
Abstract
There is preclinical and recent epidemiological evidence that thyroid hormone supports breast cancer. These observations raise the issue of whether management of breast cancer in certain settings should include consideration of reducing the possible contribution of thyroid hormone to the advancement of the disease. In a preliminary experience, elimination of the clinical action of endogenous L-thyroxine (T4) in patients with advanced solid tumors, including breast cancer, has favorably affected the course of the cancer, particularly when coupled with administration of exogenous 3,5,3′-triiodo-L-thyronine (T3) (euthyroid hypothyroxinemia). We discuss in the current brief review the possible clinical settings in which to consider whether endogenous thyroid hormone—or exogenous thyroid hormone in the patient with hypothyroidism and coincident breast cancer—is significantly contributing to breast cancer outcome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
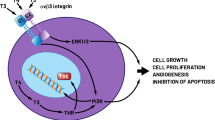
Recent epidemiologic studies have described associations between thyroid function and breast cancer [1,2,3] that indicate the hormone can support this form of cancer. Clinical evidence is also available that hypothyroidism beneficially affects the course of breast cancer [4, 5]. We have shown that the hormone, specifically L-thyroxine, T4, is a proliferative factor in vitro for breast cancer cells [6] and that, in the absence of estrogen, thyroid hormone can promote nuclear estrogen receptor-α (ERα)-dependent proliferation of breast cancer cells bearing this receptor [6]. This effect involves mitogen-activated protein kinase (MAPK)-dependent protein kinase and activation of ERα by specific serine phosphorylation. The action of thyroid hormone in this setting we now know is mediated by a thyroid hormone receptor on the extracellular domain of integrin αvβ3 expressed by tumor cells [7, 8]. Thus, an action of thyroid hormone initiated nongenomically culminates in activity in the genomic pathway of ER action, an endocrine overlap that we have reviewed elsewhere [9]. The ER degrading agent, fulvestrant (ICI 182,780), blocks the proliferative action of thyroid hormone on ERα-positive human breast cancer cells [6]. It should be noted that L-thyroxine (T4) at physiological free hormone concentrations is maximally active at this receptor site on αvβ3 [10, 11], whereas substantially higher than physiological levels of free T3 are required to achieve substantive proliferative activity via this receptor.
We have also shown that the thyroid hormone receptor on αvβ3 enables triple-negative breast cancer cells to respond differentially to T4 in physiological concentrations [12] and to a T4 derivative and antagonist, tetraiodothyroacetic acid (tetrac), with changes in transcription of a panel of cell survival genes, including those for anti-apoptosis functions, for support of angiogenesis, and for chemoresistance [8, 12,13,14]. Breast cancer cells lacking the estrogen receptor proliferate in response to thyroid hormone via αvβ3 [13]. Thus, thyroid hormone is capable of supporting breast cancer cells by two mechanisms, depending upon the absence or presence of ER. We have reported clinically that a variety of far-advanced solid tumors, including breast cancer, stabilize or are reduced in size when patient T4 is eliminated pharmacologically and replaced with 3,5,3′-triiodo-L-thyronine (T3) (euthyroid hypothyroxinemia) [15].
In the current brief review, we discuss the possibility that endogenous physiologic range T4 in breast cancer patients or replacement T4 in hypothyroid patients who have coincident breast cancer, may increase the risk of continuing/recurrent tumor. As noted above, there are ER-dependent and ER-independent mechanisms that are relevant to both premenopausal and menopausal women with breast carcinoma. Acting via the αvβ3 integrin and independently of ERα, T4 serves to support tumor cell proliferation and the cell survival functions listed above [8, 12,13,14]. Then, regardless of the menopausal status of the ER-positive patient, thyroid hormone may stimulate ER-dependent breast cancer cell proliferation. Induction of euthyroid hypothyroxinemia has conceptual attractiveness in breast cancer, regardless of ER status of the tumor.
Possible Actions of Circulating Endogenous T4 Within the Physiologic Range on Recurrent ERα-Positive Breast Cancer in the Pre- and Postmenopausal Patient
The therapy of recurrent ERα-positive breast cancer in premenopausal and postmenopausal women may include surgery of the primary or metastatic lesions, radiation, chemotherapy, and ER-targeted therapy, despite the minimized production of estrogen in the postmenopausal patient. Fulvestrant and tamoxifen are ER-targeted therapeutic agents in the setting of breast cancer. Direct comparisons of the effectiveness of these agents in breast cancer management are few in number and do not reveal clear-cut therapeutic advantages to monotherapy with either agent [16, 17]. Fulvestrant leads to degradation of ER, whereas tamoxifen stabilizes the receptor [18] and we propose that this distinction has implications for the action of T4 on ER-positive tumors. Eliminating ER with fulvestrant in ERα-positive tumors means that endogenous thyroid hormone cannot substitute for estradiol as a tumor-support agent. In contrast, T4 does not compete with steroids for the binding site on ER, but instead T4 activates MAPK to phosphorylate ER and thus we anticipated that tamoxifen would not oppose the proliferative action of T4 in ER-positive breast cancer. Preliminary studies, however, revealed that tamoxifen is capable of inhibiting the phosphorylation/activation of MAPK by T4 in breast cancer cells [19]. This novel action of tamoxifen on ER activation by T4 means that tamoxifen and fulvestrant are both capable of opposing the support of T4 for ER-positive cancer cells.
Against this background, we can ask whether tamoxifen in the pre- and postmenopausal euthyroid patient with ER-positive breast cancer has satisfactorily eliminated the possible contribution of thyroid hormone to tumor support. This issue has not been explored clinically or in animal models and does, in our opinion, require initial examination in the nude mouse bearing ER-positive human breast cancer xenografts that are exposed to several concentrations of tamoxifen. As noted above, we have shown fulvestrant to be effective anti-thyroid hormone therapy in MCF-7 breast cancer cells [6], but this requires confirmation in other ER-positive cell lines and primary cultures of breast cancer cells. Until more information on this issue is available, we propose in the patient whose ER-positive cancer remains active on regimens that include anti-ER treatment, that elimination of host T4 (euthyroid hypothyroxinemia) be considered as an option [15].
Long-term tamoxifen therapy (6 months) may alter circulating levels of thyroid hormone and TSH [20], increasing TSH and total T4 and T3, but not free hormone levels. In the absence of changes in free T4, it is unlikely that this tamoxifen effect is relevant to thyroid hormone actions on breast cancer.
Possible Actions of Endogenous T4 Within the Physiologic Range on ERα-Negative Breast Cancer
We have shown that proliferation of ERα-negative human breast cancer cells may be induced by T4 via an integrin αvβ3-dependent mechanism [8, 12]. Tetrac, a specific pharmacologic inhibitor of T4 at integrin αvβ3, has expanded our understanding of the function of this receptor in cancer cells, notably, ER-negative breast cancer cells [13].
Are the distinctive actions of T4 on support of ER-positive and ER-negative breast cancer mutually exclusive? That is, if the pro-tumor contribution of T4 can be satisfactorily eliminated by fulvestrant in the ER α-positive breast cancer patient, will the proliferative effect of T4 become apparent via the alternative αvβ3 pathway? In acute in vitro studies of fulvestrant, no such activity was apparent [19], but longer term studies are required.
Consideration of Induction of Euthyroid Hypothyroxinemia in the Setting of Breast Cancer
As noted above, there is clinical evidence that hypothyroidism may beneficially influence the course of breast cancer [1, 4, 5]. Thus, in the setting of recurrent breast cancer post-standard therapeutic interventions, we suggest that induction of euthyroid hypothyroxinemia is an alternative.
However, there is no systematic prospective study available to support this intervention, nor is there clinical information to support elimination of host T4 at the point of initial tumor diagnosis and in conjunction with standard initial therapy. In the setting of induced euthyroid hypothyroxinemia in cancer patients, there is no clinical information available about the pharmacokinetics and pharmacodynamics of standard chemotherapeutic treatments of breast cancer that might be altered with the administration of replacement T3 two or three times daily.
There is another possible advantage of euthyroid hypothyroxinemia, namely, that elimination of the action of T4 at integrin αvβ3 may serve to radiosensitize cancer cells [21, 22]. We have shown that the T4 analogue and antagonist, tetrac, impairs tumor cell repair of radiation-induced double-stranded DNA breaks [22] in glioblastoma and prostate cancer cell lines, but this property has not yet been studied preclinically in breast cancer cells.
Effect of Endogenous or Replacement T4 Within the Normal Range on Risk of Development of Breast Cancer; T3 and Breast Cancer
Certain epidemiologic studies have documented that circulating levels of T4 in the upper quartiles of the normal range are a risk factor for development of certain cancers, including breast [1, 2]. If this is indeed the case, then as an outgrowth of the discussions above, should primary care physicians monitor serum thyroid hormone levels in all women patients as a breast cancer risk factor and concentrate breast cancer screening efforts in patients with thyroid hormone levels in the upper one-half of the normal range? Such a risk analysis has not been conducted.
This possible risk issue applies additionally to patients with primary hypothyroidism who are on T4 as thyroid hormone replacement. The 2014 guidelines of the American Thyroid Associations for the treatment of hypothyroidism do not consider management of patients with hypothyroidism and nonthyroidal cancer [23]. Based on epidemiologic data currently available and cited above, thyroid hormone replacement treatment goals might include circulating thyroid hormone levels that are in the lower quartiles of the normal range. This T4-based treatment strategy may not be compatible with relief of symptoms of hypothyroidism in certain patients, and the strategy has not been tested in the hypothyroid-breast cancer patient population.
An alternative approach to be tested is T3 replacement. As noted above, the thyroid hormone receptor on integrin αvβ3 that is generously expressed by cancer cells and not by infrequently dividing nonmalignant cells is primarily a binding site for T4 [7, 8]. At this site, T4 supports breast cancer [6, 24]. Limited clinical evidence indicates that replacing systemic T4 with T3 retards breast cancer growth and growth of other solid tumors [15]. A number of preclinical studies also suggest that T3 retards breast cancer cell proliferation [25,26,27], but supraphysiologic concentrations of T3 were often involved in such studies. Additional clinical studies are needed of the possible therapeutic contribution of T3 to management of breast cancer and other solid tumors.
Is the Level of Circulating Endogenous T4 a Risk Factor for Development of Breast Cancer in Euthyroid Patients with Positive Family Histories of This Form of Cancer?
This possibility has not been examined epidemiologically. The epidemiologic data cited above that attribute cancer risk to upper-quartile-of-the-normal-range thyroid hormone levels were not designed to examine familial disease.
In Patients with Breast Cancer and Nonthyroidal Illness Syndrome, Is Elevated Circulating Free T4 and/or Total T4 Maintained Within the Normal Range a Factor Contributing to Breast Cancer Outcome?
Elevated free T4 levels in nonthyroidal illness syndrome (NTIS) are usually transient and related to acute or subacute inflammatory illness, rather than to chronic illness and cancer [28]. Thus, encounters with elevated free T4 levels in patients with cancer of any type are infrequent. However, endogenous total T4 levels may be maintained within the normal range by cancer patients who express NTIS via the hallmark of decreased circulating T3 concentration. In this setting, we raise for discussion the possibility that the persistence of aggressive cancer behavior in NTIS patients may be in part attributable to host T4 levels [19].
Summary
T4 functions as a prohormone for T3 when genomic actions of thyroid hormone that depend upon the thyroid hormone nuclear receptor are under consideration. By discrete molecular mechanisms that originate at the thyroid hormone receptor on integrin αvβ3 and are ERα-dependent or independent of ER, thyroid hormone as T4 can support breast cancer. In this setting, T4 is a hormone, not a prohormone. The affinity of the hormone receptor on the integrin is higher for T4 than for T3, and it is physiological concentrations of free T4 that we have shown to stimulate human breast cancer cell line proliferation. As noted in this review, limited clinical and preclinical evidence indicates that T3 may arrest breast cancer growth. Preclinical data may reflect supraphysiological levels of T3, however, and such levels in certain in vitro systems have been reported to stimulate breast cancer cell proliferation [25].
The current review emphasizes that we do not yet know enough about the significance of contributions of T4 to breast cancer behavior. We do need clinical studies to assess prospectively the possible contribution of endogenous T4 or replacement T4 to aggressive behavior of ERα-positive and ERα-negative breast cancer in populations both of pre- and postmenopausal patients. What is to be sought are direct correlations between levels of the hormone within the normal range and cancer behavior and, where relevant, in the absence and presence of anti-ER therapy with fulvestrant or tamoxifen. If such documentation is obtained, then the utility of pharmacological manipulation of physiologic range T4 in clinically active breast cancer becomes apparent. We do not know enough about the actions of T3 on breast cancer behavior, although euthyroid hypothyroxinemia—a therapeutic setting in which T3 replaces host circulating T4—has been shown in limited clinical examination [14] to achieve arrest of growth in the case of breast cancer and other solid tumors.
The evidence we have reviewed here does not suggest that thyroid hormone causes breast cancer.
References
Angelousi A, Diamanti-Kandarakis E, Zapanti E, Nonni A, Ktenas E, Mantzou A, Kontzoglou K, Kouraklis G (2017) Is there an association between thyroid function abnormalities and breast cancer? Arch Endocrinol Metab 61(1):54–61. https://doi.org/10.1590/2359-3997000000191
Khan SR, Chaker L, Ruiter R, Aerts JGJV, Hofman A, Dehghan A, Franco OH, Stricker BHC, Peeters RP (2016) Thyroid function and cancer risk: the Rotterdam study. J Clin Endocr Metab 101(12):5030–5036. https://doi.org/10.1210/jc.2016-2104
Journy NMY, Bernier MO, Doody MM, Alexander BH, Linet MS, Kitahara CM (2017) Hyperthyroidism, hypothyroidism, and cause-specific mortality in a large cohort of women. Thyroid 27(8):1001–1010. https://doi.org/10.1089/thy.2017.0063
Cristofanilli M, Yamamura Y, Kau SW, Bevers T, Strom S, Patangan M, Hsu L, Krishnamurthy S, Theriault RL, Hortobagyi GN (2005) Thyroid hormone and breast carcinoma. Primary hypothyroidism is associated with a reduced incidence of primary breast carcinoma. Cancer 103(6):1122–1128. https://doi.org/10.1002/cncr.20881
Sogaard M, Farkas DK, Ehrenstein V, Jorgensen JO, Dekkers OM, Sorensen HT (2016) Hypothyroidism and hyperthyroidism and breast cancer risk: a nationwide cohort study. Eur J Endocrinol 174(4):409–414. https://doi.org/10.1530/EJE-15-0989
Tang HY, Lin HY, Zhang SL, Davis FB, Davis PJ (2004) Thyroid hormone causes mitogen-activated protein kinase-dependent phosphorylation of the nuclear estrogen receptor. Endocrinology 145(7):3265–3272. https://doi.org/10.1210/en.2004-0308
Cheng SY, Leonard JL, Davis PJ (2010) Molecular aspects of thyroid hormone actions. Endocr Rev 31(2):139–170. https://doi.org/10.1210/er.2009-0007
Davis PJ, Goglia F, Leonard JL (2016) Nongenomic actions of thyroid hormone. Nat Rev Endocrinol 12(2):111–121. https://doi.org/10.1038/nrendo.2015.205
Hammes SR, Davis PJ (2015) Overlapping nongenomic and genomic actions of thyroid hormone and steroids. Best Pract Res Clin Endocrinol Metab 29(4):581–593. https://doi.org/10.1016/j.beem.2015.04.001
Bergh JJ, Lin HY, Lansing L, Mohamed SN, Davis FB, Mousa S, Davis PJ (2005) Integrin αVβ3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology 146(7):2864–2871. https://doi.org/10.1210/en.2005-0102
Lin HY, Sun M, Tang HY, Lin C, Luidens MK, Mousa SA, Incerpi S, Drusano GL, Davis FB, Davis PJ (2009) L-thyroxine vs. 3,5,3′-triiodo-L-thyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Am J Physiol Cell Physiol 296(5):C980–C991. https://doi.org/10.1152/ajpcell.00305.2008
Davis PJ, Glinsky GV, Lin H-Y, Leith JT, Hercbergs A, Tang H-Y, Ashur-Fabian O, Incerpi S, Mousa SA (2014) Cancer cell gene expression modulated from plasma membrane integrin αvβ3 by thyroid hormone and nanoparticulate tetrac. Front Endocrinol 5:240. https://doi.org/10.3389/fendo.2014.00240
Glinskii AB, Glinsky GV, Lin HY, Tang HY, Sun M, Davis FB, Luidens MK, Mousa SA, Hercbergs AH, Davis PJ (2009) Modification of survival pathway gene expression in human breast cancer cells by tetraiodothyroacetic acid (tetrac). Cell Cycle 8(21):3562–3570. https://doi.org/10.4161/cc.8.21.9963
Davis PJ, Incerpi S, Lin HY, Tang HY, Sudha T, Mousa SA (2015) Thyroid hormone and P-glycoprotein in tumor cells. Biomed Res Int 2015:168427. https://doi.org/10.1155/2015/168427
Hercbergs A, Johnson RE, Ashur-Fabian O, Garfield DH, Davis PJ (2015) Medically induced euthyroid hypothyroxinemia may extend survival in compassionate need cancer patients: an observational study. Oncologist 20(1):72–76. https://doi.org/10.1634/theoncologist.2014-0308
Zhang J, Huang Y, Wang C, He Y, Zheng S, Wu K (2017) Efficacy and safety of endocrine monotherapy as first-line treatment for hormone-sensitive advanced breast cancer: a network meta-analysis. Medicine (Baltimore) 96(33):e7846. https://doi.org/10.1097/MD.0000000000007846
Howell A, Robertson JF, Abram P, Lichinitser MR, Elledge R, Bajetta E, Watanabe T, Morris C, Webster A, Dimery I, Osborne CK (2004) Comparison of fulvestrant versus tamoxifen for the treatment of advanced breast cancer in postmenopausal women previously untreated with endocrine therapy: a multinational, double-blind, randomized trial. J Clin Oncol 22(9):1605–1613. https://doi.org/10.1200/JCO.2004.02.112
Wijayaratne AL, McDonnell DP (2001) The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J Biol Chem 276(38):35684–35692. https://doi.org/10.1074/jbc.M101097200
Meng R, Tang HY, Westfall J, London D, Cao JH, Mousa SA, Luidens M, Hercbergs A, Davis FB, Davis PJ, Lin HY (2011) Crosstalk between integrin αvβ3 and estrogen receptor-α is involved in thyroid hormone-induced proliferation in human lung carcinoma cells. PLoS One 6(11):e27547. https://doi.org/10.1371/journal.pone.0027547
Kostoglou-Athanassiou I, Ntalles K, Markopoulos C, Athanassiou P, Gogas J, Proukakis C (1998) Thyroid function in postmenopausal women with breast cancer on tamoxifen. Eur J Gynaecol Oncol 19(2):150–154
Hercbergs A, Davis PJ, Davis FB, Ciesielski MJ, Leith JT (2009) Radiosensitization of GL261 glioma cells by tetraiodothyroacetic acid (tetrac). Cell Cycle 8(16):2586–2591. https://doi.org/10.4161/cc.8.16.9328
Hercbergs AH, Lin HY, Davis FB, Davis PJ, Leith JT (2011) Radiosensitization and production of DNA double-strand breaks in U87MG brain tumor cells induced by tetraiodothyroacetic acid (tetrac). Cell Cycle 10(2):352–357. https://doi.org/10.4161/cc.10.2.14641
Jonklaas J, Bianco AC, Bauer AJ, Burman KD, Cappola AR, Celi FS, Cooper DS, Kim BW, Peeters RP, Rosenthal MS, Sawka AM, American Thyroid Association Task Force on Thyroid Hormone R (2014) Guidelines for the treatment of hypothyroidism: prepared by the American Thyroid Association task force on thyroid hormone replacement. Thyroid 24(12):1670–1751. https://doi.org/10.1089/thy.2014.0028
Lin HY, Chin YT, Nana AW, Shih YJ, Lai HY, Tang HY, Leinung M, Mousa SA, Davis PJ (2016) Actions of L-thyroxine and nano-diamino-tetrac (nanotetrac) on PD-L1 in cancer cells. Steroids 114:59–67. https://doi.org/10.1016/j.steroids.2016.05.006
Martinez MB, Ruan M, Fitzpatrick LA (2000) Altered response to thyroid hormones by breast and ovarian cancer cells. Anticancer Res 20(6B):4141–4146
Cestari SH, Figueiredo NB, Conde SJ, Clara S, Katayama ML, Padovani CR, Brentani MM, Nogueira CR (2009) Influence of estradiol and triiodothyronine on breast cancer cell lines proliferation and expression of estrogen and thyroid hormone receptors. Arq Bras Endocrinol Metabol 53(7):859–864. https://doi.org/10.1590/S0004-27302009000700010
Sar P, Peter R, Rath B, Das Mohapatra A, Mishra SK (2011) 3, 3 ′ 5 triiodo L thyronine induces apoptosis in human breast cancer MCF-7cells, repressing SMP30 expression through negative thyroid response elements. Plos One 6(6):e20861. https://doi.org/10.1371/journal.pone.0020861
Farwell AP (2013) Nonthyroidal illness syndrome. Curr Opin Endocrinol Diabetes Obes 20(5):478–484. https://doi.org/10.1097/01.med.0000433069.09294.e8
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Co-authors Davis and Mousa are stockholders in NanoPharmaceuticals LLC that is commercially developing tetrac analogues, and Davis is an officer of the company. All remaining authors have no conflicts to declare.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hercbergs, A., Mousa, S.A., Leinung, M. et al. Thyroid Hormone in the Clinic and Breast Cancer. HORM CANC 9, 139–143 (2018). https://doi.org/10.1007/s12672-018-0326-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-018-0326-9