
Abstract
According to the latest epidemiology of the US, B-cell cancers account for > 3% of all new cancer cases and > 80% of non-Hodgkin lymphomas. However, the disease-modifying small molecular drug suitable for most B-cell cancers is still lacking. RIPK1 (receptor-interacting serine/threonine-protein kinase 1) has been observed to be dysregulated and implicated in the pathogenesis of multiple solid cancers, of which, however, the roles in blood cancers are quite unclear. In our study, to identify multi-function targets for B-cell cancer treatment, we reanalyzed a public transcriptomic dataset from the database of Gene Expression Omnibus, which includes CD19+ B-cell populations from 6 normal donors and patients of 5 CLL, 10 FL, and 8 DLBCL. After overlapping three groups (CLL vs. normal, FL vs. normal, and DLBCL vs. normal) of differentially expressed genes (DEGs), we obtained 69 common DEGs, of which 3 were validated by real-time quantitative PCR, including RIPK3, IGSF3, TGFBI. Interestingly, we found that the loss function of RIPK1 significantly increases the proliferation and viability of GM12878 cells (a normal human B lymphocyte cell line). Consistently, overexpression of RIPK1 in TMD8 and U2932 cells effectively inhibited cell proliferation and growth. More importantly, modifying RIPK1 kinase activity by a small molecule (such as necrostain-1, HOIPIN-1, etc.) alters the cell growth status of B-cell lymphoma, showing that RIPK1 exhibits anti-tumor activity in the context of B-cell lymphoma. Taken together, we consider that RIPK1 may be a potential target in the clinical application of B-cell lymphoma (including CLL, DLBCL, and FL) treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The development of B lymphocytes derived from lymphoid progenitors is a complex cellular phenotypic change and differentiation progress, which involves a web of cell fate decisions in response to internal and external cues drives. During the maturation stage, B-cells with aberration in fate decisions should execute programmed cell death, but rare cells may escape from the quality control system to develop into cancer cells (the so-called B-cell cancers/lymphomas). According to the latest epidemiology of the US, B-cell cancers account for > 3% of all new cancer cases and > 80% of non-Hodgkin lymphomas (the most subcategory of hematological malignancies) [1, 2]. Importantly, the top three B-cell cancers, chronic lymphocytic leukemia (CLL), follicular lymphoma (FL), and diffusion large B-cell lymphoma (DLBCL), account for close to 70% of all non-Hodgkin lymphomas [3]. Although these three lymphomas originate from mature B-cells, they are distinct in the underlying pathogenesis and clinical presentation [1, 3]. Recently, Wong et al. constructed a BAFF (B-cell-activating factor) ligand-based chimeric antigen receptor T cell by targeting three receptors (BAFF-R, BCMA, and TACI), which was demonstrated to be effective in killing multiple B-cell cancers [4]. However, the disease-modifying small molecular drug that is suitable for most B-cell cancers is still lacking.
Receptor-interacting serine/threonine-protein kinase 1(RIPK1), a core component (scaffold function) of (tumor necrosis factor receptor 1) TNFR1 complex, serves as a vital cellular signaling hub that participates in TNF-mediated apoptosis and inflammatory pathways [5, 6]. Also, RIPK1 directly interacts with RIPK3 to promote the activation of necroptosis in a kinase activity-dependent manner [7]. In mouse models, RIPK1 depletion leads to multi-organ inflammation and abnormal cell death in animals [8, 9]. Meanwhile, RIPK1 deficiency in humans also causes severe immunodeficiency, intestinal inflammation, arthritis, etc., showing its evolutionary conserved functions among mammals [10, 11]. In the case of cancers, plenty of studies have recently demonstrated that RIPK1 plays a critical role in cancer immune checkpoint blockade (ICB) by modulating inflammation or immunity-related signaling pathway [12,13,14]. For instance, Cucolo et al. observed the expression level of RIPK1 is strongly associated with interferon-mediated resistance, and genetic deletion of RIPK1 in cancer cells leads to ICB failure by compromising chemokine secretion to decrease ARG1+ suppressive myeloid cells [14]. The vital functions of RIPK1 have been extensively investigated in solid tumors, such as hepatocellular carcinoma, soft-tissue sarcoma, melanoma, cervical Cancer, etc. [7, 15,16,17] However, whether and how RIPK1 is implicated in hematological malignancies (especially B-cell cancers) is still not fully explored.
To identify multi-function targets for B-cell cancer treatment, we reanalyzed a public transcriptomic dataset from the database of Gene Expression Omnibus, which includes CD19+ B-cell populations from 6 normal donors and patients of 5 CCL, 10 FL, and 8 DLBCL. After overlapping three groups (CLL vs. normal, FL vs. normal, and DLBCL vs. normal) of differentially expressed genes (DEGs), we obtained 69 common DEGs, of which 3 were validated using real-time quantitative PCR, including RIPK3, IGSF3, TGFBI, etc. Interestingly, we found that the knockdown of RIPK1 in GM12878 cells (a normal human B lymphocyte cell line) significantly increased cell proliferation and viability. Consistently, overexpression of RIPK1 in TMD8 and U2932 cells effectively inhibited cell proliferation and growth. More importantly, restoration of RIPK1 using HOIPIN-1 treatment significantly inhibited cell proliferation of TMD8 and U2932 cells. Altogether, RIPK1 is a promising target for B-cell lymphoma treatment.
2 Methods and materials
2.1 Human specimen collection and processing
In this study, 17 healthy donors and 55 B-cell lymphoma patients (including 14 children and 41 adults) were enrolled, consisting of 19 chronic lymphocytic leukemia (CLL), 15 diffuse large B-cell patients (DLBCL), and 21 follicular lymphomas (FL). The basic clinical information of patients was summarized in Table 1. The peripheral blood, lymph node, or tonsil tissues were collected (before pharmacological interventions) for the purification of CD19+ B-cells. The written informed consent was obtained from the participants or their families, and all the protocols used in our experiments were approved by the ethics committee of Xuzhou children’s hospital, Xuzhou Medical University.
For the purification of CD19+ B-cells, the tissues of the lymph node or tonsil were mechanically dissociated and washed with PBS buffer containing 1% fetal bovine serum (FBS) and 2 mM EDTA. To remove the red blood cells, the grated tissues were incubated with lysis buffer containing 10 mM KHCO3, 150 mM NH4Cl, and 0.12 mM EDTA for 30 min at 4 °C. Then, the CD19+ B-cells were isolated from the treated tissues (consisting of PBMCs) using CD19 MicroBeads from Miltenyi Biotec company (Cat: 130-050-301, Shanghai, China) following the protocol provided by the manufacturer. The obtained CD19+ B-cell was subjected to total RNA isolation.
2.2 Cell culture
GM12878 cells were purchased from Coriell Institute for Medical Research (NJ, USA), and cultured in RPMI-1640 containing 2 mM L-glutamine and 15%FBS. OCI-LY3 (ACC 761), RAJI (ACC 319), JEKO-1 (ACC 553), U2932 (ACC 633), WSU-FSCCL(ACC 612), and L428(ACC 197) cells were purchased from DSMZ-German Collection of Microorganisms and Cell Cultures (Braunschweig, GERMANY), and cultured in 85%RPMI-1640 plus 15%FBS. TMD8 (CBP600693) cells were purchased from COBIOER biosciences company (Nanjing China), and cultured in RPMI-1640 containing 10%FBS and 0.05 mM 2-mercaptoethanol. Human embryonic kidney (HEK) 293 T cells used for lentivirus packaging were purchased from American Type Culture Collection and were cultured in Gibco Dulbecco’s Modified Eagle Medium (DMEM) containing 10%FBS. All cells were maintained in a 5% CO2 incubator with an inside 37 °C condition.
2.3 Construct stable cell lines
To construct RIPK1 knockdown and overexpression stable cell lines, home-made lentivirus vectors pLKO.1-puro (based on plasmid #8453 backbone from Addgene) and pLVX-M-puro (based on plasmid #125,839 from Addgene) were employed, respectively. For the packaging transgene lentivirus, HEK293T cells were co-transfected with helper plasmids psPAX.2 and pMD2.G with lentivirus backbone vector (carrying RIPK1 coding sequences or shRNA mimics) using Sinofection transfection reagent (STF02, shanghai, China) following manufacturer’s instructions. After 2 days of transfection, lentivirus supernatant was collected and filtered using a 0.45-mm filter. Next, target cells were directly infected using the obtained virus supernatant. After 12 h of infection, the virus-containing medium was replaced with a fresh culture medium and supplied with puromycin (ST551, Beyotime, Shanghai, China) at a dose of 1.0 μg/ml for positive selection. The oligos for the shRNA construct were listed as follows:shRIPK1_#1: Forward, 5ʹ—CCGG CAGC ACAA ATAC GAAC TTC AACT CGAG TTGA AGTT CGTA TTTG TGCT GTTT TTC—3ʹ; Reverse,5ʹ—AATT GAAA AACA GCAC AAAT ACGA ACTT CAAC TCGA GTTG AAGT TCGT ATTT GTGC TG—3ʹ shRIPK1_#2: Forward,5ʹ—CCGG AGGT CATG TTCT TTCA GCTT ACTC GAGT AAGC TGAA AGAA CATG ACCT TTTT TC-3ʹ; Reverse,5ʹ—AATT GAAA AAAG GTCA TGTT CTTT CAGC TTAC TCGA GTAA GCTG AAAG AACA TGAC CT—3ʹ; shRIPK1_#3: Forward,5ʹ—CCGG CCTT GTTG ATAA TGAC TTCC ACTC GAGT GGAA GTCA TTAT CAAC AAGG TTTT TC—3ʹ; Reverse,5ʹ—AATT GAAA AACC TTGT TGAT AATG ACTT CCAC TCGA GTGG AAGT CATT ATCA ACAA GG-3ʹ.
2.4 Quantitative real-time PCR (qPCR)
The total RNA of B-cells was isolated using the RNAsimple kit from the Tiangen company (DP419, Beijing, China). Next, cDNA was synthesized from 1 μg total RNA using Quantscript Reversely transcription Kit (KR103, Tiangen, Beijing, China). Finally, qPCR was performed using FastFire qPCR PreMix (Cat: FP207, Tiangen, Beijing, China) on CFX Opus 96 Real-Time PCR System (Bio-rad, USA), and each sample was replicated at least three times. GAPDH served as the internal control, and the relative quantification of genes was calculated based on the ΔΔCt method. The oligos used in the study were listed in Table 2.
2.5 Cell proliferation, viability assay
Cell proliferation was determined using Cell Count Kit-8 (CCK-8, Cat: 40203ES60, YEASEN, Shanghai, China). Cell viability was determined using CellQuanti-Blue™ Cell Viability Assay Kit (CQBL-05 K, BioAssay systems). About 5000 cells (including RIPK1-knockdown cells GM12878/L428 and RIPK1 overexpression cells TMD8/U2932) were seeded in a 96-well microplate containing a culture growth medium for 12 h. Then, cells were subjected to a cell proliferation/viability assay every 12 h (0, 12, 24, 36, and 48 h) following the manufacturer’s instruction. Each sample was detected with at least three replications.
2.6 Cell cycle analysis
Propidium Iodide (P1304MP, ThermoFisher Scientific, USA) staining was employed to determine cell cycles. In detail, the collected cells were aliquoted to 1 × 106 cells/100 μL into FACS tubes, followed by twice washing using 1xPBS. Then, cells were centrifuged for 5 min at a speed of 300×g. Furthermore, cells were resuspended using 100 μL of Flow Cytometry Staining Buffer (Cat: # FC001, R&D systems). 10 μL of PI staining solution was added to a control tube of otherwise unstained cells to adjust flow cytometer settings for PI. Cells were mixed gently and incubated for 5 min in the dark. PI fluorescence of samples was determined (using the FL-2 or FL-3 channel) with a FACScan™ instrument. Each detection was repeated at least 3 times. For cell death ratio detection, Annexin V-FITC/PI double-labeled FACS was carried out following the manufacturer’s protocol (Cat: 40302ES20, Yeasen, Shanghai). Flowjo software was used for gate setting FACS original data, and the ggcyto R package (https://bioconductor.org/packages/release/bioc/html/ ggcyto.html) was used for visualization.
2.7 Public data analysis
The public high throughput sequencing data were downloaded from the Gene Expression Omnibus database, which includes CD19+ B-cells from primary peripheral blood or lymph node biopsies of normal participants (N = 6), chronic lymphocytic leukemia (CLL, N = 5), follicular lymphoma (FL, N = 10), and diffuse large b cell patients (DLBCL, N = 8).
(GSE145842, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE145842).
The analysis of differentially expressed genes (DEGs) was performed using an R package of DEseq2 (version-1.36, https://bioconductor.org/packages/release/bioc/html/DESeq2.html), with a cutoff of p-adjust < 0.001 and log2 fold changes > 3. The DEGs identified were visualized by volcano plot using an R package of EnhancedVolcano (version-1.14, https://bioconductor.org/packages/release/bioc/html/EnhancedVolcano.html).
2.8 Statistical analysis
In this study, data were presented as mean ± (SD), and the replications of all the samples were presented in the figure legends. The software of GraphPad Prism9 (San Diego, USA) was employed for statistical analyses. The student’s t-test was utilized for comparisons between two groups, and a one- or two-way ANOVA test for comparisons between three or more groups. A P value < 0.05 was set as the cutoff of statistical significance.
3 Results
3.1 RIPK1 is down-regulated in tumor cells of lymphoma patients
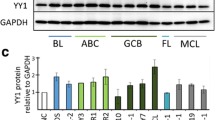
To identify a common target for B-cell lymphoma in treatment, we analyzed a CD19+ B-cell transcriptomic dataset from the Gene Expression Omnibus (GEO) database, which consists of samples from normal participants (N = 6), chronic lymphocytic leukemia (CLL, N = 5), follicular lymphoma (FL, N = 10), and diffuse large B-cell lymphoma patients (DLBCL, N = 8). Firstly, we identified three groups of differentially expressed genes (DEGs) by comparing CLL, DLBCL, and FL with normal controls, respectively, and then calculated the common DEGs (Fig. 1A and Additional file 1: S1A, B). Fortunately, we obtained 69 common DEGs (Fig. 1B, Table 3). Then, we selected the top 14 high-expressed DEGs and validated the expression in our B-cell lymphoma samples (19 CLL, 21 FL, and 15 DLBCL) using real-time PCR (Fig. 1C). In the pooled samples' real-time PCR, 64.2% (9/14) DEGs were successfully confirmed, such as FOS, RIPK1, SPRR3, APOE, etc. (Fig. 1C). Furthermore, we detected the RNA level of these 9 DEGs in individual samples mentioned above, three of which were further validated, including RIPK1, IGSF3, and TGFB1 (Fig. 1D). In detail, the expression level of RIPK1 was dramatically down-regulated both in the CD19+ B-cells of CLL, DLBCL, and FL when compared with that in normal donors, and in contrast, IGSF3 and TGFB1 expression levels were up-regulated (Fig. 1D). Besides, we estimated the expression of these three genes in 8 lymphocyte lines, including a normal human B lymphocyte cell line (GM12878), and 6 B-cell non-Hodgkin lymphoma (OCI-LY3, JEKO-1, RAJI, TMD8, U2932, WSU-FSCCL) and a Hodgkin's lymphoma cell line (L428). Interestingly, we observed that RIPK1 mRNA levels were decreased in B-cell lymphoma cell lines when compared with GM12878 normal B-cells (Fig. 1E). Taken together, our results indicated that RIPK1 is commonly down-regulated in B-cell lymphoma cells.

RIPK1 is down-regulated in tumor cells of lymphoma patients. A Volcano plot of DEGs of CLL and DLBCL vs. normal donors, respectively. Chronic lymphocytic leukemia (CCL, N = 5) and diffuse large B-cell patients (DLBCL, N = 8) vs. normal donors (N = 6). The cutoff was set at p-adjust < 0.001 and log2 fold change > 3. B Venn gram of three groups of DEGs. CLL and DLBCL vs. normal donor from 1A, FL vs. normal donor from S1B, all the genes in detail were listed in Table 3. C Real-time PCR estimated the mRNA level of the top 14 highly expressed DEGs in B. The CD19+ B-cells of 17 normal donors, 19 CCL, 15 DLBCL, and 21 FL were pooled together, respectively, and then were subjected to real-time PCR. The color shows the mean of the relative expression of each group. N = 3. **P < 0.01 and ***P < 0.001 by one-way ANOVA. D Real-time PCR estimated the mRNA level of 3 DEGs in CD19+ B-cells from the indicated participants. Samples are the same with C. **P < 0.01 and ***P < 0.001 by student’s t-test. E Real-time PCR estimated the mRNA level of RIPK1 in 8 lines of B-cells. Normal human B lymphocyte cell line (GM12878), B-cell non-Hodgkin lymphoma (OCI-LY3, JEKO-1, RAJI, TMD8, U2932, WSU-FSCCL), and a Hodgkin's lymphoma cell line (L428). N = 6, ***P < 0.001 by student’s t-test
3.2 Loss-function of RIPK1 contributes to the cell growth of lymphocytes
The aberration of RIPK1 has been extensively established to be implicated in the pathogenesis of multiple categories of solid cancers by altering cell survival, such as pancreatic, colorectal, hepatocellular cancers, etc. [18,19,20]. Therefore, we speculated that the down-regulation of RIPK1 may also play a pathogenic role in B-cell lymphomas. Then, we knock down the expression of RIPK1 in GM12878 (a normal human B lymphocyte cell line) and L428 (a Hodgkin's lymphoma cell line with a relatively high expression of RIPK1) cells using the shRNA vector (Fig. 2A). In the CCK8 assay, we observed that knockdown of RIPK1 in GM12878 cells significantly promoted cell proliferation after 24 h of seeding, and consistently, L428 cells of the shRIPK1 group also dramatically faster than that of the scramble group (Fig. 2B). These results indicate that RIPK1 possesses an inhibitory function in the cell proliferation of B lymphocytes. In the cell viability assay, our results showed that the cell viability of the shRIPK1 group (both in GM12878 and L428 cells) was elevated after 24 h of seeding when compared with that of the scramble group (Fig. 2C). Furthermore, the cell cycle was analyzed using propidium iodide (PI) staining assay. We observed that the loss function of RIPK1 using shRNA in GM12878 cells significantly increased the G2 stage percentage, from 14.3% (in ~ 5000 cells) in the scramble group to 19.8% in the shRNA group (Fig. 2D, E). Consistently, similar results were also found when comparing the shRIPK1 L428 with the scramble one (Fig. 2D, E). These results indicated that RIPK1 possesses an inhibitory function in the proliferative ability of B lymphocytes. Besides, we observed that the loss-function of RIPK1 using shRNA inhibited cell death, evidenced by a reduction of 37.6% and 35.9% of cell death in GM12878 and L428 cells, respectively (Fig. 2F and Additional file 1: S2E). Altogether, the loss function of RIPK1 contributes to the cell growth of lymphocytes.

Loss-function of RIPK1 contributes to the cell growth of lymphocytes A Real-time PCR estimated the RIPK1 knockdown efficiency in GM12878 and L428 cells. Scramble, scramble shRNA without a specific target; shRIPK1, a pool of three independent shRNAs targeting RIPK1. N = 6, ***P < 0.001 by student’s t-test. B CCK8 assay determined the cell growth of GM12878 and L428 cell lines. Cells in A were seeded onto 96-well plates, and 12 h later, cell growth status was detected every 12 h. N = 3, *P < 0.05, **P < 0.01, ***P < 0.001 by two-way ANOVA. C Cell viability of GM12878 and L428 cell lines. Cells in A were seeded onto 96-well plates, and 12 h later, cell viability was detected every 12 h. N = 3, *P < 0.05, **P < 0.01 by two-way ANOVA. D–E. PI staining determined the cell cycle distribution of GM12878 and L428 cell lines. ~ 5000 cells cultured for 36 h were collected for PI staining. shRIPK1#1/2/3 indicates independent shRNA targets of RIPK1. Scr, scramble shRNA; N = 6, ***P < 0.001 by student’s t-test. E Annexin V-FITC/PI double-labeled FACS determined the cell death of GM12878 cell lines. Cells in D were used for experiments
3.3 Ectopic RIPK1 in lymphocytes suppresses cell growth
To further validate RIPK1’s inhibitory function in B-cell proliferation, we utilized a lentivirus vector to construct RIPK1 stably overexpressed cell lines in TMD8 and U2932 cells, two RIPK1 low-expressed DLBCL cell lines (Fig. 1E). After quantification of RIPK1 using real-time PCR, we found RIPK1 expression was elevated to 9.5-fold and 6.8-fold in TMD8 and U2932, respectively (Fig. 3A). In the CCK8 assay, our results showed that, after 24 h of seeding, the cell growth of RIPK1-overexpressed TMD8 cells was significantly slower than that of the empty vector group (Fig. 3B). Consistently, similar results also were observed in RIPK1-overexpressed U2932 cells (Fig. 3B). These results directly reveal that RIPK1 possesses an inhibitory function in the cell proliferation of B lymphocytes. Meanwhile, we evaluated the cell viability assay for RIPK1-overexpressed TMD8 and U2932 cell lines (Fig. 3C). Our results indicated that the cell viability of RIPK1-overexpressed U2932 cells was decreased after 24 h of seeding, which is similar to that of TMD8 cells (since 36 h) (Fig. 3C). Furthermore, we carried out cell cycle analysis for RIPK1-overexpressed TMD8 and U2932 cell lines using PI staining assay. We observed that ectopic RIPK1 in TMD8 cells significantly decreased the G2 stage percentage, from 19.3% (in ~ 5000 cells) in the empty vector group to 13.4% in the RIPK1 group (Fig. 3D, E). Similarly, we also observed G2 stage percentage decreased from 23.7% to 19.4% in RIPK1 overexpressed U2932 cells. In addition, the cell death ratio was increased by 1.1 and 0.8 folds after overexpression of RIPK1 in U2932 and TMD8 cells, respectively (Fig. 3F and Additional file 1: S3E). Altogether, the loss function of RIPK1 contributes to the cell growth of lymphocytes. Therefore, our findings collectively revealed that RIPK1 possesses inhibitory functions in the proliferative ability of B lymphocytes.

Ectopic RIPK1 in lymphocytes suppresses cell growth A Real-time PCR estimated the RIPK1 knockdown efficiency in TMD8 and U2932 cells. N = 6, ***P < 0.001 by student’s t-test. B CCK8 assay determined cell proliferation of TMD8 and U2932 cell lines. Cells in A were seeded onto 96-well plates, and 12 h later, cell growth status was detected every 12 h. Vector, empty vector; oeRIPK1, RIPK1 overexpression. N = 3, *P < 0.05, **P < 0.01, ***P < 0.001 by two-way ANOVA. C Cell viability of TMD8 and U2932 cell lines. Cells in A were seeded onto 96-well plates, and 12 h later, cell growth status was detected every 12 h. Vector, empty vector; oeRIPK1, RIPK1 overexpression. N = 3, **P < 0.01, ***P < 0.001 by two-way ANOVA. D–E. PI staining determined the cell cycle distribution of TMD8 and U2932 cell lines. ~ 5000 cells cultured for 36 h were collected for PI staining. oeRIPK1#1/2/3 indicates three duplications of RIPK1 overexpressed cell lines. Vector, empty vector; oeRIPK1, RIPK1 overexpression. N = 6, *P < 0.05, **P < 0.01, ***P < 0.001 by student’s t-test F. Annexin V-FITC/PI double-labeled FACS determined the cell death of U2932 cell lines. Cells in B were used for experiments.
3.4 RIPK1 inhibitor promotes the proliferation of lymphoma cells
A bulk of studies have indicated that RIPK1 has a dual biological role: 1) promoting cell apoptosis and necroptosis in a kinase activity-dependent manner, and 2) promoting cell survival via its scaffolding function in TNFR1 (tumor necrosis factor receptor 1) signaling [21, 22]. Necrostatin-1 (Nec), as the first specific inhibitor of RIPK1, has been extensively demonstrated to possess noticeable effects on inhibiting necroptosis in various diseases, including inflammatory cardiovascular, neurological diseases, etc. [23]. Therefore, we explored the effects of Nec on B-cell lymphoma. In the cell proliferation assay, we observed that Nec treatment (30 μM, based on the previous report [24]) on GM12878/L428 cells significantly promoted cell growth, which was partially blocked by downregulated RIPK1 expression using shRNA (Fig. 4A and Additional file 1: S2A). Meanwhile, cell viability also was increased by Nec treatment on GM12878/L428 cells, and these effects were largely attenuated in the RIPK1 knockdown group (Fig. 4B and Additional file 1: S2B). Furthermore, we observed that Nec treatment elevated G2 percentage both on GM12878 and L428 cells in a dose-dependent manner, and effects were partially abated in RIPK1 knockdown groups (Fig. 4C–D and Additional file 1: S2C). These results indicate that Nec promotes cell proliferation of B-cell lymphoma largely via targeting RIPK1. Besides, we treated GM12878 and L428 cells using another RIPK1 inhibitor, RIPK1-IN-7 (IN-7), which has a low enzymatic IC50 of 11 nM [25]. In our experiments, we found that treatment of IN-7 (5 nM) increased G2 percentage both in GM12878 and L428 cells, which were largely attenuated in RIPK1 knockdown cells (Additional file 1: Fig. S2D). Altogether, inhibition of RIPK1 kinase activity by small molecule inhibitors can efficiently promote cell proliferation.

RIPK1 inhibitor promotes the proliferation of lymphoma cells A CCK8 assay determined cell proliferation of GM12878 lines. Cells were seeded onto 96-well plates and pre-treated with Nec (30 μM) for 24 h, and then, cell growth status was detected by CCK8 assay every 12 h. N = 3, *P < 0.05, **P < 0.01, ***P < 0.001 by two-way ANOVA. B Cell viability of GM12878 cell lines. Cells in A were seeded onto 96-well plates and pre-treated with Nec (30 μM) for 24 h, and then, cell growth status was detected every 12 h. N = 3, *P < 0.05, **P < 0.01, ***P < 0.001 by two-way ANOVA. C-D. PI staining C determined the cell cycle distribution of GM12878 cell lines and the statistical results D. ~ 5000 cells were pre-treated with Nec (30 μM) for 24 h and then collected for PI staining. N = 6, *P < 0.05, ***P < 0.001 by student’s t-test
3.5 HOIPIN-1 inhibits the proliferation of lymphoma cells by enhancing RIPK1 function
Due to the down-regulation of RIPK1 expression of kinase activity promoting cell proliferation, we speculated that activation of RIPK1 by small molecules may lead to cell death of B-cell lymphoma. However, to date, there is still no effective and specific RIPK1 agonist available to activate this enzyme in experiments. Numerous previous studies showed that there is a balance between the kinase activity and scaffold function of RIPK1 [9, 26, 27]. Moreover, modulation of RIPK1 scaffold functions via ubiquitination-related enzymes (such as IAP, LUBAC, A20, and CYLD) can largely influence RIPK1-mediated signaling [28]. Therefore, we hypothesized that LUBAC inhibitors, such as HOIPIN-1 (with an IC50 of 2.8 μM [29]) may enhance RIPK1 kinase-mediated cell death by abating its ubiquitination. To this end, we treated TMD8 and U2932 cells with HOIPIN-1 (HOI) and checked the growth status of the cells. Indeed, we observed that HOI treatment (10 μM) on TMD8 and U2932 cells dramatically suppressed cell growth, and these effects were largely attenuated by the RIPK1-knockdown group (Fig. 5A and Additional file 1: S3A). Consistently, cell viability also decreased after HOI treatment on GM12878/L428 cells, which were abated by the knockdown of RIPK1 expression (Fig. 5B and Additional file 1: S3B). In the cell cycle analysis, we observed that HOI treatment decreased G2 percentage both in TMD8 and U2932 cells in a dose-dependent manner (Fig. 5C–D and Additional file 1: S3C–D). More importantly, these effects were effectively attenuated after the knockdown of RIPK1 using shRNA (Fig. 5C–D and Additional file 1: S3C–D). These results indicate that 1) HOI treatment inhibits cell proliferation of B-cell lymphoma largely via targeting RIPK1; 2) restoration of RIPK1 by small molecule inhibitors can efficiently promote cell death of B-cell lymphoma.

HOIPIN-1 inhibits the proliferation of lymphoma cells by blocking RIPK1 function A. CCK8 assay determined cell proliferation of TMD8 lines. Cells were seeded onto 96-well plates and pre-treated with HOIPIN-1 (HOI, 10 μM) for 24 h, and then, cell growth status was detected by CCK8 assay every 12 h. N = 3, *P < 0.05, **P < 0.01, ***P < 0.001 by two-way ANOVA. B Cell viability of TMD8 cell lines. Cells in A were seeded onto 96-well plates and pre-treated with HOI (10 μM) for 24 h, and then, cell viability status was detected every 12 h. N = 3, *P < 0.05, **P < 0.01, ***P < 0.001 by two-way ANOVA. C–D. PI staining (C) determined the cell cycle distribution of TMD8 cell lines and the statistical results (D). ~ 5000 cells were pre-treated with HOI at the indicated dose for 24 h and then collected for PI staining. N = 6, *P < 0.05, ***P < 0.001 one-way ANOVA
4 Discussion and conclusion
A bulk of reports have demonstrated that RIPK1 generally functions as a critical scaffold molecule of the TNFR1 complex, and achieves various inflammatory and pro-survival biological activities by modulating the activation of the MAPK and NF-κB signaling pathways [10, 22]. Meanwhile, as a kinase, RIPK1 physically interacts with RIPK3 to paradoxically promote apoptosis, necroptosis, etc. by catalyzing the phosphorylation of MLKL [30,31,32]. The dual role of RIPK1 in cell survival emphasizes that the unbalance of RIPK1 expression or activity may cause a serious outcome for cells. In our study, we observed that the mRNA level of RIPK1 was significantly down-regulated in the main categories of B-cell lymphoma, including CLL, DLBCL, and FL by re-analyzing the public dataset in the GEO database. Meanwhile, we also validated these results using biopsies we collected by real-time quantitative PCR. These results suggest that characterizing the roles of RIPK1 in B-cell lymphoma may identify a common target for potential therapy of these diseases.
Firstly, we estimated the expression of RIPK1 in 8 lymphocyte lines, including a normal human B lymphocyte cell line (GM12878), and 6 B-cell non-Hodgkin lymphomas (OCI-LY3, JEKO-1, RAJI, TMD8, U2932, WSU-FSCCL) and a Hodgkin's lymphoma cell line (L428). Indeed, we observed that RIPK1 mRNA levels were decreased in 6 B-cell lymphoma cell lines when compared with normal B-cell GM12878. However, RIPK1 in L428 is slightly (but not significantly) lower than that of GM12878 cells. These findings suggest that the down-regulation of RIPK1 may be common in B-cell but not all subtypes of lymphoma cells. However, this conclusion needs more evidence to support. In 2021, Yin et al. reported that RIPK1 depletion occurs in triple-negative breast carcinoma, and the loss function of RIPK1 significantly aggravates the AQP1-driven TNBC progression and metastasis [33]. More recently, Yu et al. demonstrated that SMYD2 contributes to colorectal cancer by targeting RIPK1 and inhibiting the phosphorylation of RIPK1 [34]. Combined with our observations, it is obvious that the aberration of RIPK1 kinase activity contributes to the pathogenesis of multiple cancers, both solid and blood cancers.
To further investigate the roles of RIPK1 in B-cell lymphoma, we knock down the expression of RIPK1 in GM12878 and L428 cells. Interestingly, our results indicate that the knockdown of RIPK1 significantly promotes cell growth and viability, and inhibits cell death both in GM12878 and L428 cells. Meanwhile, we also observed that the G2 percentage is elevated after RIPK1 down-regulation. In contrast, overexpression of RIPK1 in TMD8 and U2932 cells inhibits cell growth and viability and decreases G2 percentage, and promotes cell death. These results support the previously established kinase-dependent pro-apoptosis and -necroptosis functions of RIPK1. Furthermore, we employed two different RIPK1 inhibitors, Necrostatin-1 and RIPK1-IN-7, to confirm that the kinase activity of RIPK1 plays a critical role in the cell growth of normal B lymphocytes and other types of lymphoma cell lines. Our data reveal that inhibition of RIPK1 kinase activity contributes to cell growth of normal B-cells, which is in favor of the RIPK1 knockdown results mentioned above. Noticeably, necrostatin-1, the inhibitor of necroptosis, has been explored to be developed into a small molecule drug for inflammation-, apoptosis- or necroptosis-related diseases, such as cardiovascular diseases, neonatal hypoxia–ischemia, Alzheimer’s disease, Parkinson’s disease, coronavirus disease 2019 (COVID-19), etc. [23, 35,36,37]. According to our evidence, necrostatin-1 (or RIPK1 inhibitors) may increase the risk of B-cell lymphoma occurrence.
On the other hand, we speculated that the RIPK1 agonist may exhibit an effect against the cell growth of B-cell lymphoma. However, no effective and specific RIPK1 agonist is available to date. It has been well established that the ubiquitination of RIPK1 is mainly mediated by E3 ubiquitin ligases LUBAC, by which the kinase activity of RIPK1 is indirectly arrested [38]. To this end, we used HOI to treat TMD8 and U2932 cells. Interestingly, we observed that HOI treatment dramatically suppressed cell growth of TMD8 and U2932, and increased the G1 arrest of these two cell lines. These findings reveal that restoration of RIPK1 by small molecule inhibitors can efficiently promote cell death of B-cell lymphoma. Notably, HOI is well-known as an inhibitor of the canonical NF-kB by suppressing LUBAC ubiquitin ligase activity [39]. In our recent results (data not shown), we also observed that the p65 phosphorylation (the activated NF-kB subunit) level was significantly decreased by HOI treatment in both TMD8 and U2932 cells. Meanwhile, we found overexpression of RIPK1 inhibited NF-kB activation, which is similar to HOI treatment. These results suggest that HOI is versatile in suppressing cell apoptosis, via both NF-kB and RIPK1-dependent mechanisms. Besides, considering the dramatic inhibitory effect of RIPK1 specific inhibitor on cell death, we hold that RIPK1 promotes cell death by both kinase- and scaffold-mediated function.
In summary, our study reveals that RIPK1 is down-regulated in B-cell lymphoma, including CLL, DLBCL, and FL. Meanwhile, RIPK1 exhibits anti-tumor activity in a kinase-dependent manner in the context of B-cell lymphoma. More importantly, modifying RIPK1 kinase activity by a small molecule (such as necrostain-1, HOIPIN-1, etc.) alters the cell growth status of B-cell lymphoma. Taken together, we consider that RIPK1 may be a potential target in the clinical application of B-cell lymphoma treatment.
References
Armitage JO, et al. Non-HODGKIN LYMPHOMA. The Lancet. 2017;390(10091):298–310.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30.
Hanel W, Epperla N. Evolving therapeutic landscape in follicular lymphoma: a look at emerging and investigational therapies. J Hematol Oncol. 2021;14(1):104.
Wong DP, et al. A BAFF ligand-based CAR-T cell targeting three receptors and multiple B cell cancers. Nat Commun. 2022;13(1):217.
Ashida H, Sasakawa C, Suzuki T. A unique bacterial tactic to circumvent the cell death crosstalk induced by blockade of caspase-8. EMBO J. 2020;39(17): e104469.
Tao P, et al. A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature. 2020;577(7788):109–14.
Yamamoto S, Iwakuma T. RIPK1-TRAF2 interplay on the TNF/NF-κB signaling, cell death, and cancer development in the liver. Transl Cancer Res. 2017;6(Suppl 3):94–109.
Vanlangenakker N, et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 2011;18(4):656–65.
Newton K. Multitasking kinase RIPK1 regulates cell death and inflammation. Cold Spring Harb Perspect Biol. 2020. https://doi.org/10.1101/cshperspect.a036368.
Takahashi N, et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature. 2014;513(7516):95–9.
Shutinoski B, et al. K45A mutation of RIPK1 results in poor necroptosis and cytokine signaling in macrophages, which impacts inflammatory responses in vivo. Cell Death Differ. 2016;23(10):1628–37.
Vredevoogd DW, et al. Augmenting immunotherapy impact by lowering tumor TNF cytotoxicity threshold. Cell. 2019;178(3):585–99.
Sriram G, et al. The injury response to DNA damage in live tumor cells promotes antitumor immunity. Sci Signal. 2021;14(705):eabc4764.
Cucolo L, et al. The interferon-stimulated gene RIPK1 regulates cancer cell intrinsic and extrinsic resistance to immune checkpoint blockade. Immunity. 2022;55(4):671-685.e10.
Smith HG, et al. RIPK1-mediated immunogenic cell death promotes anti-tumour immunity against soft-tissue sarcoma. EMBO Mol Med. 2020;12(6): e10979.
Luan Q, et al. RIPK1 regulates survival of human melanoma cells upon endoplasmic reticulum stress through autophagy. Autophagy. 2015;11(7):975–94.
Tuoheti Z, et al. RIPK1 polymorphisms alter the susceptibility to cervical Cancer among the Uyghur population in China. BMC Cancer. 2020;20(1):299.
Akimoto M, et al. Antidiabetic adiponectin receptor agonist AdipoRon suppresses tumour growth of pancreatic cancer by inducing RIPK1/ERK-dependent necroptosis. Cell Death Dis. 2018;9(8):804.
Di Grazia A, et al. The fragile X mental retardation protein regulates RIPK1 and colorectal cancer resistance to necroptosis. Cell Mol Gastroenterol Hepatol. 2021;11(2):639–58.
Nicolè L, et al. Necroptosis-driving genes RIPK1, RIPK3 and MLKL-p are associated with intratumoral CD3(+) and CD8(+) T cell density and predict prognosis in hepatocellular carcinoma. J Immunother Cancer. 2022. https://doi.org/10.1136/jitc-2021-004031.
Kondylis V, Pasparakis M. RIP Kinases in liver cell death, inflammation and cancer. Trends Mol Med. 2019;25(1):47–63.
Ting AT, Bertrand MJM. More to Life than NF-kappaB in TNFR1 Signaling. Trends Immunol. 2016;37(8):535–45.
Cao L, Mu W. Necrostatin-1 and necroptosis inhibition: pathophysiology and therapeutic implications. Pharmacol Res. 2021;163: 105297.
Feyen D, et al. Increasing short-term cardiomyocyte progenitor cell (CMPC) survival by necrostatin-1 did not further preserve cardiac function. Cardiovasc Res. 2013;99(1):83–91.
Liang S, Nian Z, Shi K. Inhibition of RIPK1/RIPK3 ameliorates osteoclastogenesis through regulating NLRP3-dependent NF-kappaB and MAPKs signaling pathways. Biochem Biophys Res Commun. 2020;526(4):1028–35.
Annibaldi A, et al. Ubiquitin-mediated regulation of RIPK1 kinase activity independent of IKK and MK2. Mol Cell. 2018;69(4):566-580.e5.
Darding M, Meier P. IAPs: guardians of RIPK1. Cell Death Differ. 2012;19(1):58–66.
Varfolomeev E, Vucic D. RIP1 post-translational modifications. Biochem J. 2022;479(9):929–51.
Katsuya K, et al. High-throughput screening for linear ubiquitin chain assembly complex (LUBAC) selective inhibitors using homogenous time-resolved fluorescence (HTRF)-based assay system. SLAS Discov. 2018;23(10):1018–29.
Tanzer MC, et al. Combination of IAP antagonist and IFNγ activates novel caspase-10- and RIPK1-dependent cell death pathways. Cell Death Differ. 2017;24(3):481–91.
Zhang X, Dowling JP, Zhang J. RIPK1 can mediate apoptosis in addition to necroptosis during embryonic development. Cell Death Dis. 2019;10(3):245.
Zhang T, et al. Necroptosis pathways in tumorigenesis. Semin Cancer Biol. 2022. https://doi.org/10.1016/j.semcancer.2022.07.007.
Yin Z, et al. RIPK1 is a negative mediator in Aquaporin 1-driven triple-negative breast carcinoma progression and metastasis. NPJ Breast Cancer. 2021;7(1):53.
Yu YQ, et al. SMYD2 targets RIPK1 and restricts TNF-induced apoptosis and necroptosis to support colon tumor growth. Cell Death Dis. 2022;13(1):52.
Fang T, et al. Alterations in necroptosis during ALDH2mediated protection against high glucoseinduced H9c2 cardiac cell injury. Mol Med Rep. 2018;18(3):2807–15.
Liang W, et al. A novel damage mechanism: contribution of the interaction between necroptosis and ROS to high glucose-induced injury and inflammation in H9c2 cardiac cells. Int J Mol Med. 2017;40(1):201–8.
Yang SH, et al. Nec-1 alleviates cognitive impairment with reduction of Abeta and tau abnormalities in APP/PS1 mice. EMBO Mol Med. 2017;9(1):61–77.
Delanghe T, Dondelinger Y, Bertrand MJM. RIPK1 Kinase-dependent death: a symphony of phosphorylation events. Trends Cell Biol. 2020;30(3):189–200.
Katsuya K, et al. Small-molecule inhibitors of linear ubiquitin chain assembly complex (LUBAC), HOIPINs, suppress NF-kappaB signaling. Biochem Biophys Res Commun. 2019;509(3):700–6.
Acknowledgements
We thank the individuals and their families who participated in this project. This study was funded by Grants from the Applied Basic Research Program of Xuzhou (Nos. KC19031). The funders had no role in study design, data collection and analysis, manuscript preparation, or decision to publish.
Funding
This work was supported by Grants from the Applied Basic Research Program of Xuzhou (Nos. KC19031).
Author information
Authors and Affiliations
Contributions
BW conceived of this study and obtained financial support. BW wrote and revised the manuscript, JL, HW, JL, JL, FS, and DF performed research and analyzed data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Fig S1. RIPK1 is down-regulated in tumor cells of lymphoma patients. A Heatmap of 3 groups of DEGs (CLL, DLBCL, and FL vs. normal, respectively). Chronic lymphocytic leukemia (CCL, N=5), follicular lymphoma (FL, N=10), and diffuse large B-cell patients (DLBCL, N=8) vs. normal donors (N=6). The cutoff was set at p-adjust < 0.001 and log2 fold change > 3. B Volcano plot of DEGs of FL vs. normal. Follicular lymphoma (FL, N=10) vs. normal donors (N=6). A cutoff of p-adjust < 0.001 and log2 fold change > 3. Fig S2. RIPK1 inhibitor promotes the proliferation of lymphoma cells A CCK8 assay determined cell proliferation of L428 lines. Cells were seeded onto 96-well plates and pre-treated with Nec (30 μM) for 24 h, and then, cell growth status was detected by CCK8 assay every 12 h. N=3, *P<0.05, **P<0.01, ***P<0.001 by two-way ANOVA. B Cell viability of L428 cell lines. Cells in A were seeded onto 96-well plates and pre-treated with Nec (30 μM) for 24 h, and then, cell growth status was detected every 12 h. N=3, *P<0.05, ***P<0.001 by two-way ANOVA. C PI staining determined the cell cycle distribution of L428 cell lines and the statistical results. ~5000 cells were pre-treated with Nec (30 μM) for 24 h and then collected for PI staining. D PI staining determined the cell cycle distribution of GM12878/L428 cell lines and the statistical results. ~5000 cells were pre-treated with RIPK1-IN-7 (IN-7, 5 nM) for 24 h and then collected for PI staining. N=6, ***P<0.001 by student’s t-test. E Annexin V-FITC/PI double-labeled FACS determined the cell death of L428 cell lines. Cells in A were used for experiments. Fig S3. HOIPIN-1 inhibits the proliferation of lymphoma cells by blocking RIPK1 function. A CCK8 assay determined cell proliferation of U2932 lines. Cells were seeded onto 96-well plates and pre-treated with HOIPIN-1 (HOI, 10 μM) for 24 h, and then, cell growth status was detected by CCK8 assay every 12 h. N=3, *P<0.05, **P<0.01, ***P<0.001 by two-way ANOVA. B Cell viability of U2932 cell lines. Cells in A were seeded onto 96-well plates and pre-treated with HOI (10 μM) for 24 h, and then, cell growth status was detected every 12 h. N=3, *P<0.05, ***P<0.001 by two-way ANOVA. C–D. PI staining determined the cell cycle distribution of U2932 cell lines and the statistical results D. ~5000 cells were pre-treated with HOI at the indicated dose for 24 h and then collected for PI staining. N=6, *P<0.05, ***P<0.001 by one-way ANOVA.E. Annexin V-FITC/PI double-labeled FACS determined the cell death of TMD8 cell lines. Cells in Fig. 3B were used for experiments.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, B., Li, J., Wang, H. et al. RIPK1 is aberrantly expressed in multiple B-cell cancers and implicated in the underlying pathogenesis. Discov Onc 14, 131 (2023). https://doi.org/10.1007/s12672-023-00725-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-023-00725-z