
Abstract
Estrogen metabolites may have different genotoxic and mitogenic properties yet their relationship with endometrial and ovarian cancer risk remains unclear. Within the Breast and Bone Follow-up to the Fracture Intervention Trial (B∼FIT, n = 15,595), we conducted a case-cohort study to evaluate 15 pre-diagnostic serum estrogens and estrogen metabolites with risk of incident endometrial and ovarian cancer among postmenopausal women not on hormone therapy. Participants included 66 endometrial and 67 ovarian cancer cases diagnosed during follow-up (∼10 years) and subcohorts of 346 and 416 women, respectively, after relevant exclusions. Serum concentrations were measured by liquid chromatography-tandem mass spectrometry. Hazard ratios (HRs) and 95 % confidence intervals (CIs) were estimated using Cox proportional hazard regression. Exposures were categorized in tertiles (T) and analyzed individually, as metabolic pathways (C-2, -4, or -16) and as ratios to parent estrogens (estradiol, estrone). Estradiol was significantly associated with increased endometrial cancer risk (BMI-adjusted HRT3vsT1 = 4.09, 95 % CI 1.70, 9.85; p trend = 0.003). 2-Hydroxyestrone and 16α-hydroxyestrone were not associated with endometrial risk after estradiol adjustment (2-OHE1:HRT3vsT1 = 1.97, 95 % CI 0.78, 4.94; 16-OHE1:HRT3vsT1 = 1.50, 95 % CI 0.65, 3.46; p trend = 0.16 and 0.36, respectively). Ratios of 2- and 4-pathway catechol-to-methylated estrogens remained positively associated with endometrial cancer after BMI or estradiol adjustment (2-pathway catechols-to-methylated: HRT3vsT1 = 4.02, 95 % CI 1.60, 10.1; 4-pathway catechols-to-methylated: HRT3vsT1 = 4.59, 95 % CI 1.64, 12.9; p trend = 0.002 for both). Estrogens and estrogen metabolites were not associated with ovarian cancer risk; however, larger studies are needed to better evaluate these relationships. Estrogen metabolism may be important in endometrial carcinogenesis, particularly with less extensive methylation of 2- or 4-pathway catechols associated with elevated endometrial cancer risk.
Similar content being viewed by others

Introduction
Endometrial cancer has long been considered an estrogen-driven malignancy due to observed positive associations with circulating estrogen [1], the use of unopposed estrogen therapy [2], and estrogen-related exposures such as obesity [3, 4]. Although the postulated relationship between estrogen and ovarian cancer remains less clear [5], recent findings from a reanalysis of epidemiological data suggest an elevated risk of ovarian cancer, specifically serous or endometroid tumors, with estrogen only or estrogen and progesterone therapy [6]. Despite prior research on endogenous and exogenous estrogens in relation to gynecological cancers, little is known regarding the relationship between estrogen metabolism and the risk of endometrial [7, 8] or ovarian cancer.
The metabolism of estradiol or estrone begins with irreversible hydroxylation at the C-2, -4, or -16 positions of the steroid ring [9], resulting in metabolites (Online Resource 1) with potential genotoxic and mitogenic properties [10, 11]. Earlier research suggested an elevated ratio of two estrogen metabolites, 2-hydroxyestrone (2-OHE1) and 16α-hydroxyestrone (16α-OHE1), may be inversely related to estrogen-mediated cancer risk [12, 13]. Both metabolites bind to the estrogen receptor, however, with different affinities. Based on laboratory studies, 16α-OHE1 is suggested to covalently bind to the estrogen receptor [14], induce cell proliferation [15, 16], and subsequently, increase cancer risk. Additionally, estrogen metabolism may lead to genotoxic derivatives [10], as hydroxylation at the C-2 and C-4 positions of the steroid ring produces catechol estrogens (2-OHE1, 2-hydroxyestradiol (2-OHE2), and 4-hydroxyestrone (4-OHE1)), characterized by the addition of a second hydroxyl group. Oxidation of these catechol estrogens can lead to the formation of mutagenic quinone products [17] while methylation prevents this process [18]. Given these biological properties, an elevated ratio of catechol to methylated catechols is hypothesized to increase cancer risk.
Most epidemiological studies have explored these hypotheses in relation to breast cancer and not other estrogen-mediated cancers [19–23]. To date, only one prospective study has evaluated circulating 2-OHE1 and 16α-OHE1, as measured by enzyme immunoassay, in relation to endometrial cancer risk among postmenopausal women [7]. Additionally, two case-control studies [8, 24] have reported differences in estrogen metabolite profiles when comparing endometrial cancer patients with healthy controls. With regard to ovarian cancer, only one prior prospective study [25] has evaluated associations with circulating estrone among pre- and postmenopausal women while a recent prospective study examined early pregnancy sex steroids in relation to maternal epithelial cancer risk by histological subtype [26]. No prior studies have examined the role of serum estrogen metabolites. Given the lack of studies, we conducted a case-cohort study within the Breast and Bone Follow-up to the Fracture Intervention Trial [22, 27], to evaluate associations between 15 pre-diagnostic serum estrogens and estrogen metabolites and endometrial and ovarian cancer risk among postmenopausal women not currently using postmenopausal estrogens.
Materials and Methods
We conducted a prospective case-cohort study within B∼FIT, a longitudinal cohort of participants screened for the Fracture Intervention Trial (FIT), which has previously been described [27]. The case-cohort design was selected to assess the relationship between estrogen metabolism and multiple cancer endpoints.
In brief, FIT was a randomized, placebo-controlled trial designed to test the efficacy of alendronate an oral bispohosphonate, in reducing the rate of fractures in women with low bone mineral density [27]. In 1992–1993, 22,695 postmenopausal women (ages 55–80) were screened for participation at 11 clinical centers in the USA. Potential participants underwent a bone mineral density scan (dual-energy X-ray absorptiometry), donated a baseline serum sample, provided clinical examination data including measured anthropometrics, and completed an extensive health history questionnaire that ascertained information on demographic, lifestyle, hormonal, and reproductive factors. Serum samples were originally stored at −20 °C for 3 years and then transferred to −70 °C for long-term storage. Primary results from FIT were reported in 1996 [27] and 1998 [28], and a subset of participants who had used alendronate for at least 3 years were invited to participate in the FIT Long-Term Extension Trial (FLEX) [29].
B∼FIT (N = 15,595) is comprised of female volunteers originally screened for FIT at 10 of the original 11 FIT clinical centers; one clinic declined to participate in the follow-up study. Women who refused or withdrew were excluded (n = 7100). Vital status and cause of death was determined using the National Death Index Plus (NDI+). From 2001 to 2004, surviving women were contacted by mail and/or telephone and invited to complete a follow-up questionnaire (64 % of eligible women completed the BFIT questionnaire), which asked about cancer diagnoses, other health outcomes and reproductive surgeries that occurred since they were screened for FIT, family history of cancer, detailed hormone use, and preventive screening procedures. Women who reported an incident cancer or fracture were asked to give permission for medical record review of those events. In addition, all B∼FIT women from clinical sites located in states with cancer registries (Florida, Maryland, North Carolina, Oregon, and Tennessee) or in Surveillance Epidemiology and End Results (SEER) registry areas (Northern California, Washington, and Iowa) were linked to the registry to identify and confirm cancer diagnoses (73 % of subjects resided in areas with registry linkage, of which 29 % were SEER registry areas). Approximately 93 % of the endometrial and 70 % of the ovarian cancer cases reported among B∼FIT participants were confirmed by medical record or linkage. All women provided written informed consent. B∼FIT was approved by the Institutional Review Board (IRB) of each participating site and the University of California, San Francisco Coordinating Center, as well as the National Cancer Institute.
Eligibility Criteria and Subcohort Selection
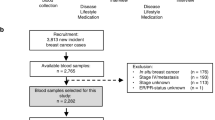
Details on the case-cohort design (including study flowchart) and findings from the breast cancer analysis have previously been published [22]. Prior to selecting the subcohort from the overall B∼FIT cohort (N = 15,595), the following exclusion criteria were applied to determine the eligible population: (1) no available baseline unthawed serum sample (n = 872), (2) missing age at screening (n = 13), (3) ineligible for breast cancer analysis due to history of bilateral mastectomy (n = 623), (4) reported use of postmenopausal estrogens (oral, injection, or patch) within 4 months of their FIT interview/blood draw (n = 45), and (5) a previous history of any cancer other than non-melanoma before FIT baseline (n = 258). Thus, 13,784 participants were eligible for selection for the case-cohort study. These included women who were randomized in the FIT trial, as well as women who were screened for but not included in this trial. The subcohort (N = 515) was randomly selected from among these eligible women (N = 13,784), within 10-year age and geographical clinic strata, irrespective of case status. In determining the analytic population for the analysis of endometrial and ovarian cancer risk, additional exclusion criteria were applied to this subcohort (N = 515). Women who reported a history of hysterectomy at baseline or unknown hysterectomy status (n = 153) or a bilateral oophorectomy or unknown oopherectomy status at baseline (n = 75) were excluded from the subcohort, respectively; one ovary cancer case with unknown oophorectomy status at baseline was also excluded. Additionally, women were excluded from the analytic population based on the following: missing dates for calculation of follow-up time (endometrial—n = 1 case, 3 non-cases; ovarian—13 non-cases) and issues with sample vials (endometrial—13 non-cases; ovarian—11 non-cases).
The final study population for the endometrial cancer analysis includes 412 postmenopausal women, including 66 incident endometrial cases, of which 4 occurred in women sampled as part of the subcohort, and 342 subcohort members who did not develop endometrial cancer during follow-up (total subcohort = 346, 4 cases + 342 non-cases). The final study population for the ovarian cancer analysis includes 67 incident ovarian cancer cases and 416 subcohort members; no ovarian cancer cases were sampled as part of the subcohort.
Laboratory Assays
Assays were conducted at the Cancer Research Technology Program, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research, Frederick, Maryland. Stable isotope dilution liquid chromatography mass spectrometry (LC-MS/MS) was used to simultaneously measure 15 serum estrogens and estrogen metabolites including parent estrogens (estrogen and estradiol) and estrogen metabolites in the 2-hydroxylation pathway (2-hydroxyestrone, 2-hydroxyestradiol, 2-hydroxyestrone-3-methyl ether, 2-methoxyestrone, and 2-methoxyestradiol), the 4-hydroxylation pathway (4-hydroxyestrone, 4-methoxyestrone, and 4-methoxyestradiol), and the 16-hydroxylation pathway (16α-hydroxyestrone, 17-epiestriol, estriol, 16-epiestriol, and 16-ketoestradiol). The total estrogens and estrogen metabolites measured included both conjugated (attached to sulfate or glucuronide moieties) and unconjugated forms. Details on this method have previously been published [30]. In brief, six stable isotopically labeled standards were used including deuterated 2-hydroxyestradiol, 2-methoxyestradiol, and estriol (C/D/N Isotopes Inc, Pointe-Claire, QC, Canada); deuterated 16-epiestriol (Medical Isotopes Inc, Pelham, NH); and 13C-labeled estrone and estradiol (Cambridge Isotope Laboratories, Andover, MA). These standards were added to 0.5 ml of serum, followed by enzymatic hydrolysis, using a preparation from Helix pomatia with β-glucuronidase and sulfatase activity (Sigma Chemical Co, St Louis, MO), to enable the measurement of the sum of the unconjugated and conjugated forms of each estrogen or estrogen metabolite.
Samples were randomized across the batches irrespective of case status; three blinded quality control samples were included within each batch. Coefficients of variation (within and between-batch) from masked quality control samples were <3 % for all analytes. While the published lower limit of quantitation for these serum estrogens is 26.5–29.6 pmol/L [30], our results suggest this assay can detect estrogens below this limit. Additionally, there were no samples with undetectable levels of any of the estrogens or estrogen metabolites.
Statistical Analysis
Baseline characteristics between cases and subcohort members were compared using t tests or chi-square tests as appropriate. Hazard ratios (HRs) and 95 % confidence intervals (CIs) for the relationship between each estrogen exposure and endometrial or ovarian cancer risk were estimated using Cox proportional hazard regression with robust variance adjustment (to account for the case-cohort design) [31]. The time scale was defined as age at baseline (entry) and age at event or censoring (exit). For endometrial cases (n = 62) or ovarian cases (n = 67) not sampled in the subcohort, follow-up started 6 months prior to their age of endometrial or ovarian cancer diagnosis, contributing information only to their risk set [31]. Women in the subcohort were censored at the first of the following events: (1) diagnosis of endometrial or ovarian cancer, (2) death, or (3) end of follow-up. To assess the appropriateness of the proportional hazards assumption, we tested for deviations by including interactions between follow-up time and estrogens.
Tertile categories for each estrogen and estrogen metabolite were determined based on the distribution among the subcohort with the lowest tertile (T1) as the referent group. Estrogens and estrogen metabolites were analyzed individually, as metabolic pathways (C-2, -4, or -16), as ratios of metabolic pathways and as ratios of metabolic pathways relative to the parent estrogens (estradiol and estrone). Summary variables for each metabolic pathway (C-2, -4, -16) were created by summing the individual EM within the respective pathway. Tests for trend were performed by modeling the tertiles of each estrogen or EM exposure as an ordinal variable. All models for the analysis of endometrial cancer were adjusted for study design variables, clinic (ten geographical sites) and trial participation status [screenee only (no trial participation), FIT only, or FIT and FLEX]. We additionally adjusted models for factors known to influence endometrial cancer risk including body mass index (BMI, categorical <25, 25–29.9, ≥30 kg/m2, unknown) and circulating estradiol (continuous). The ovarian models only adjusted for study design variables, as neither BMI nor estradiol, were associated with risk in this study population. Additional adjustment for other baseline covariates known to affect either endometrial or ovarian cancer risk did not change the HR estimates by more than 10 % and were not included in the final models. Potential confounders considered included ethnicity, education, age at menarche, parity, prior estrogen therapy use, smoking status, self-reported history of diabetes, and first-degree breast cancer family history. Sensitivity analyses were performed restricting to never users of estrogen therapy. Given the exploratory nature of this analysis, p values were not adjusted for multiple comparisons. All statistical analyses were performed using the SAS software package, version 9.2 (SAS Institute, Cary, NC).
Results
Endometrial Cancer
Endometrial cases and subcohort members were mostly Caucasian, with a mean age of 67 years at blood draw and 73 years at age at diagnosis. The average time between blood draw and diagnosis among the cases was 6 years (SD 3.2). Cases were more likely to be screenees only (p = 0.02), nulliparous (p = 0.01), with a higher bone mineral density (neck or total hip, p < 0.02) and BMI (≥35 kg/m2, p = 0.002), and to have self-reported history of diabetes (p = 0.02) (Table 1).
Median levels of circulating estrogens, and the majority of estrogen metabolites, were significantly elevated among endometrial cases as compared with the subcohort (Table 2). Estradiol was significantly higher among the cases with a median (10th, 90th) of 53.8 (30.0, 138.5) versus 38.2 (18.1, 91.4) pmol/L among the subcohort (p < .05). Catechol estrogens in the 2-pathway and 4-pathway and 16α-hydroxyestrone were also significantly elevated among cases compared with controls (p < .05). No significant differences between cases and subcohort members were observed for methylated catechols, estrone, or other individual metabolites in the 16-pathway.
Increasing levels of estradiol were significantly associated with an increased risk of endometrial cancer (Table 3; p trend < .001), with an approximate fourfold increase in risk when comparing women in the highest tertile to those in the lowest (HRT3vsT1 = 4.38, 95 % CI 1.82, 10.5; p trend < .001). This relationship persisted after adjustment for BMI. When examining each hydroxylation pathway as a whole, positive associations were observed with the C-2, 4-, and 16-pathways (2-pathway: HRT3vsT1 = 2.21, 95 % CI 1.02, 4.79, p trend = 0.05; 4-pathway: HRT3vsT1 = 2.29, 95 % CI 1.08, 4.89, p trend = 0.03; 16-pathway: HRT3vsT1 = 2.24, 95 % CI 1.07, 4.68, p trend = 0.03). However, these associations were attenuated and no longer significant with adjustment for BMI or estradiol.
Individual metabolites within each hydroxylation pathway were also examined (Tables 4 and 5). Levels of 2-hydroxyestrone and 4-hydroxyestrone (Table 4) were significantly associated with increased endometrial cancer risk (2-OHE1: HRT3vsT1 = 3.24, 95 % CI 1.38, 7.59; 4-OHE1: HRT3vsT1 = 2.83, 95 % CI 1.26, 6.36; p trend = 0.005 and 0.01, respectively). Additionally, the significant dose response observed with 2-OHE1 and 4-OHE1 persisted after adjustment for BMI (p trend = 0.03 and 0.04, respectively) but not with after adjustment for estradiol (p trend = 0.16 and 0.27, respectively). With regard to methylated catechols, no significant associations were observed with either 2-methoxyestrone or 4-methyoxyestrone. Similarly, neither 2-methoxyestradiol nor 4-methoxyestradiol was associated with risk in models adjusted only for study design variables or additionally for BMI; however, these methylated catechols were significantly associated with reduced risk in models adjusted for estradiol (2-methoxyestradiol: HRT3vsT1 = 0.45, 95 % CI 0.21, 0.98, p trend = 0.04; 4-methoxyestradiol: HRT3vsT1 = 0.38, 95 % CI 0.16, 0.94, p trend = 0.03).
Within the 16-pathway (Table 5), elevated levels of 16α-hydroxyestrone were significantly associated with increasing risk (p trend = 0.009), even after adjustment for BMI (p trend = 0.04). However, adjustment for estradiol attenuated these results and the dose response was no longer observed (p trend = 0.36). Significant reductions in risk were observed with higher levels of 17-epiestriol only when estradiol was included in the model (HRT3vsT1 = 0.37, 95 % CI 0.16, 0.88, p trend = 0.03). No other significant associations were observed with individual metabolites in the 16-pathway.
Ratio measures of pathways and individual EM were also assessed (Table 6). When evaluating each hydroxylation pathway as a ratio to the parent estrogens, no significant associations were observed. Increasing levels of 2-OHE1:16-OHE1 were significantly associated with risk after adjustment for estradiol (p trend = 0.03). Women in the highest tertile of 2-OHE1:16-OHE1 were two times as likely to develop endometrial cancer risk as compared to those in the referent group. Additionally, an increase in the ratio of catechol to methylated catechol estrogens in both the 2- and 4-pathways was significantly associated with increased risk, even after adjustment for either BMI (p trend ≤ 0.002) or estradiol (p trend = 0.002 for both). In estradiol adjusted models, the magnitude of risk was similar with either ratio measure, with an approximate fourfold increase in endometrial cancer risk when comparing the highest to lowest tertile (2-pathway catechols: methylated HRT3vsT1 = 4.02, 95 % CI 1.60, 10.1; 4-pathway catechols: methylated HRT3vsT1 = 4.59, 95 % CI 1.64, 12.9). No other ratio measures were significantly associated with endometrial cancer risk.
Ovarian Cancer
Ovarian cases and subcohort members were mostly Caucasian and were similar with regard to mean age at blood draw (cases 68.5 ± 5.7; subcohort 67.0 ± 6.3 years) and other baseline descriptive characteristics (p > 0.05) (Table 1). Similar to the endometrial cases, the mean age at diagnosis for ovarian cases was 74.9 years of age (SD 6.3) with an average of 6 years between diagnosis and blood draw (SD 2.9).
Median levels of estrogens and EM were also compared by case status (Table 2). While levels of all analytes were higher among ovarian cases versus subcohort members, significant differences were only observed with 2-methoxyestrone, 4-methoxyestrone, and the summary measures for 2-pathway methylated catechols and 4-pathway methylated catechols (p < 0.05 for all) (Table 2).
Neither estradiol nor estrone was associated with risk of ovarian cancer (Table 7). Analyses of specific hydroxylation pathways (Table 7) and individual metabolites (Tables 8 and 9) within each pathway also yielded null results, with the exception of the ratio of catechol to methylated catechols in both the 2- and 4-pathways (Table 10). An elevated ratio of 2-pathway catechols to methylated catechols (HRT3vsT1 = 0.42, 95 % CI 0.21, 0.83; p trend = 0.01) and 4-pathway catechols to methylated catechols (HRT3vsT1 = 0.48, 95 % CI 0.24, 0.99; p trend = 0.05) was significantly associated with ovarian cancer risk, although a dose-response relationship was only observed within the 2-pathway. When other ratio measures were examined, no significant associations were observed.
Discussion
Findings from this prospective study of a comprehensive profile of estrogen metabolites in relation to endometrial and ovarian cancer support a role of estrogen metabolism in the development of endometrial cancer, while the relationships with ovarian cancer are less clear. Endogenous estrogens have long been implicated in endometrial carcinogenesis, whereas limited studies [25, 26] have investigated sex steroids in relation to ovarian cancer [32]. In our prospective study of postmenopausal women, higher levels of circulating pre-diagnostic estradiol were significantly associated with increased endometrial cancer risk, consistent with prior reports [1, 7, 33, 34]. However, serum estradiol and estrone were not associated with risk of ovarian cancer, similar to findings from one prior prospective study of estrone and ovarian cancer risk [25].
Prior laboratory and epidemiological studies of estrogen metabolism have focused on the evaluation of 2-hydroxyestrone, 16α-hydroxyestrone, and the ratio of these metabolites, mainly in relation to breast cancer [19, 20, 22, 23] and recently in relation to endometrial cancer [7]. These studies tested the hypothesis that a higher 2-OHE1:16-OHE1 ratio would be associated with a reduction in risk of estrogen-mediated cancers. Contrary to this hypothesis, in our study, higher levels of both these individual metabolites were associated with increased endometrial cancer risk, although the estimates were no longer statistically significant after adjusting for estradiol. To date, only one nested case-control study has evaluated 2-OHE1, 16-OHE1, and their ratio, in relation to endometrial cancer among postmenopausal women, using serum samples from three prospective studies [7]. Findings from this prior study (n = 179 cases) do not support the hypothesis of a reduction in endometrial cancer risk with an elevated 2-OHE1:16-OHE1 ratio; however, higher levels of the individual metabolites were associated with an increased risk of endometrial cancer in models unadjusted for estrone or estradiol [7], similar to the findings reported herein.
The strongest findings observed in the endometrial cancer analysis were those of the ratio of catechol estrogens to methylated estrogens in both the 2- and 4-hydroxylation pathways. Significant increases in risk were observed across all models, including those with adjustment for either BMI or estradiol. This is consistent with the hypothesis that catechol estrogens may increase risk through the production of estrogen quinones, whereas methylated catechols may reduce risk by blocking the formation of these genotoxic derivatives [17, 18]. However, given our limited sample size, particularly in the referent group of this ratio measure, our results should be interpreted with caution and replicated in larger prospective studies of endometrial cancer.
Two prior case-control studies measured select catechol and methylated catechol estrogen metabolites in either post-diagnostic serum [8] or urine [24] samples from endometrial cases. Audet-Walsh et al. assessed serum estradiol and estrone, measured by gas chromatography-MS, and glucoronidated estrogen metabolites (including 2-methoxyestrone-3-glucoronide and 2-methoxyestradiol-3-glucoronide), measured by LC-MS/MS, in relation to postmenopausal endometrial cancer (n = 126 cases). Higher post-diagnostic levels of 2-methoxy-estrone-3-glucoronide and 2-methoxy-estradiol-3-glucoronide were associated with increased endometrial risk [8]. Zhao et al. reported higher urinary levels of 4-hydroxyestradiol and lower levels of 2-methoxyestrone and 2-methoxyestradiol among 23 endometrial cases versus 23 controls [24]. While these prior findings are not directly comparable to our results, given the differences in sample type, timing of measurement (post-diagnostic), and estrogen metabolite measured, taken together with our findings, they lend support for the further investigation of catechol estrogens and methylated catechol estrogens in relation to endometrial cancer risk.
In our prior analysis of estrogen metabolism and breast cancer [22], estradiol was positively associated with risk whereas elevated ratios of the 2-pathway to parents and 4-pathway to parents reduced risk. In the endometrial analysis conducted within the same B∼FIT case-cohort, significant reductions were not observed with these ratios; however, due to our limited sample size for the endometrial cancer analysis, it is difficult to compare results for the breast and endometrial analysis of estrogen metabolites.
Although the evidence in support of estrogen-mediated carcinogenesis is stronger for endometrial cancer, estrogen receptors are also expressed in ovarian tissue [5], providing a biological rationale for the potential role of estrogen and/or estrogen metabolism in ovarian carcinogenesis. However, we found no statistically significant associations with the exception of a reduction in risk with a higher ratio of catechol estrogens to methylated catechols in the 2- and 4-hydroxylation pathways. This finding with the ratio of catechol estrogens to methylated catechols is in contrast to that observed in the analysis of endometrial cancer and with the hypothesis that higher levels of catechol estrogens may confer an elevated risk. Possible explanations for this finding are unclear but may include differences in the activation of catechol-O-methyl-transferase. This result is also somewhat surprising given that the parent estrogens were not associated with risk of ovarian cancer in this study population. However, it is important to note that many of the estrogens and estrogen metabolites associated with ovarian cancer in our study were of weaker magnitude (HR < 2.0) and perhaps not statistically significant due to the limited power to detect these estimates. Based on post hoc power calculations, we had approximately 80 % power to detect a significant trend across tertiles, with a minimum detectable HR of 2.6 in the third tertile (type I error = 0.05, two-sided test). Therefore, our findings from the ovarian cancer analysis may have been limited by the small sample size. This limitation is also an important consideration for the interpretation of findings from the endometrial cancer analysis, with virtually a similar number of cases and subcohort members, in which only very strong associations were statistically significant.
While no prior studies of ovarian cancer have evaluated estrogen metabolites in circulation or using pre-diagnostic serum, one prior case-control study (n = 33 ovary cases, 34 controls) assessed 38 urinary estrogen metabolites, conjugates, and DNA adducts [35] as measured by LC-MS/MS. A higher ratio of depurinating estrogen-DNA adducts to estrogen metabolites was reported among cases as compared to controls, suggesting unbalanced estrogen metabolism among ovarian cancer cases [35]. Given that our study is one of two prospective studies of circulating estrogens and ovarian cancer risk [25, 26], and the only prospective study of estrogen metabolism, further research is needed to determine whether estrogen metabolism is an important factor in the development of ovarian cancer. Furthermore, relationships between circulating estrogens and estrogen metabolites and ovarian cancer risk may vary by histologic subtype. Prior studies have reported differences by histology when examining menopausal hormone therapy [6] and circulating estrogens during early pregnancy [26]. In the present analysis, we were unable to assess potential differences by histological subtype.
Strengths to this analysis include the use of pre-diagnostic serum to assess a comprehensive profile of estrogens and estrogen metabolites and the approximate 10-year follow-up period. Additionally, the use of an LC-MS/MS assay with high specificity and sensitivity [30] allowed for the measurement of estrogens among all postmenopausal women in our study population, even those with relatively low levels. Given the reported low coefficients of variation from prior studies [19, 20] that utilized the LC-MS/MS assay for circulating estrogens and estrogen metabolites, and our interest in studying multiple outcomes, we selected the case-cohort design for this analysis. All samples were assayed during the same time period to ensure similar quality of exposure measurement in the subcohort and the case series, and samples from cases and subcohort members were randomized across all batches. Lastly, in analysis, age was used as the time scale whereby risk sets were created based on age at blood collection. Therefore, it is unlikely that the absence of matching on these factors would have affected our results.
Limitations to our study include the relatively small number of cases, the lack of information on the histology of endometrial and ovarian cases and select confounders on the baseline questionnaire such as history of oral contraceptive use and type of prior menopausal therapy. Type of estrogen therapy may have differential effects on endometrial cancer risk; however, in sensitivity analyses among never users of estrogen therapy, similar patterns were observed. If estrogen and estrogen metabolite levels are associated with future hysterectomy or bilateral oophorectomy, it is possible that the risk estimates presented may be biased as some women may have undergone these procedures after completing the baseline questionnaire. Additionally, 70 % of ovarian cases were confirmed by medical records or linkage; it is possible that some ovarian cases may have been misclassified. It is also possible that some of the women residing in non-registry states who did not complete the follow-up questionnaire may have been misclassified as non-cases. While we could not ascertain case status information on these women, this group comprised only 7.4 % of the B∼FIT study population.
In summary, understanding estrogen metabolism may provide additional insight to the mechanisms underlying endometrial cancer. The potential role of estrogen metabolism in ovary cancer, however, is less clear. Accumulating evidence suggests that estrogen metabolism may be an important component of estrogen-mediated carcinogenesis; further investigations in larger prospective studies of endometrial and ovarian cancer, overall and by histological subtype, are needed.
References
Allen NE, Key TJ, Dossus L, Rinaldi S, Cust A, Lukanova A, Peeters PH et al (2008) Endogenous sex hormones and endometrial cancer risk in women in the European Prospective Investigation into Cancer and Nutrition (EPIC). Endocr Relat Cancer 15(2):485–497. doi:10.1677/ERC-07-0064
Brinton LA, Felix AS (2014) Menopausal hormone therapy and risk of endometrial cancer. J Steroid Biochem Mol Biol 142:83–89. doi:10.1016/j.jsbmb.2013.05.001
Calle EE, Kaaks R (2004) Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer 4(8):579–591. doi:10.1038/nrc1408
Kaaks R, Lukanova A, Kurzer MS (2002) Obesity, endogenous hormones, and endometrial cancer risk: a synthetic review. Cancer Epidemiol Biomarkers Prev 11(12):1531–1543
Lukanova A, Kaaks R (2005) Endogenous hormones and ovarian cancer: epidemiology and current hypotheses. Cancer Epidemiol Biomarkers Prev 14(1):98–107
Collaborative Group on Epidemiological Studies of Ovarian, Cancer (2015) Menopausal hormone use and ovarian cancer risk: individual participant meta-analysis of 52 epidemiological studies. Lancet. doi:10.1016/S0140-6736(14)61687-1
Zeleniuch-Jacquotte A, Shore RE, Afanasyeva Y, Lukanova A, Sieri S, Koenig KL, Idahl A et al (2011) Postmenopausal circulating levels of 2- and 16α-hydroxyestrone and risk of endometrial cancer. Br J Cancer 105(9):1458–1464. doi:10.1038/bjc.2011.381
Audet-Walsh E, Lepine J, Gregoire J, Plante M, Caron P, Tetu B, Ayotte P et al (2011) Profiling of endogenous estrogens, their precursors, and metabolites in endometrial cancer patients: association with risk and relationship to clinical characteristics. J Clin Endocrinol Metab 96(2):E330–E339. doi:10.1210/jc.2010-2050
Lippert TH, Seeger H, Mueck AO (2000) The impact of endogenous estradiol metabolites on carcinogenesis. Steroids 65(7):357–369
Cavalieri E, Chakravarti D, Guttenplan J, Hart E, Ingle J, Jankowiak R, Muti P et al (2006) Catechol estrogen quinones as initiators of breast and other human cancers: implications for biomarkers of susceptibility and cancer prevention. Biochim Biophys Acta 1766(1):63–78. doi:10.1016/j.bbcan.2006.03.001
Mueck AO, Seeger H, Lippert TH (2002) Estradiol metabolism and malignant disease. Maturitas 43(1):1–10
Fishman J, Schneider J, Hershcope RJ, Bradlow HL (1984) Increased estrogen-16 alpha-hydroxylase activity in women with breast and endometrial cancer. J Steroid Biochem 20(4B):1077–1081
Bradlow HL, Sepkovic DW, Klug T, Osborne MP (1998) Application of an improved ELISA assay to the analysis of urinary estrogen metabolites. Steroids 63(7–8):406–413
Swaneck GE, Fishman J (1988) Covalent binding of the endogenous estrogen 16 alpha-hydroxyestrone to estradiol receptor in human breast cancer cells: characterization and intranuclear localization. Proc Natl Acad Sci U S A 85(21):7831–7835
Seeger H, Wallwiener D, Kraemer E, Mueck AO (2006) Comparison of possible carcinogenic estradiol metabolites: effects on proliferation, apoptosis and metastasis of human breast cancer cells. Maturitas 54(1):72–77. doi:10.1016/j.maturitas.2005.08.010
Lippert C, Seeger H, Mueck AO (2003) The effect of endogenous estradiol metabolites on the proliferation of human breast cancer cells. Life Sci 72(8):877–883
Liehr JG (1990) Genotoxic effects of estrogens. Mutat Res 238(3):269–276
Zhang Y, Gaikwad NW, Olson K, Zahid M, Cavalieri EL, Rogan EG (2007) Cytochrome P450 isoforms catalyze formation of catechol estrogen quinones that react with DNA. Metabolism 56(7):887–894. doi:10.1016/j.metabol.2007.03.001
Fuhrman BJ, Schairer C, Gail MH, Boyd-Morin J, Xu X, Sue LY, Buys SS et al (2012) Estrogen metabolism and risk of breast cancer in postmenopausal women. J Natl Cancer Inst 104(4):326–339. doi:10.1093/jnci/djr531
Falk RT, Brinton LA, Dorgan JF, Fuhrman BJ, Veenstra TD, Xu X, Gierach GL (2013) Relationship of serum estrogens and estrogen metabolites to postmenopausal breast cancer risk: a nested case-control study. Breast Cancer Res 15(2):R34. doi:10.1186/bcr3416
Samavat H, Kurzer MS (2014) Estrogen metabolism and breast cancer. Cancer Lett. doi:10.1016/j.canlet.2014.04.018
Dallal CM, Tice JA, Buist DS, Bauer DC, Lacey JV Jr, Cauley JA, Hue TF et al (2014) Estrogen metabolism and breast cancer risk among postmenopausal women: a case-cohort study within B∼FIT. Carcinogenesis 35(2):346–355. doi:10.1093/carcin/bgt367
Dallal CM, Stone RA, Cauley JA, Ness RB, Vogel VG, Fentiman IS, Fowke JH et al (2012) Urinary estrogen metabolites and breast cancer: a combined analysis of individual level data. Int J Biol Markers. doi:10.5301/JBM.2012.9353
Zhao H, Jiang Y, Liu Y, Yun C, Li L (2014) Endogenous estrogen metabolites as biomarkers for endometrial cancer via a novel method of liquid chromatography-mass spectrometry with hollow fiber liquid-phase microextraction. Horm Metab Res. doi:10.1055/s-0034-1371865
Lukanova A, Lundin E, Akhmedkhanov A, Micheli A, Rinaldi S, Zeleniuch-Jacquotte A, Lenner P et al (2003) Circulating levels of sex steroid hormones and risk of ovarian cancer. Int J Cancer 104(5):636–642. doi:10.1002/ijc.10990
Schock H, Surcel HM, Zeleniuch-Jacquotte A, Grankvist K, Lakso HA, Fortner RT, Kaaks R et al (2014) Early pregnancy sex steroids and maternal risk of epithelial ovarian cancer. Endocr Relat Cancer 21(6):831–844. doi:10.1530/ERC-14-0282
Black DM, Reiss TF, Nevitt MC, Cauley J, Karpf D, Cummings SR (1993) Design of the Fracture Intervention Trial. Osteoporos Int 3(Suppl 3):S29–S39
Cummings SR, Black DM, Thompson DE, Applegate WB, Barrett-Connor E, Musliner TA, Palermo L et al (1998) Effect of alendronate on risk of fracture in women with low bone density but without vertebral fractures: results from the Fracture Intervention Trial. JAMA 280(24):2077–2082
Black DM, Schwartz AV, Ensrud KE, Cauley JA, Levis S, Quandt SA, Satterfield S et al (2006) Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA 296(24):2927–2938. doi:10.1001/jama.296.24.2927
Xu X, Roman JM, Issaq HJ, Keefer LK, Veenstra TD, Ziegler RG (2007) Quantitative measurement of endogenous estrogens and estrogen metabolites in human serum by liquid chromatography-tandem mass spectrometry. Anal Chem 79(20):7813–7821. doi:10.1021/ac070494j
Barlow WE, Ichikawa L, Rosner D, Izumi S (1999) Analysis of case-cohort designs. J Clin Epidemiol 52(12):1165–1172
Eliassen AH, Hankinson SE (2008) Endogenous hormone levels and risk of breast, endometrial and ovarian cancers: prospective studies. Adv Exp Med Biol 630:148–165
Lukanova A, Lundin E, Micheli A, Arslan A, Ferrari P, Rinaldi S, Krogh V et al (2004) Circulating levels of sex steroid hormones and risk of endometrial cancer in postmenopausal women. Int J Cancer 108(3):425–432. doi:10.1002/ijc.11529
Zeleniuch-Jacquotte A, Akhmedkhanov A, Kato I, Koenig KL, Shore RE, Kim MY, Levitz M et al (2001) Postmenopausal endogenous oestrogens and risk of endometrial cancer: results of a prospective study. Br J Cancer 84(7):975–981. doi:10.1054/bjoc.2001.1704
Zahid M, Beseler CL, Hall JB, LeVan T, Cavalieri EL, Rogan EG (2014) Unbalanced estrogen metabolism in ovarian cancer. Int J Cancer 134(10):2414–2423. doi:10.1002/ijc.28565
Acknowledgments
We thank Stephanie Litwack-Harrison, MPH (UCSF, San Francisco, CA), Eric Boyd (IMS, Silver Spring, MD), and Vicky Chia, PhD, for their invaluable assistance with study and data management and the B∼FIT investigators and participants for their contributions to this study.
B∼FIT Research Group members: (1) Coordinating Center—University of California, San Francisco: Douglas C. Bauer MD, Trisha F. Hue PhD MPH, Stephanie Litwack-Harrison MPH, Susan Rubin MPH, Jeffrey A. Tice, MD; (2) Clinical Centers—Group Health Cooperative, Seattle: Diana Buist, PhD and Andrea. Z. LaCroix PhD; Kaiser Permanente Center for Health Research, Portland: Emily Harris PhD; Stanford Medical Center, Palo Alto: William. L. Haskell PhD; University of California, San Diego: Elizabeth Barrett-Connor MD; University of Iowa, Iowa City: James C. Torner PhD; University of Maryland, Baltimore: Marc C. Hochberg MD; University of Miami Medical School: Silvina Levis MD; University of Pittsburgh: Jane Cauley DrPH; University of Tennessee, Memphis: Suzanne Satterfield MD MPH; Wake Forest University, Winston-Salem: Sara. A. Quandt PhD; (3) National Cancer Institute: Louise Brinton PhD; James V. Lacey Jr. PhD, MPH (current affiliation: City of Hope).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Compliance with Ethical Standards
All women provided written informed consent. B∼FIT was approved by the Institutional Review Board (IRB) of each participating site and the University of California, San Francisco Coordinating Center, as well as the National Cancer Institute.
Funding
The original FIT study was funded by Merck Research Laboratories. B∼FIT was funded by the National Cancer Institute (contract #N02-CP-01019), Department of Health and Human Services, USA. Dr. Cher Dallal was supported by the Cancer Prevention Fellowship Program, Division of Cancer Prevention, National Cancer Institute.
Conflict of Interest
The authors declare that they have no competing interests.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 367 kb)
Rights and permissions
About this article

Cite this article
Dallal, C.M., Lacey, J.V., Pfeiffer, R.M. et al. Estrogen Metabolism and Risk of Postmenopausal Endometrial and Ovarian Cancer: the B∼FIT Cohort. HORM CANC 7, 49–64 (2016). https://doi.org/10.1007/s12672-015-0237-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-015-0237-y