yao F, Deng Y, Zhao Y, Mei Y, Zhang Y, Liu X, Martinez C, Su X, Rosato RR, Teng H, et al. A targetable LIFR-NF-κB-LCN2 axis controls liver tumorigenesis and vulnerability to ferroptosis. Nat Commun. 2021;12(1):7333.
Li D, Wang Y, Dong C, Chen T, Dong A, Ren J, Li W, Shu G, Yang J, Shen W, et al. CST1 inhibits ferroptosis and promotes gastric cancer metastasis by regulating GPX4 protein stability via OTUB1. Oncogene. 2023;42(2):83–98.
Belakhoua SM, Rodriguez FJ. Diagnostic pathology of tumors of peripheral nerve. Neurosurgery. 2021;88(3):443–56.
Glendining KA, Campbell RE. Recent advances in emerging PCOS therapies. Curr Opin Pharmacol. 2023;68: 102345.
Feng M, Pan Y, Kong R, Shu S. Therapy of primary liver cancer. Innovation (Cambridge, Mass). 2020;1(2): 100032.
Lin S, Hoffmann K, Schemmer P. Treatment of hepatocellular carcinoma: a systematic review. Liver Cancer. 2012;1(3–4):144–58.
Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther. 2021;221: 107753.
Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019;30(1):36–50.
Du T, Gao J, Li P, Wang Y, Qi Q, Liu X, Li J, Wang C, Du L. Pyroptosis, metabolism, and tumor immune microenvironment. Clin Transl Med. 2021;11(8): e492.
Bader JE, Voss K, Rathmell JC. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol Cell. 2020;78(6):1019–33.
Li T, Feng Y, Liu Y, He C, Liu J, Chen H, Deng Y, Li M, Li W, Song J, et al. A novel ABHD12 nonsense variant in Usher syndrome type 3 family with genotype-phenotype spectrum review. Gene. 2019;704:113–20.
Kind L, Kursula P. Structural properties and role of the endocannabinoid lipases ABHD6 and ABHD12 in lipid signalling and disease. Amino Acids. 2019;51(2):151–74.
Ogasawara D, Ichu TA, Vartabedian VF, Benthuysen J, Jing H, Reed A, Ulanovskaya OA, Hulce JJ, Roberts A, Brown S, et al. Selective blockade of the lyso-PS lipase ABHD12 stimulates immune responses in vivo. Nat Chem Biol. 2018;14(12):1099–108.
Ichu TA, Reed A, Ogasawara D, Ulanovskaya O, Roberts A, Aguirre CA, Bar-Peled L, Gao J, Germain J, Barbas S, et al. ABHD12 and LPCAT3 interplay regulates a lyso-phosphatidylserine-C20:4 phosphatidylserine lipid network implicated in neurological disease. Biochemistry. 2020;59(19):1793–9.
Jun S, Kim SW, Lim JY, Park SJ. ABHD12 knockdown suppresses breast cancer cell proliferation. Migrat Invasion Anticancer Res. 2020;40(5):2601–11.
Kathman SG, Boshart J, Jing H, Cravatt BF. Blockade of the Lysophosphatidylserine Lipase ABHD12 Potentiates Ferroptosis in Cancer Cells. ACS Chem Biol. 2020;15(4):871–7.
Acknowledgements
Not applicable.
Funding
This work did not receive specific supports.
Author information
Authors and Affiliations
Contributions
Tiantao Mao, Tianyu Li and Chengshu Liang developed a major research plan. Tiantao Mao, Maosong Zhang, Zupei Peng and Min Tang performed experiments, analyze data, draw charts and write manuscripts. Tianyu Li helped collect data and references and provided technical support. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mao, T., Zhang, M., Peng, Z. et al. Integrative analysis of ferroptosis-related genes reveals that ABHD12 is a novel prognostic biomarker and facilitates hepatocellular carcinoma tumorigenesis. Discov Onc 15, 330 (2024). https://doi.org/10.1007/s12672-024-01211-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-024-01211-w