
Abstract
The US Food and Drug Administration-approved BRAF inhibitors, vemurafenib and dabrafenib, have demonstrated superior efficacy in patients with BRAF-mutant melanomas but have limited efficacy in BRAF-mutant colorectal cancer. Little is known at this time regarding BRAF inhibitors in thyroid cancer. Initial reports in patients with progressive, radioactive iodine–refractory BRAF-mutant papillary thyroid cancer suggest response rates of approximately 30–40 %. In this review, we discuss BRAF inhibitors in the context of thyroid cancer, the toxicities associated with BRAF inhibitors, and the suggested management of those toxicities. The management of vemurafenib and dabrafenib toxicities is applicable across all tumor types and may serve as a practical guide to their use.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Thyroid cancer, the most common endocrine malignancy, is estimated to be diagnosed in nearly 63,000 new patients and cause almost 1900 deaths in the USA in 2014 [1]. Differentiated thyroid cancer (DTC), a broad category that includes papillary thyroid cancer (PTC), follicular thyroid cancer, Hürthle cell thyroid cancer, and thyroid cancers with poorly differentiated histologies, accounts for more than 90 % of all thyroid tumors. Most DTC patients can be cured with standard primary treatment, but the 15 % of DTC patients who develop metastatic disease have significantly shorter survival. The 10-year median survival rate after the discovery of metastatic DTC is 42 %, but these patients’ prognoses vary widely according to many factors, including the age of the patient; the histology, location, and size of distant metastases; and whether the disease takes up radioactive iodine (RAI). For example, patients with RAI–refractory, progressive DTC who develop macroscopic metastases in the lungs or bones have a poor prognosis, with a 10-year overall survival rate of approximately 10 % [2]. Although much progress has been made against DTC in the last decade, largely owing to the increased use of kinase inhibitors, relatively little progress has been made against anaplastic thyroid cancer (ATC), a rare and aggressive form of thyroid cancer.
Many studies have focused on identifying the molecular mechanisms that contribute to thyroid cancer tumorigenesis and progression. Because vascular endothelial growth factor receptor (VEGFR) is upregulated in DTC patients, VEGFR-targeting drugs have been studied extensively, and small molecule inhibitors of tyrosine kinases have shown promising activity against DTC in phase 2 [3–11] and phase 3 [12,13] clinical trials. Furthermore, oncogenic mutations in the BRAF, RAS, and RET genes, in addition to RET/PTC gene rearrangements, have prognostic implications for DTC patients, and understanding these mutations represents an important step towards developing molecularly targeted therapies against thyroid cancer. One signaling pathway that plays a key role in the development and progression of DTC is the RAS-RAF-MEK-MAP-ERK, or mitogen-activated protein kinase (MAPK), signaling pathway. The most potent activators of the MAPK pathway are BRAF mutations, the most common genetic alteration in PTC. In particular, the BRAF V600E mutation, which occurs in approximately 40 % of primary PTCs, up to 80 % of recurrent PTCs, and approximately 25 % of ATCs [14], is correlated with aggressive tumor characteristics (e.g., extrathyroidal extension, advanced tumor stage at presentation, metastasis to the lymph nodes or distant sites) [15–19] and possibly increased mortality [20]. A recent review suggests that the association between the BRAF V600E mutation and poor prognosis in patients with metastatic PTC must be reexamined; however, doing so has proven challenging owing to the inherent limitations of retrospective studies and difficulties in identifying a sufficient number of patients with clinically aggressive PTC in prospective studies [21]. The BRAF V600E mutation is also associated with decreased ability of these tumors to take up RAI [22], which is the only agent known to cure patients who have distant metastatic disease.
Given these considerations, BRAF kinase inhibition may be an important treatment strategy for patients with BRAF-mutant thyroid cancer. In this review, we discuss the role of BRAF mutations in thyroid cancer, the efficacy of the selective BRAF inhibitors against thyroid cancer and other BRAF-driven malignancies, mechanisms of resistance to BRAF-inhibition-based treatment, and possible combination strategies that may overcome such resistance. We will also describe the toxicity profile of the BRAF inhibitors, which are currently US Food and Drug Administration (FDA)-approved for melanoma (vemurafenib and dabrafenib) and the underlying mechanisms and suggested management of BRAF-inhibitor-induced toxicity.
Advanced Thyroid Cancer Management
The management of ATC and that of DTC are vastly different. A clinical suspicion or pathological diagnosis of ATC is an urgent medical situation that requires rapid evaluation for airway stability, disease staging, and tumor resectability. Expert thyroid pathological analysis to confirm the diagnosis is also advisable. Although the management of ATC is beyond the scope of this review, the American Thyroid Association (ATA) offers excellent guidelines for treatment [23].
The initial standard treatment of advanced DTC is more straightforward and includes surgery with or without RAI and thyroid hormone suppression therapy. Surgery is the primary mode of therapy; the extent of surgery varies and largely depends on the size of the primary tumor, presence of extrathyroidal extension, extension into the surrounding structures, or presence of nodal metastases in the central and/or lateral compartment. The most effective adjuvant treatment for DTC is RAI but should be reserved for intermediate- and high-risk patients per the ATA guidelines, which are an excellent resource. Post-thyroidectomy RAI has three uses: (1) ablation of the remaining thyroid tissue and any possible residual cancer, (2) treatment of known residual or metastatic disease, and (3) imaging to evaluate for possible metastatic disease. Treatment with thyroid hormone is required for all patients, not only prevent hypothyroidism but also reduce thyroid-stimulating hormone-driven stimulation of tumor growth. The levothyroxine dose should be adjusted according to the extent of the disease and the likelihood of recurrence.
Of the DTC patients, 7–23 % develop distant metastases during their disease course, and 1–4 % of DTC patients present with distant metastases. DTC patients who present with distant metastasis should undergo surgery to remove the source of large RAI uptake, followed by RAI to eliminate any disease that remains. Of special consideration are patients with BRAF-mutant tumors, which typically do not take up RAI. Documentation of RAI avidity on pre- or post-treatment whole-body scan is imperative for further treatment decision making. If the distant disease is RAI non-avid or RAI–refractory, RAI treatment is not recommended by most specialists, and monitoring for the pace of disease progression is warranted because metastatic DTC tends to be indolent in many cases. This “watch-and-wait” approach and monitoring with cross-sectional images is appropriate in patients who are asymptomatic, have a low tumor burden, and/or have a slow pace of disease progression. A watch-and-wait approach is also advocated because systemic therapies, which can be given indefinitely if the patient is receiving benefit, have a broad range of toxicities that may negatively affect these patients’ quality of life [24].
Local or systemic treatments may be considered once clinically significant progression has been documented or the patient has developed disease-burden-related symptoms or is at significant risk of developing disease-related morbidity (e.g., spinal cord compression). However, identifying the subpopulation of DTC patients who may benefit from systemic therapies and choosing the optimal drug remains a challenge. Until recently, doxorubicin was the only drug approved by the FDA for metastatic, RAI–refractory DTC, but it yielded a low response rate. Sorafenib is the first oral tyrosine kinase inhibitor approved for progressive RAI–refractory metastatic DTC. A double-blind, randomized, multicenter phase 3 trial was conducted to assess sorafenib’s efficacy and safety versus placebo in patients with progressive RAI–refractory DTC [25]. The progression-free survival (PFS) duration of the sorafenib-treated patients (10.8 months) was significantly longer than that of the placebo-treated patients (5.8 months; hazard ratio, 0.59; 95 % confidence interval, 0.45–0.76; p < 0.0001). The partial response rates in the sorafenib arm and placebo arm were 12.2 and 0.5 %, respectively, and the rates of stable disease lasting at least 6 months were 42–33 %, respectively. Given its inhibition profile, sorafenib has antitumor activity that is most likely exerted through VEGFR inhibition. While sorafenib can inhibit RAF family members in vitro, it does not appear to sufficiently inhibit the kinase activity of BRAF in patients, as demonstrated by the lack of benefit in melanoma. Similarly, in an analysis of DTC patients whose tumors were tested for BRAF mutation, there was no difference in the PFS with sorafenib for those patients with or without a BRAF mutation [12]. In the subgroup of patients with BRAF-mutant tumors, the PFS duration of those who received sorafenib was substantial (20.8 months) but not significantly different from that of those who received placebo (9.4 months); however, this could be explained by the different prognoses of the differing subtypes of DTC [26].
Role of BRAF in Oncogenesis
The MAPK pathway is responsible for transformational phenotypes in many cancers, including thyroid cancers. Under normal conditions, the activation of the MAPK cascade is initiated through ligand activated receptor tyrosine kinases (RTKs) followed by guanosine-triphosphate-bound RAS binding to RAF kinase family members, BRAF and/or CRAF (serine-threonine kinases). This interaction repositions the RAF kinase “activator” to the plasma membrane, where conformational changes and subsequent phosphorylation induces the activator RAF kinase to form a heterodimer or homodimer with a “receiver” RAF kinase. The activator RAF (primarily BRAF) transactivates the bound receiver RAF (primarily CRAF), enabling it to phosphorylate MEK [27]. ARAF can also dimerize with its self and the other RAF molecules; however, it has weak kinase activity relative to the other two. It appears to be more of a scaffolding molecule in some cells, stabilizing the interactions between BRAF and CRAF independent of its own binding of RAS [28,29]. These RAF dimers are integral to the activation of the MAPK signaling cascade; however, their interactions with RAS are consequently disrupted by an ERK-mediated feedback loop [30,31]. BRAF is believed to be the key RAF molecule in this cascade, as its participation has been shown to be essential; depletion of B-Raf in HeLa cells has been shown to reduce epidermal growth factor-induced CRAF kinase activity by 90 %, whereas CRAF depletion reduces BRAF activity by only 50 %, and ARAF depletion has no significant effect [32]. In addition, BRAF can constitutively homodimerize to some degree and thus has significantly higher basal kinase activity than ARAF and CRAF do.
In contrast to wild-type BRAF, activating mutants of BRAF are constitutively active and have been shown to bypass the dimerization requirement. In melanoma, the BRAF V600E mutation enables BRAF to signal as a monomeric enzyme in the absence of activated Ras and upstream RTK inputs to potentiate dimerization [33]. Activating BRAF mutations occur frequently in melanoma and PTC and less frequently in colorectal cancer, non-small-cell lung cancer, and ovarian cancer. Interestingly, activating BRAF and RAS mutations are mutually exclusive in all of these cancers, including well-differentiated thyroid cancer [34]. In addition, the stabilization of the BRAF V600E and CRAF heterodimer by oncogenic RAS inhibits BRAF V600E-mediated activation of the MAPK pathway in melanoma cells [35]. However, the coexistence of mutations that activate both the MAPK and phosphoinositide 3-kinase (PI3K)-protein kinase B pathways is believed to contribute to the dedifferentiation and progression of thyroid cancer [36,37].
Selective Braf Inhibitors in Cancer Treatment
The prevalence of the BRAF V600E mutation in PTC, melanoma, and colorectal cancer and the aggressiveness of these tumors make the BRAF V600E kinase a therapeutic target of interest. Vemurafenib (PLX4032) and dabrafenib (GSK2118436) are two small-molecule RAF inhibitors that have been developed as V600 mutant specific inhibitors. Both molecules work by competing for the modified adenosine triphosphate binding site in the active forms of the BRAF V600E kinase, thereby inhibiting its ability to participate in MAPK pathway activation. In this respect, vemurafenib and dabrafenib have been shown to be highly selective for BRAF V600E-mutant cells that are 100- and 500-fold higher, respectively, than those for cells with wild-type BRAF [38,39]. Other BRAF inhibitors are also being tested in the clinic, but have not been evaluated in thyroid cancer or FDA approved for other indications [40].
Melanoma and Colorectal Cancer
Of the cutaneous melanomas, 40–60% have BRAF mutations [41,42]. Vemurafenib, owing to strong preclinical data and phase 1/2 clinical trial findings, was the first selective BRAF inhibitor to be tested. In a phase 3 trial comparing vemurafenib against what was then the standard of care—dacarbazine—in previously untreated unresectable stage IIIC or IV melanoma with the BRAF V600E mutation, vemurafenib was associated with reductions in the relative risks for death and tumor progression of 63 % and 74 %, respectively. Response rates were 48 % with vemurafenib [43] and 5 % with dacarbazine. On the basis of these results, vemurafenib was approved for the treatment of advanced melanoma. In another phase 3 clinical trial, patients treated with dabrafenib had a PFS duration (5.1 months) that was significantly longer than that of patients treated with dacarbazine (2.7 months). The response rate was 50 % with dabrafenib [44] and only 6 % with dacarbazine. The results of these two phase 3 clinical trials demonstrated the important role of selective BRAF inhibitors in the treatment of BRAF-mutant melanoma. Subsequent studies demonstrated the added benefit of further inhibition of the MAPK pathway with dual BRAF and MEK inhibition. A randomized phase II study of dabrafenib with or without the MEK inhibitor, trametinib, resulted in higher response rates and PFS (76 vs 54 %) and prolonged PFS (9.4 vs 5.8 months) for the combination [45].
In contrast to melanoma patients, colorectal cancer patients harboring the BRAF V600E mutation have a very limited response to vemurafenib. In colorectal cancer, 5–10 % of tumors have BRAF mutations [46,47]. The efficacy of BRAF inhibitors in BRAF-mutant colorectal cancer has been quite disappointing, with a response rate of only about 5 % [47]. Similarly, dabrafenib and trametinib failed to demonstrate a meaningful improvement in response rates [48].
Thyroid Cancer
The first description of using a selective BRAF inhibitor to treat thyroid cancer was from the first-in-human phase 1 trial of vemurafenib [49,50]. The drug elicited a partial response in 1 patient’s BRAF-mutant PTC and stable disease with tumor regression in two other patients. These preliminary observations led to the development of an open-label phase 2 trial of vemurafenib in patients with BRAF-mutant PTC, the results of which were presented at the European Cancer Congress Annual Meeting in 2013 [51]. The trial enrolled 51 patients in the USA and Europe. Eligible patients had RAI–refractory recurrent, unresectable, or metastatic progressive PTC with a BRAF V600 mutation. Patients were analyzed separately based on whether they had previously been treated with a VEGFR inhibitor. The starting vemurafenib dose was 960 mg twice per day, and patients were treated until disease progression or unacceptable toxicity. The primary endpoint was the response rate in VEGFR-inhibitor-naive patients. Of the 26 patients in the VEGFR-inhibitor-naive cohort, 9 (35 %) had a partial response to vemurafenib, and 6 (23 %) had a best response of stable disease for more than 6 months. There were no complete responses. The median PFS duration was 15.6 months [95 % confidence interval, 11.20—not reached (NR)], and the median overall survival duration had not been reached at the time the results were reported. Of the 21 patients who had previously received VEGFR inhibitor therapy, 6 (29 %) had a partial response, and 2 (10 %) had stable disease for 6 months or more. Compared with the VEGFR-inhibitor-naive patients, these patients had a significantly shorter median PFS duration (6.3 months; 95 % confidence interval, 5.38—NR). Their median overall survival duration was 9.8 months. The authors concluded that vemurafenib had activity in VEGFR-inhibitor-naive patients and warranted further study. The adverse events (AEs) were similar to those reported in melanoma patients, with the most common being rash, fatigue, weight loss, taste alteration, and alopecia. Of the 51 patients in the study, 11 (22 %) developed a cutaneous squamous cell carcinoma (SCC), and 1 was diagnosed with an SCC of the distal trachea.
Dadu et al. retrospectively studied a population of thyroid cancer patients who discontinued first-line sorafenib either because of disease progression or drug toxicity and subsequently received salvage treatment with a kinase inhibitor [52]. Of the four patients treated with salvage vemurafenib, three had a partial response. All three patients had discontinued first-line sorafenib treatment because progressive disease. Dadu et al. [53] also reported on their retrospective, off-label experience with vemurafenib in 15 PTC patients; 7 patients (41 %) had partial responses, and 8 patients (47 %), many of whom had tumor regression, had stable disease. The median PFS duration was 13 months. (An updated report is forthcoming.)
There has been 1 case report of a patient with BRAF-mutant ATC being successfully treated with vemurafenib [54]. Vemurafenib is not being studied in ATC at this time, and this report should be interpreted with caution because ATC is often mistaken for poorly differentiated thyroid cancer. Furthermore, ATC patients often have tumors with more than one mutation; thus, targeting a single mutation in this setting may not be fruitful.
Falchook et al. enrolled 14 patients with BRAF-mutant thyroid cancer in the first-in-human phase 1 trial of dabrafenib [55]. The thyroid cancer cohort was results were recently published. The cohort consisted of 13 BRAF mutant PTC and 1 BRAF mutant ATC patients. Four (29 %) partial responses were observed, and six (45 %) achieved a stable disease. The ATC patient had a best response of progressive disease. Median PFS was 11.3 months.
A clinical trial of patients with BRAF-mutated PTC randomized to receive dabrafenib with or without trametinib is ongoing, and therefore, it remains to be determined if combined BRAF and MEK inhibition will increase the activity of single agent BRAF inhibition, as seen in melanoma.
Treatment with BRAF inhibitors results in tumor redifferentiation and RAI reuptake in animal studies of BRAFV600E-mutant thyroid cancer [56]. Thus, several studies in humans with drugs targeting the BRAF pathway (such as selective BRAF inhibitors and MEK inhibitors [57]) in hopes of restoring RAI uptake have been developed. The preliminary results of a pilot trial using dabrafenib to restore RAI uptake in patients with RAI–refractory, BRAF V600E-mutant PTC were reported at the 2013 American Society of Clinical Oncology Annual Meeting [58]. Of the nine patients in the study, five (56 %) had tumors that were RAI-avid on a diagnostic scan after 42 days of dabrafenib treatment. All five were treated with 150 mCi of I-131 (RAI); of these patients, one had a complete response, and three had stable disease.
Other ongoing trials of selective BRAF inhibitors in thyroid cancer patients are listed in Table 1.
Mechanisms of Resistance to Braf Inhibitors
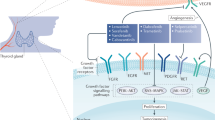
Although BRAF inhibitors are initially effective, resistance is inevitably acquired in most patients as the cells develop alternative mechanisms to pathway activation. Several potential mechanisms of acquired and intrinsic resistance within the context of each cancer have been described (Fig. 1). In melanoma, resistance mechanisms include NRAS mutations, activation of upstream RTKs [e.g., insulin-like growth factor 1 receptor, platelet-derived growth factor receptor β (PDGFRβ), epidermal growth factor receptor (EGFR)], BRAF V600E kinase splice variants that cannot be inhibited by BRAF inhibitors, transactivation of an uninhibited RAF dimer partner by the inhibited BRAF V600 mutant, acquisition of MEK-activating mutations, and overexpression of COT (MAP3K8). These mechanisms result in continued signaling along the MAPK pathway or an alternative pro-survival pathway such as the PI3K pathway [33,59–62]. Response to BRAF inhibition differs among cell types; for example, colon and thyroid cancer cells harboring the BRAF V600E mutation have intrinsic resistance mechanisms to BRAF inhibitors. EGFR expression level is a determinant in sensitivity to BRAF V600E kinase inhibitors. Colon and thyroid cancer cells expressing high levels of EGFR responded well to the combination of vemurafenib and an EGFR inhibitor, but poorly to vemurafenib alone. These cells appear to respond to vemurafenib by deactivating EGFR-negative feedback loops and consequently rapidly activate this RTK [63,64]. When these colon cancer cells are treated with a combination of vemurafenib and PI3K inhibitors, they become sensitive to BRAF V600E kinase inhibitors, resulting in growth inhibition [65,66].

Mechanisms of selective B-Raf (V600E) inhibitor resistance. a In melanoma and colon cancer, cells are known to acquire resistance to B-Raf (V600E) inhibitors through several mechanisms. The RAS-RAF-MEK-ERK or PI3K-AKT pathways can be driven by (1) upregulation or activation of receptor tyrosine kinases (RTK) such as IGF-1R, PDGFRβ, and EGFR or (2) acquired RAS-activating mutations. (3) BRAF splice variants with truncated RAS binding domains permit RAS-independent activator-receiver dimerization. (4) RAS-dependent transactivation of RAF receivers (BRAF or CRAF) by inhibitor-bound wild-type BRAF or CRAF activators. (5) Acquired MEK-activating mutations can act on ERK1/2 independently of RAS and RAF activity. (6) Increased activity of downstream kinases, such as COT, can bypass the inhibition of BRAF and directly phosphorylate MEK. b In papillary thyroid cancer, a mechanism of resistance has been demonstrated through neuregulin-1 (NRG1)-dependent activation of HER2/HER3. Activation of HER2/HER3 then drives both the RAS-RAF-MEK-ERK and/or PI3K-AKT pathways
Much of the work on the mechanisms of mutant BRAF inhibitor resistance in thyroid cancer has been published by Fagin et al. That group reported that, unlike in colon cancer, EGFR activation in response to vemurafenib treatment is not detectable in thyroid cancer cell lines harboring the BRAF V600E mutation. Instead, most cell lines showed a decrease in EGFR phosphorylation (4 of 6 lines) and an 11-fold increase in HER3 phosphorylation and heterodimerization with HER2, thereby increasing the activation of both the MAPK and PI3K pathways. HER2/HER3 activation was found to be dependent on autocrine production of neuregulin-1 (NRG1), which was expressed at much higher levels in BRAF V600E-mutant thyroid cell lines than in BRAF V600E-mutant melanoma or colorectal cell lines. In addition, unlike in melanoma, activation of PDGFRβ resulted in only modest increases in the activation of the MAPK and PI3K pathways [67]. A more recent study demonstrated that BRAF dependent viability did not correlate with primary BRAF inhibitor sensitivity in a panel of BRAF V600E-mutant cell lines derived from thyroid, colorectal, and melanoma tissues. Transient knockdown of BRAF, but not CRAF, led to significant decrease in cellular viability for all cell lines regardless of inhibitor sensitivity. Interestingly, the study also found increased levels of extracellular IL-6 in BRAF inhibitor resistant lines compared to sensitive lines, suggesting an additional autocrine activation loop mechanism of resistance [68].
Many clinical trials in thyroid cancer patients are designed to determine how to overcome resistance to BRAF inhibition (Table 1).
Adverse Effects of BRAF Inhibitors and Suggested Management
The most common AEs related to dabrafenib and vemurafenib, the two selective BRAF inhibitors that are commercially available, are dermatologic AEs. Other common toxicities associated with these drugs are gastrointestinal or constitutional AEs that include headache, pyrexia, fatigue, nausea, and arthralgia.
Dermatologic AEs
Selective BRAF inhibitors have significant cutaneous side effects whose prevention and treatment require a multidisciplinary approach. These side effects can be divided into non-neoplastic and neoplastic dermatologic AEs. Keratosis-pilaris-like eruptions, panniculitides, and photosensitization are among the most common non-neoplastic AEs, whereas actinic and verrucous keratoses and SCC are among the most common neoplastic dermatologic AEs. These dermatologic side effects result in dose cessation or reduction in <10 % of patients [69].
Non-neoplastic Dermatologic AEs
Superficial keratotic plugging of the follicle results in a keratosis-pilaris-like eruption (Fig. 2a), which is seen frequently and more often asymptomatic than pruritic. In phase 2 and 3 trials of vemurafenib, 5–9 % of patients had keratosis-pilaris-like eruptions [43,70]; however, the true incidence may be underreported [71–73]. A case of multiple eruptive milia on the face of a patient receiving vemurafenib therapy has also been reported [74]. In these lesions, the follicular plugging is more prominent and leads to the formation of tiny follicular cysts. These lesions are often asymptomatic and left untreated. If symptomatic, a bland emollient or emollient with urea, salicylic acid, or lactic acid can be used.

Examples of skin toxicities: a keratosis pilaris-like eruptions, b hyperkeratotic punctate lesion on the hand, c verrucous keratoses, and d melanocytic nevi
Hair follicle changes are a common AE of both dabrafenib and vemurafenib. Alopecia, hair changing from straight to curly, and changes in hair color during dabrafenib treatment have all been reported [75]. In phase 2 and 3 trials of vemurafenib, the incidence of alopecia—typically grade 1—ranged from 8 to 36 % [43,70].
Palmoplantar hyperkeratosis or keratoderma is the thickening of the epidermis without inflammation. It has been observed in 6–19 % [76,77] of patients treated with vemurafenib and 13 % [44] of patients treated with dabrafenib. Affected patients develop thick yellow plaques, similar to large calluses, on the palms of their hands and soles of their feet. The keratoderma is most commonly seen on the feet at pressure points, without vesiculation. When these plaques start to vesiculate, particularly in areas of friction, the side effect is classified as a hand–foot skin reaction. Although more common in patients treated with multi-kinase inhibitors such as sorafenib and sunitinib, these lesions have been reported in patients treated with selective BRAF inhibitors [73]. We recommend that patients who develop keratoderma use a urea-based moisturizer; in some cases, referral to a podiatrist to have the calloused areas on the feet reduced is appropriate. Punctate hyperkeratotic lesions (Fig. 2b) may be treated with cryotherapy followed by topical retinoids.
Neutrophilic dermatoses encompass a number of cutaneous conditions with similar histologic features. These conditions include acute febrile neutrophilic dermatosis (Sweet’s syndrome), pustular vasculitis, neutrophilic dermatosis of the dorsal hand, pyoderma gangrenosum, and neutrophilic panniculitis. Sweet’s syndrome [78,79], mainly a dermal process, and neutrophilic panniculitis, mainly a subcutaneous process, have been attributed to the use of BRAF inhibitors [78–84].
All reported cases of neutrophilic panniculitis in patients treated with BRAF inhibitors occurred in patients with metastatic melanoma. In our practice, we have also seen this skin condition in thyroid cancer patients treated with the agents. Patients present with tender, erythematous nodules on the legs and occasionally arms that have a histology consistent with a neutrophilic lobular panniculitis [80–84]. Of the eight patients reported, four had arthralgias [81,83,84]. The lesions appeared 1 day to 7 weeks after drug initiation, with a median time to occurrence of close to 4 weeks. Of the seven patients with known outcomes, two had self-resolution of symptoms, one had resolution with oral non-steroidal anti-inflammatories (NSAIDs), two had resolution with NSAIDs and a short cessation of the BRAF inhibitor treatment, and one patient had resolution with dose cessation, dose reduction, oral steroids, and oral NSAIDs [80,82–84]. The final patient had resolution only after dose cessation [84]. In our practice, we treat panniculitis symptomatically with NSAIDs, but these lesions tend to disappear spontaneously over time.
Photosensitivity sunburn is commonly seen in patients treated with vemurafenib but has not been reported in patients treated with dabrafenib. In one trial, photosensitivity sunburn was seen in 31 % of vemurafenib-treated melanoma patients [77]. This phototoxic reaction has been shown to be caused by exposure to ultraviolet A rays [85]. Patients should be advised to use a broad-spectrum sunscreen (protecting against both ultraviolet A and ultraviolet B rays) and protective barriers (e.g., long sleeves, hat) when exposed to the sun. The sunburn due to vemurafenib can happen within minutes of sun exposure.
Radiation recall dermatitis occurs when patients develop a dermatitis triggered by a drug that is limited to previously irradiated body surface areas. Radiation recall dermatitis, which is usually caused by taxanes and anthracyclines, is thought to reveal subclinical radiation damage that occurred previously. These areas could have been sites of severe sunburns or radiation therapy [86]. Boussemart et al. reported two patients with metastatic melanoma who had vesicular and eczematous eruptions limited to the sites of radiation 10 and 7 days after initiating vemurafenib therapy, respectively. In one patient, radiation therapy concluded 1 day before starting vemurafenib, and the second patient had a 23-day latency period. Both patients’ dermatitis resolved with topical steroid cream [87]. Forschner described three patients with metastatic melanoma, two of whom had a radiation recall pneumonitis and one of whom had radiation recall dermatitis. All three patients were treated with steroids (topical steroids for the dermatitis and systemic steroids for the pneumonitis), and no dose cessation was necessary. Latency periods were 2–4 weeks after starting the vemurafenib [86].
Pulvirenti et al. described five patients who were treated with BRAF inhibitors and radiation therapy simultaneously. They had severe, quickly occurring acute radiation dermatitis that was disproportionate to the low radiotherapy doses they received [88]. Similarly, Satzger et al. suggested that, in patients receiving concomitant radiation therapy and BRAF inhibitor therapy, BRAF inhibition exacerbates radiation dermatitis. The authors suggested using skin controls to better delineate the severity of the dermatitis. Prophylactic cutaneous therapies may be useful in these cases [89].
Increased photosensitivity, particularly in patients who are undergoing or have undergone radiation therapy, should be considered in patients receiving BRAF inhibitor therapy. They can be treated prophylactically or reactively with topical steroids for acute radiation or radiation recall dermatitides.
To date, dermatitis associated with stereotactic radiation of brain metastases has not been reported and remains only a theoretical concern. In our clinical experience, we typically hold BRAF inhibitor therapy for two days before and after stereotactic brain radiation, and we have not observed subsequent radiation dermatitis.
Traditional low-grade morbilliform drug reactions have been seen in 4–21 % of patients receiving vemurafenib [69]. Patients may be treated with topical steroids if no skin blistering occurs and the mucous membranes are not affected. These symptoms would suggest traditional high-grade drug reactions, which have been seen in patients treated with selective BRAF inhibitors, such as Stevens–Johnson syndrome [90] and toxic epidermal necrolysis [91], which require immediate discontinuation of the BRAF inhibitor and medical attention.
Vitiligo [92], sarcoidosis [93,94], and exacerbation of transient acantholytic dermatosis (Grover’s disease) [95] have also been reported in patients treated with BRAF inhibitors.
Neoplastic Dermatologic AEs
The most common neoplastic cutaneous toxicities of the BRAF inhibitors, dabrafenib and vemurafenib, include actinic keratosis, verrucous keratosis, and SCC of the skin. SCC and keratoacanthomas have also been seen in patients treated with sorafenib, another compound with inhibitory activity against RAF kinases [26,96,97]. Squamous cell tumors in patients treated with the BRAF inhibitors vemurafenib and sorafenib have a distinct mutational profile that indicates a mechanism of therapy-induced tumorigenesis in RAS-primed cells [98,99]. The molecular mechanism of this tumorigenesis is consistent with the paradoxical activation of MAPK signaling and accelerates the growth of these lesions [100–102]. Therefore, co-targeting of MEK and RAF may reduce or prevent the formation of these tumors. The combination of dabrafenib and the MEK inhibitor, trametinib, has been assessed in melanoma patients [45], and one study revealed that the proliferative skin lesions commonly seen in patients treated with dabrafenib monotherapy, including cutaneous SCC, papillomas, and hyperkeratosis, were less frequently observed patients treated with a combination of dabrafenib and trametinib [81]. Recent results implicate the suppression of JNK signaling independent of the ERK pathway as an additional mechanism of sorafenib-induced cutaneous SCC [103]. Noncutaneous SCCs of the head and neck and other malignancies associated with RAS activation may occur in patients receiving vemurafenib.
Actinic keratoses are precancerous epithelial lesions typically associated with chronic sun damage. The incidence of actinic keratoses is 6–16 % in vemurafenib-treated patients [43,70,72] and 5–10 % in dabrafenib-treated patients [55,75,104]. Verrucous keratoses (Fig. 2c) are papillated, hyperkeratotic, well-demarcated papules that are often inflamed and appear in an eruptive nature 3–4 months after BRAF inhibitor therapy [75]. Because these lesions demonstrate mild epidermal dysplasia [105], they are treated as precancerous lesions. Verrucous keratoses are not true verruca, as multiple reports have noted that the lesions are negative for human papilloma virus [106,107]. Prompt treatment with cryotherapy, photodynamic therapy, curettage, and/or topical 5-fluorouracil helps prevent both actinic and verrucous keratoses from forming SCC.
SCC of the skin usually manifests as dome-shaped, well-demarcated, hyperkeratotic, erythematous papules and nodules. They grow quickly and are more prevalent in older patients with chronic sun damage [108]. Brose et al. reported cutaneous SCCs in 11 of 51 (22 %) thyroid cancer patients enrolled in a phase 2 trial of vemurafenib [51]. The incidence of SCCs reported in other trials is 4–31 % in vemurafenib-treated patients [43,49,70] and 6–11 % in dabrafenib-treated patients [44,55,104,109]. Sosman et al. showed that most SCCs that occur in dabrafenib- or vemurafenib-treated patients are well-differentiated or keratoacanthoma-type SCCs, which are less aggressive than the normal array of sun-induced SCCs are and have a median time to occurrence of 8 weeks [70]. These lesions are not limited to sun-exposed areas, even though the majority of patients (78 %) in one study had a history or signs of chronic sun damage [99]. Chu et al. found that there was solar elastosis in all BRAF-inhibitor-induced SCCs in their study. No studies of vemurafenib or dabrafenib have reported either drug giving rise to metastatic SCC [110].
In two case reports, systemic retinoid therapy with acitretin was found to reduce the incidence of benign and malignant keratotic lesions in vemurafenib-treated patients [105,111]. Another study found that photodynamic therapy given in three sessions over 5 months was effective against keratoacanthoma and well-differentiated SCC in patients receiving vemurafenib. The lesions were gently curetted, and the keratoacanthomas were treated with 5-aminolevulinic acid for 3 h before undergoing red light activation [112]. Other studies have reported combination therapies of topical 5-fluorouracil, photodynamic therapy, and surgical excision [113] and intralesional 5-fluorouracil plus acitretin [114] to be effective against BRAF-inhibitor-induced SCCs and keratoacanthomas. This type of multimodal therapy as well as early therapy may help reduce the number of surgical excisions that patients receiving BRAF inhibitors must undergo.
Changes in pigmented lesions, such as involution of nevi and new and darkening nevi (Fig. 2d), have also been reported in patients receiving BRAF inhibitors. New nevi have wild-type BRAF and lack the V600E mutation and appear in 8–14 weeks [115]. These lesions have been confirmed with biopsy to be common nevi, dysplastic nevi, and new primary cutaneous melanoma. In phase 2 and 3 trials of vemurafenib, 5 of 464 patients had new primary melanoma. Zimmer et al. [116] reported 11 new melanomas in 10 of 19 patients; 2 of the lesions were >1 mm in thickness and all were wild-type BRAF. Dalle et al. reported 25 new melanoma lesions in 16 of 120 patients treated with vemurafenib; all tumors were <1 mm in thickness and were wild-type BRAF [117]. In phase 3 clinical trials of dabrafenib, 3 of 187 patients had new primary melanoma [44]. The reported incidence of BRAF-inhibitor-associated melanoma varies [43,44,84,117,118]. Longer follow-up will provide more accurate incidence rates.
The numerous cutaneous side effects detailed above highlight the importance of dermatologic care in patients receiving BRAF inhibitor therapy. Overall, dabrafenib-treated patients have a lower incidence of severe cutaneous reactions than vemurafenib-treated patients do [119], but both medications have been implicated in causing the above toxicities. Closely monitoring patients for new and changing lesions can facilitate the early diagnosis of atypical keratinocytic and melanocytic lesions and the early treatment of new cutaneous SCCs and melanomas.
Gastrointestinal AEs
Nausea, dysgeusia, anorexia, and diarrhea are common gastrointestinal AEs associated with vemurafenib, occurring in 35, 14, 18, and 28 % of patients, respectively [76], but these AEs are much less commonly associated with dabrafenib, occurring in fewer than 10 % of patients who take the drug [120]. In one trial, 12 % of dabrafenib-treated melanoma patients had constipation [120]. There are no specific guidelines for managing the gastrointestinal AEs of BRAF inhibitors. However, diarrhea can typically be easily managed with loperamide or diphenoxylate and atropine.
Elevations in liver function tests have been reported to occur in 5 % of vemurafenib-treated patients [77] but have not been reported to occur in dabrafenib-treated patients. Transaminase, alkaline phosphatase, and bilirubin levels should be measured at baseline and monthly thereafter in vemurafenib-treated patients [76]. Recommended dose modifications in response to changes in liver function markers are listed in Table 2.
Constitutional AEs
Arthralgias are more common in patients treated with vemurafenib than in patients treated with dabrafenib; in one trial of vemurafenib, 53 % of treatment-naive melanoma patients had arthralgias [76], and in one trial of dabrafenib, 27 % of patients had arthralgias [120]. There are no clear guidelines for the management of arthralgias in such patients, but in our experience, patients may benefit from a trial of NSAIDs or cyclooxygenase-2 inhibitors and from resting the affected joints. If these actions do not diminish the joint pain and swelling, one should consider temporarily stopping the drug and restarting it at a reduced dose (Table 2).
Headache is a common AE associated with BRAF inhibitors, affecting 32 % of melanoma patients in one trial of dabrafenib [120] and 23 % of patients in a trial of vemurafenib [76]. Less common AEs include back pain and myalgia, which have been reported to occur in 12–11 %, respectively, of dabrafenib-treated patients [120], and in 8–13 %, respectively, of vemurafenib-treated patients [76]. Pain can be managed with standard pain management protocols, starting with non-narcotic analgesics.
Fatigue is a common problem in melanoma patients treated with vemurafenib, with one trial reporting this AE in 38 % of patients [76]. Fatigue is a much less frequently reported in dabrafenib-treated patients, affecting only 6 % in one trial, but is listed in the package insert as the reason for discontinuation in 2 % of melanoma patients in the phase 3 trial of single-agent dabrafenib [120].
Fever is a common AE of dabrafenib, occurring in 28 % of melanoma patients who received single-agent dabrafenib in a phase 3 trial [120]. In that trial, fever and chills were cited as the reason for discontinuation for 9 and 3 % of patients, respectively. Fever is not common in vemurafenib-treated patients. Fever in patients receiving dabrafenib should not be immediately assumed to be drug-related; rather, fever should first prompt a consideration of infectious causes. The dabrafenib package insert offers vague management guidelines for fever, and little information on fever management in dabrafenib-treated patients has been published. On the basis of our experience with the drug, we propose guidelines for treating dabrafenib-related fever according to whether the fever is uncomplicated (temperature <104 °F with no hypotension, dehydration, dizziness, arrhythmia, rigors, or grade 3 related symptoms) or complicated (temperature >104 °F with hypotension, dehydration, dizziness, arrhythmia, rigors, and/or grade 3 related symptoms) (Table 3).
Hematologic AEs
Anemia and leukopenia have been reported in dabrafenib-treated melanoma patients at rates of 28 and 21 %, respectively. Forty percent of dabrafenib-treated melanoma patients had lymphopenia (6 % were grade 3 or 4), and 9 % experienced neutropenia (2 % were grade 3 or 4). Thrombocytopenia was rare, reported in only 8 % of patients [120].
Although anemia is not among the more commonly reported vemurafenib-related AEs, in phase 2 trial of the drug in thyroid cancer patients, 42 % of the treatment-naive cohort had anemia [51]. Information regarding the frequency of leukopenia among the trial’s participants is not available.
Ocular Toxicities
Ocular toxicities rarely occur in patients treated with BRAF inhibitors. One ocular AE associated with BRAF inhibitor use is uveitis, which has been reported to occur in 2 % of vemurafenib-treated patients [76,121] and 1 % of dabrafenib-treated patients [120,122]. Patients receiving these BRAF inhibitors should be monitored for symptoms of uveitis, which include blurred vision, photophobia, and ocular pain, and referred to an ophthalmologist if uveitis is suspected or if any vision changes occur. Ophthalmic steroid drops may be required to control uveitis, and patients should be monitored for complications of topical steroid treatment, which include glaucoma, infection, and cataract progression.
Noncutaneous Malignancies
Both vemurafenib and dabrafenib have been associated with the development of new non-cutaneous tumors, including SCC of the lung and trachea as well as leukemia, gastrointestinal polyps, adenocarcinoma of the colon, and pancreatic cancer. Kim et al. [50] reported a case of a patient with BRAF-mutant PTC with squamoid changes who had a partial response to vemurafenib but also developed a rapidly enlarging lung mass. Biopsy revealed a BRAF-mutant squamous carcinoma, thought to be a progressive dedifferentiated metastasis from the primary PTC. Many clinicians in the thyroid cancer community consider squamous carcinomas of the thyroid to be synonymous with ATC. It is not clear if the progression to a dedifferentiated cancer is the natural progression of the thyroid cancer or if it is caused or accelerated by the BRAF inhibition. Targeted therapies other than those targeting BRAF should be considered in PTC patients with histologic changes consistent with squamous metaplasia.
There is one report of a vemurafenib-treated melanoma patient who developed NRAS-mutant chronic myelomonocytic leukemia [123]. His white blood cell and monocyte counts decreased after vemurafenib was discontinued. Further evaluation of monocytes before, during, and after vemurafenib treatment were consistent with increased activation of ERK.
The development of colonic and gastric polyps has been reported in vemurafenib-treated melanoma patients [124]. Of eight melanoma patients in a phase 1 trial who were treated with vemurafenib for more than 2 years, four underwent endoscopic evaluations; three of these patients were found to have colonic adenomas and/or hyperplastic gastric polyps. There has been one reported case of a melanoma patient treated with dabrafenib plus trametinib who developed a cerebral metastasis consistent with KRAS-mutant adenocarcinoma of the colon [125]. The patient had a previous history of stage II adenocarcinoma of the colon; brain magnetic resonance imaging findings were normal prior to the patient’s starting dabrafenib. More recently, the first case of a new KRAS-mutant pancreatic adenocarcinoma arising during dabrafenib plus trametinib treatment in a melanoma patient was reported [126]. The latter two cases suggest that the addition of a MEK inhibitor does not abrogate the risk of BRAF-inhibitor-induced second malignancy.
From these case studies, it appears that the paradoxical activation of the ERK pathway by RAF inhibitors in cells containing wild-type BRAF is not sufficient for the development of new non-cutaneous tumors following BRAF inhibitor treatment. The preexisting or novel RAS mutations detected in these tumors suggest that an activating mutation upstream of ERK is required for the transformation of secondary disease. In addition to NRAS and KRAS mutations, other upstream MEK-activating events could also be responsible for this transformation. These events may involve the activation of RTKs, such as ERBB family members and PDGFRβ, as they have been described to participate in the development of resistance to BRAF V600E kinase inhibition [59,60,62,127,128].
Future Directions
BRAF inhibitors have shown promise in patients with BRAF-mutant PTC who have advanced or metastatic disease. However, like other kinase inhibitors, these drugs do not offer a cure, and patients inevitably die from their disease. The patients at the highest risk of developing metastatic disease and dying from their disease are those who have gross residual disease after surgery. Thus, employing neoadjuvant approaches to achieve a complete resection—such as using BRAF inhibitors to shrink tumors prior to surgery—is a logical strategy in this patient population. BRAF inhibitors, as opposed to anti-angiogenics, do not impair wound healing and can likely be used safely in the perioperative setting. We have previously reported one patient with BRAF-mutant PTC who was successfully treated with neoadjuvant vemurafenib [129]. A trial to assess the pharmacodynamics of vemurafenib in such patients is ongoing (NCT01709292).
Other areas of interest for future study should include investigating the utility of circulating BRAF-mutant cells as an early marker of response and determining whether modifying surgical approaches based on BRAF mutational status leads to better outcomes.
Summary
BRAF inhibitors bring new hope to patients with BRAF-mutant PTC but should be used with caution, or not at all, in patients whose tumors contain squamous metaplasia. These drugs have a unique AE profile, and most of their AEs are manageable. Physicians and patients alike should be aware of the risk of developing secondary malignancies due to these drugs’ paradoxical activation of the MAPK pathway. Whether these drugs will benefit patients who have BRAF-mutant ATC or poorly differentiated thyroid cancer, which can have multiple mutations, remains unclear; therefore, single-agent BRAF inhibitors are not yet recommended for these indications outside of clinical trials.
References
Siegel R, Ma J, Zou Z et al (2014) Cancer statistics, 2014. CA Cancer J Clin 64:9–29
Durante C, Haddy N, Baudin E et al (2006) Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab 91:2892–2899
Bible KC, Suman VJ, Molina JR et al (2010) Efficacy of pazopanib in progressive, radioiodine-refractory, metastatic differentiated thyroid cancers: results of a phase 2 consortium study. Lancet Oncol 11:962–972
Ahmed M, Barbachano Y, Riddell A et al (2011) Analysis of the efficacy and toxicity of sorafenib in thyroid cancer: a phase II study in a UK based population. Eur J Endocrinol 165:315–322
Hoftijzer H, Heemstra KA, Morreau H et al (2009) Beneficial effects of sorafenib on tumor progression, but not on radioiodine uptake, in patients with differentiated thyroid carcinoma. Eur J Endocrinol 161:923–931
Hong DS, Cabanillas ME, Wheler J et al (2011) Inhibition of the Ras/Raf/MEK/ERK and RET kinase pathways with the combination of the multikinase inhibitor sorafenib and the farnesyltransferase inhibitor tipifarnib in medullary and differentiated thyroid malignancies. J Clin Endocrinol Metab 96:997–1005
Kloos RT, Ringel MD, Knopp MV et al (2009) Phase II trial of sorafenib in metastatic thyroid cancer. J Clin Oncol 27:1675–1684
Gupta-Abramson V, Troxel AB, Nellore A et al (2008) Phase II trial of sorafenib in advanced thyroid cancer. J Clin Oncol 26:4714–4719
Sherman SI, Wirth LJ, Droz JP et al (2008) Motesanib diphosphate in progressive differentiated thyroid cancer. N Engl J Med 359:31–42
Cohen EE, Rosen LS, Vokes EE et al (2008) Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. J Clin Oncol 26:4708–4713
Sherman SI, Jarzab B, Cabanillas ME et al (2011) A phase II trial of the multitargeted kinase inhibitor E7080 in advanced radioiodine (RAI)-refractory differentiated thyroid cancer (DTC). J Clin Oncol 29:5503
Brose MS, Nutting CM, Jarzab B et al. (2014) Sorafenib in radioactive iodine–refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet 384(9940):319–28.
Schlumberger M, Tahara M, Wirth LJ et al. (2014) A phase 3, multicenter, double-blind, placebo-controlled trial of lenvatinib (E7080) in patients with 131I-refractory differentiated thyroid cancer (SELECT). J Clin Oncol 32 (5s; suppl): abstr LBA6008
Nikiforova MN, Wald AI, Roy S et al (2013) Targeted next-generation sequencing panel (ThyroSeq) for detection of mutations in thyroid cancer. J Clin Endocrinol Metab 98:E1852–E1860
Elisei R, Viola D, Torregrossa L et al (2012) The BRAF (V600E) mutation is an independent, poor prognostic factor for the outcome of patients with low-risk intrathyroid papillary thyroid carcinoma: single-institution results from a large cohort study. J Clin Endocrinol Metab 97:4390–4398
Ricarte-Filho J, Ganly I, Rivera M et al (2012) Papillary thyroid carcinomas with cervical lymph node metastases can be stratified into clinically relevant prognostic categories using oncogenic BRAF, the number of nodal metastases, and extra-nodal extension. Thyroid 22:575–584
Elisei R, Ugolini C, Viola D et al (2008) BRAF (V600E) mutation and outcome of patients with papillary thyroid carcinoma: a 15-year median follow-up study. J Clin Endocrinol Metab 93:3943–3949
Ugolini C, Giannini R, Lupi C et al (2007) Presence of BRAF V600E in very early stages of papillary thyroid carcinoma. Thyroid 17:381–388
Li C, Aragon Han P, Lee KC et al (2013) Does BRAF V600E mutation predict aggressive features in papillary thyroid cancer? Results from four endocrine surgery centers. J Clin Endocrinol Metab 98:3702–3712
Xing M, Alzahrani AS, Carson KA et al (2013) Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA 309:1493–1501
Gandolfi G, Sancisi V, Piana S et al. (2014) Time to re-consider the meaning of BRAF V600E mutation in papillary thyroid carcinoma. Int J Cancer doi:10.1002/ijc.28976
Durante C, Puxeddu E, Ferretti E et al (2007) BRAF mutations in papillary thyroid carcinomas inhibit genes involved in iodine metabolism. J Clin Endocrinol Metab 92:2840–2843
Smallridge RC, Ain KB, Asa SL et al (2012) American thyroid association guidelines for management of patients with anaplastic thyroid cancer. Thyroid 22:1104–1139
Schlumberger M, Jarzab B, Elisei R, et al. (2013) Phase III randomized, double-blinded, placebo-controlled trial of sorafenib in locally advanced or metastatic patients with radioactive iodine (RAI)–refractory differentiated thyroid cancer (Dtc)—exploratory analyses of patient-reported outcomes, 83rd Annual American Thyroid Association Meeting. San Juan, PR, 2013
Brose M NC, Jarzab B, Elisei R, Siena S, Bastholt L, de la Fouchardiere C, Pacini F, Paschke R, Shong YK, Sherman SI, Smit J, Chung JW, Siedentop H, Molnar I, Schlumberger M (2013) Sorafenib in locally advanced or metastatic patients with radioactive iodine–refractory differentiated thyroid cancer: the phase III DECISION trial. J Clin Oncol 31 (suppl): abstr 4
Williams VL, Cohen PR, Stewart DJ (2011) Sorafenib-induced premalignant and malignant skin lesions. Int J Dermatol 50:396–402
Hu J, Stites EC, Yu H et al (2013) Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154:1036–1046
Rebocho AP, Marais R (2013) ARAF acts as a scaffold to stabilize BRAF: CRAF heterodimers. Oncogene 32:3207–3212
Mooz J, Oberoi-Khanuja TK, Harms GS et al (2014) Dimerization of the kinase ARAF promotes MAPK pathway activation and cell migration. Sci Signal 7:ra73
Dougherty MK, Muller J, Ritt DA et al (2005) Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell 17:215–224
Avraham R, Yarden Y (2011) Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol 12:104–117
Freeman AK, Ritt DA, Morrison DK (2013) Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol Cell 49:751–758
Poulikakos PI, Persaud Y, Janakiraman M et al (2011) RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF (V600E). Nature 480:387–390
Kimura ET, Nikiforova MN, Zhu Z et al (2003) High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res 63:1454–1457
Karreth FA, DeNicola GM, Winter SP et al (2009) C-Raf inhibits MAPK activation and transformation by B-Raf (V600E). Mol Cell 36:477–486
Hou P, Liu D, Shan Y et al (2007) Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clin Cancer Res 13:1161–1170
Liu Z, Hou P, Ji M et al (2008) Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J Clin Endocrinol Metab 93:3106–3116
Tsai J, Lee JT, Wang W et al (2008) Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A 105:3041–3046
Rheault TR, Stellwagen JC, Adjabeng GM et al (2013) Discovery of dabrafenib: a selective inhibitor of raf kinases with antitumor activity against B-Raf-driven tumors. ACS Med Chem Lett 4:358–362
Morris V, Kopetz S: BRAF inhibitors in clinical oncology. F1000Prime Rep 5:11, 2013
Houben R, Becker JC, Kappel A et al (2004) Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog 3:6
Davies H, Bignell GR, Cox C et al (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954
Chapman PB, Hauschild A, Robert C et al (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364:2507–2516
Hauschild A, Grob JJ, Demidov LV et al (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380:358–365
Flaherty KT, Infante JR, Daud A et al (2012) Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med 367:1694–1703
Richman SD, Seymour MT, Chambers P et al (2009) KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol 27:5931–5937
Kopetz S, Desai J, Chan E et al. (2010) PLX4032 in metastatic colon cancer patients with mutant BRAF tumors. J Clin Oncol 28 (15s; suppl): abstr 3534
Corcoran RB, Atreya CE, Falchook G, et al.: Phase 1–2 trial of the BRAF inhibitor dabrafenib (D) plus MEK inhibitor trametinib (T) in BRAF V600 mutant colorectal cancer (CRC): updated efficacy and biomarker analysis. J Clin Oncol 32 (5s): abstr 3517
Flaherty KT, Puzanov I, Kim KB et al (2010) Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 363:809–819
Kim KB, Cabanillas ME, Lazar AJ et al. (2013) Clinical responses to vemurafenib in patients with metastatic papillary thyroid cancer harboring BRAF mutation. Thyroid 23(10):1277–83
Brose MS, Cabanillas ME, Cohen EE et al. (2013) An open-label, multi-center phase 2 study of the BRAF inhibitor vemurafenib in patients with metastatic or unresectable papillary thyroid cancer positive for the BRAF V600 mutation and resistant to radioactive iodine. Proc European Cancer Congress, Amsterdam, 2013 oral abstr 28
Dadu R, Devine C, Hernandez M et al. (2014) Role of salvage targeted therapy in differentiated thyroid cancer patients who failed first-line sorafenib. J Clin Endocrinol Metab 99(6):2086–94
Dadu R, Shah K, Waguespack SG et al. (2013) Efficacy and tolerability of vemurafenib in BRAFV600E positive papillary thyroid cancer. Proc 83rd Annual Meeting of the American Thyroid Association, San Juan, PR poster 227
Rosove MH, Peddi PF, Glaspy JA (2013) BRAF V600E inhibition in anaplastic thyroid cancer. N Engl J Med 368:684–685
Falchook GS, Long GV, Kurzrock R et al (2012) Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet 379:1893–1901 doi:10.1089/thy.2014.0123
Chakravarty D, Santos E, Ryder M et al (2011) Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. J Clin Invest 121:4700–4711
Ho AL, Grewal RK, Leboeuf R et al (2013) Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med 368:623–632
Rothenberg SM, McFadden DG, Palmer E et al. (2013) Re-differentiation of radioiodine-refractory BRAF V600E-mutant thyroid carcinoma with dabrafenib: a pilot study. J Clin Oncol 31 (suppl): abstr 6025
Villanueva J, Vultur A, Herlyn M (2011) Resistance to BRAF inhibitors: unraveling mechanisms and future treatment options. Cancer Res 71:7137–7140
Nazarian R, Shi H, Wang Q et al (2010) Melanomas acquire resistance to B-RAF (V600E) inhibition by RTK or N-RAS upregulation. Nature 468:973–977
Johannessen CM, Boehm JS, Kim SY et al (2010) COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468:968–972
Greger JG, Eastman SD, Zhang V et al (2012) Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther 11:909–920
Prahallad A, Sun C, Huang S et al (2012) Unresponsiveness of colon cancer to BRAF (V600E) inhibition through feedback activation of EGFR. Nature 483:100–103
Corcoran RB, Ebi H, Turke AB et al (2012) EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2:227–235
Mao M, Tian F, Mariadason JM et al (2013) Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res 19:657–667
Yang H, Higgins B, Kolinsky K et al (2012) Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res 72:779–789
Montero-Conde C, Ruiz-Llorente S, Dominguez JM et al. (2013) Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF mutant thyroid carcinomas. Cancer Discov 3(5):520–33
Sos ML, Levin RS, Gordan JD et al (2014) Oncogene mimicry as a mechanism of primary resistance to BRAF inhibitors. Plant Cell Rep 8:1037–1048
Lacouture ME, Duvic M, Hauschild A et al (2013) Analysis of dermatologic events in vemurafenib-treated patients with melanoma. Oncologist 18:314–322
Sosman JA, Kim KB, Schuchter L et al (2012) Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 366:707–714
Wang CM, Fleming KF, Hsu S (2012) A case of vemurafenib-induced keratosis pilaris-like eruption. Dermatol Online J 18:7
Huang V, Hepper D, Anadkat M et al (2012) Cutaneous toxic effects associated with vemurafenib and inhibition of the BRAF pathway. Arch Dermatol 148:628–633
Boyd KP, Vincent B, Andea A et al (2012) Nonmalignant cutaneous findings associated with vemurafenib use in patients with metastatic melanoma. J Am Acad Dermatol 67:1375–1379
Gebhardt C, Staub J, Schmieder A et al. (2014) Multiple white cysts on face and trunk of a melanoma patient treated with vemurafenib. Acta Derm Venereol doi:10.2340/00015555-1869
Anforth RM, Blumetti TC, Kefford RF et al (2012) Cutaneous manifestations of dabrafenib (GSK2118436): a selective inhibitor of mutant BRAF in patients with metastatic melanoma. Br J Dermatol 167:1153–1160
Package insert vemurafenib (Zelboraf), Genentech
Larkin J, Del Vecchio M, Ascierto PA et al (2014) Vemurafenib in patients with BRAF (V600) mutated metastatic melanoma: an open-label, multicentre, safety study. Lancet Oncol 15:436–444
Yorio JT, Mays SR, Ciurea AM et al. (2014) Case of vemurafenib-induced Sweet’s syndrome. J Dermatol 41(9):817–20
Pattanaprichakul P, Tetzlaff MT, Lapolla WJ et al (2014) Sweet syndrome following vemurafenib therapy for recurrent cholangiocarcinoma. J Cutan Pathol 41:326–328
Maldonado-Seral C, Berros-Fombella JP, Vivanco-Allende B et al (2013) Vemurafenib-associated neutrophilic panniculitis: an emergent adverse effect of variable severity. Dermatol Online J 19:16
Infante JR, Falchook G, Lawrence DP et al. (2011) Phase I/II study to assess safety, pharmacokinetics, and efficacy of the oral MEK 1/2 inhibitor GSK1120212 (GSK212) dosed in combination with the oral BRAF inhibitor GSK2118436 (GSK436). J Clin Oncol 29 (suppl): abstr CRA8503
Sinha R, Edmonds K, Newton-Bishop J et al (2013) Erythema nodosum-like panniculitis in patients with melanoma treated with vemurafenib. J Clin Oncol 31:e320–e321
Monfort JB, Pages C, Schneider P et al (2012) Vemurafenib-induced neutrophilic panniculitis. Melanoma Res 22:399–401
Zimmer L, Livingstone E, Hillen U et al (2012) Panniculitis with arthralgia in patients with melanoma treated with selective BRAF inhibitors and its management. Arch Dermatol 148:357–361
Dummer R, Rinderknecht J, Goldinger SM (2012) Ultraviolet A and photosensitivity during vemurafenib therapy. N Engl J Med 366:480–481
Forschner A, Zips D, Schraml C et al. (2014) Radiation recall dermatitis and radiation pneumonitis during treatment with vemurafenib. Melanoma Res 24(5):512–6
Boussemart L, Boivin C, Claveau J et al (2013) Vemurafenib and radiosensitization. JAMA Dermatol 149:855–857
Pulvirenti T, Hong A, Clements A et al. (2014) Acute radiation skin toxicity associated with BRAF inhibitors. J Clin Oncol pii: JCO.2013.49.0565
Satzger I, Degen A, Asper H et al (2013) Serious skin toxicity with the combination of BRAF inhibitors and radiotherapy. J Clin Oncol 31:e220–e222
Minor DR, Rodvien R, Kashani-Sabet M (2012) Successful desensitization in a case of Stevens-Johnson syndrome due to vemurafenib. Melanoma Res 22:410–411
Wantz M, Spanoudi-Kitrimi I, Lasek A et al (2014) Vemurafenib-induced toxic epidermal necrolysis. Ann Dermatol Venereol 141:215–218
Alonso-Castro L, Rios-Buceta L, Vano-Galvan S et al (2013) Vitiligo in 2 patients receiving vemurafenib for metastatic melanoma. J Am Acad Dermatol 69:e28–e29
Adam A, Thomas L, Bories N et al (2013) Sarcoidosis associated with vemurafenib. Br J Dermatol 169:206–208
Park JJ, Hawryluk EB, Tahan SR et al (2014) Cutaneous granulomatous eruption and successful response to potent topical steroids in patients undergoing targeted BRAF inhibitor treatment for metastatic melanoma. JAMA Dermatol 150:307–311
Gupta M, Huang V, Linette G et al (2012) Unusual complication of vemurafenib treatment of metastatic melanoma: exacerbation of acantholytic dyskeratosis complicated by Kaposi varicelliform eruption. Arch Dermatol 148:966–968
Kwon EJ, Kish LS, Jaworsky C (2009) The histologic spectrum of epithelial neoplasms induced by sorafenib. J Am Acad Dermatol 61:522–527
Arnault JP, Mateus C, Escudier B et al (2012) Skin tumors induced by sorafenib; paradoxic RAS-RAF pathway activation and oncogenic mutations of HRAS, TP53, and TGFBR1. Clin Cancer Res 18:263–272
Oberholzer PA, Kee D, Dziunycz P et al (2012) RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J Clin Oncol 30:316–321
Su F, Viros A, Milagre C et al (2012) RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 366:207–215
Hatzivassiliou G, Song K, Yen I et al (2010) RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464:431–435
Poulikakos PI, Zhang C, Bollag G et al (2010) RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464:427–430
Heidorn SJ, Milagre C, Whittaker S et al (2010) Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140:209–221
Vin H, Ojeda SS, Ching G et al (2013) BRAF inhibitors suppress apoptosis through off-target inhibition of JNK signaling. Elife 2:e00969
Long GV, Trefzer U, Davies MA et al (2012) Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol 13:1087–1095
\Anforth R, Blumetti TC, Mohd Affandi A et al (2012) Systemic retinoid therapy for chemoprevention of nonmelanoma skin cancer in a patient treated with vemurafenib. J Clin Oncol 30:e165–e167
Ko CJ, McNiff JM, Iftner A et al (2013) Vemurafenib (PLX-4032)-induced keratoses: verrucous but not verrucae. J Am Acad Dermatol 69:e95–e96
Ganzenmueller T, Hage E, Yakushko Y et al (2013) No human virus sequences detected by next-generation sequencing in benign verrucous skin tumors occurring in BRAF-inhibitor-treated patients. Exp Dermatol 22:725–729
Anforth R, Fernandez-Penas P, Long GV (2013) Cutaneous toxicities of RAF inhibitors. Lancet Oncol 14:e11–e18
Ascierto PA, Minor D, Ribas A et al (2013) Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol 31:3205–3211
Chu EY, Wanat KA, Miller CJ et al (2012) Diverse cutaneous side effects associated with BRAF inhibitor therapy: a clinicopathologic study. J Am Acad Dermatol 67:1265–1272
Chon SY, Sambrano BL, Geddes ER (2014) Vemurafenib-related cutaneous side effects ameliorated by acitretin. J Drugs Dermatol 13:586–588
Alloo A, Garibyan L, LeBoeuf N et al (2012) Photodynamic therapy for multiple eruptive keratoacanthomas associated with vemurafenib treatment for metastatic melanoma. Arch Dermatol 148:363–366
Fathi R, Kamalpour L, Gammon B et al (2013) A novel treatment approach for extensive, eruptive, cutaneous squamous cell carcinomas in a patient receiving BRAF inhibitor therapy for metastatic melanoma. Dermatol Surg 39:341–344
LaPresto L, Cranmer L, Morrison L et al (2013) A novel therapeutic combination approach for treating multiple vemurafenib-induced keratoacanthomas: systemic acitretin and intralesional fluorouracil. JAMA Dermatol 149:279–281
Cohen PR, Bedikian AY, Kim KB (2013) Appearance of new vemurafenib-associated melanocytic nevi on normal-appearing skin: case series and a review of changing or new pigmented lesions in patients with metastatic malignant melanoma after initiating treatment with vemurafenib. J Clin Aesthet Dermatol 6:27–37
Zimmer L, Hillen U, Livingstone E et al (2012) Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol 30:2375–2383
Dalle S, Poulalhon N, Debarbieux S et al (2013) Tracking of second primary melanomas in vemurafenib-treated patients. JAMA Dermatol 149:488–490
Goppner D, Muller J, Kruger S, et al. (2014) High incidence of naevi-associated BRAF wild-type melanoma and dysplastic naevi under treatment with the class I BRAF inhibitor vemurafenib. Acta Derm Venereol 94(5):517–20
Trinh VA, Davis JE, Anderson JE et al (2014) Dabrafenib therapy for advanced melanoma. Ann Pharmacother 48:519–529
Package insert dabrafenib (Tafinlar), GlaxoSmithKline
Wolf SE, Meenken C, Moll AC et al (2013) Severe pan-uveitis in a patient treated with vemurafenib for metastatic melanoma. BMC Cancer 13:561
Joshi L, Karydis A, Gemenetzi M et al (2013) Uveitis as a result of MAP Kinase pathway inhibition. Case Rep Ophthalmol 4:279–282
Callahan MK, Rampal R, Harding JJ et al (2012) Progression of RAS-mutant leukemia during RAF inhibitor treatment. N Engl J Med 367:2316–2321
Chapman P, Metz D, Sepulveda AR (2012) Development of colonic adenomas and gastric polyps in BRAF mutant melanoma patients treated with vemurafenib. Society for Melanoma Research Congress, Los Angeles, CA
Andrews MC, Behren A, Chionh F et al (2013) BRAF inhibitor-driven tumor proliferation in a KRAS-mutated colon carcinoma is not overcome by MEK1/2 inhibition. J Clin Oncol 31:e448–e451
Carlino MS, Kwan V, Miller DK, et al. (2014) New RAS-mutant pancreatic adenocarcinoma with combined BRAF and MEK inhibition for metastatic melanoma. J Clin Oncol doi:10.1200/JCO.2013.51.5783
Sun C, Wang L, Huang S et al (2014) Reversible and adaptive resistance to BRAF (V600E) inhibition in melanoma. Nature 508:118–122
Wilson TR, Fridlyand J, Yan Y et al (2012) Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 487:505–509
Cabanillas ME, Dadu R, Sturgis EM (2014) Neoadjuvant vemurafenib (VEM) for locally advanced papillary thyroid cancer (PTC) harboring BRAF V600E mutation: a case report. Proc American Association of Clinical Endocrinologists 23rd Annual Scientific & Clinical Congress, abstr no. 1156
Acknowledgments
We would like to thank Joseph Munch for helping with grammatical assistance and stylistic suggestions.
Disclosures
MEC has received research funding from Roche/Genentech. GF has received research funding and travel reimbursement from GlaxoSmithKline. SK is a paid consultant for GSK, Novartis, and Roche/Genentech.
Research Support
This study was supported in part through The University of Texas MD Anderson Cancer Center’s Cancer Center Support Grant CA16672.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cabanillas, M.E., Patel, A., Danysh, B.P. et al. BRAF Inhibitors: Experience in Thyroid Cancer and General Review of Toxicity. HORM CANC 6, 21–36 (2015). https://doi.org/10.1007/s12672-014-0207-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-014-0207-9