
Abstract
We previously reviewed our study of the pharmacological properties of cardiac Na+/Ca2+ exchange (NCX1) inhibitors among cardioprotective drugs, such as amiodarone, bepridil, dronedarone, cibenzoline, azimilide, aprindine, and benzyl-oxyphenyl derivatives (Watanabe et al. in J Pharmacol Sci 102:7–16, 2006). Since then we have continued our studies further and found that some cardioprotective drugs are NCX1 stimulators. Cardiac Na+/Ca2+ exchange current (INCX1) was stimulated by nicorandil (a hybrid ATP-sensitive K+ channel opener), pinacidil (a non-selective ATP-sensitive K+ channel opener), flecainide (an antiarrhythmic drug), and sodium nitroprusside (SNP) (an NO donor). Sildenafil (a phosphodiesterase-5 inhibitor) further increased the pinacidil-induced augmentation of INCX1. In paper, here I review the NCX stimulants that enhance NCX function among the cardioprotective agents we examined such as nicorandil, pinacidil, SNP, sildenafil and flecainide, in addition to atrial natriuretic (ANP) and dofetilide, which were reported by other investigators.
Similar content being viewed by others
Introduction
The plasma membrane Na+/Ca2+ exchanger (NCX) is a bi-directional transporter that mediates the electrogenic exchange of 3Na+ for 1Ca2+. Among the three NCX subtypes, cardiac NCX (NCX1) is abundantly expressed in the heart, smooth muscle, and other tissues. NCX1 plays an important role in the regulation of intracellular Ca2+ homeostasis to maintain mechanical activity and normal electrical rhythm in the heart. In physiological conditions in the heart, NCX1 operates in either Ca2+ exit (generating an inward membrane current) mode or Ca2+ entry (outward membrane current) mode, depending on the membrane potential during the action potential (AP) and ion gradients across the plasma membrane. To maintain stable excitation–contraction coupling, the Ca2+ entry must be balanced by Ca2+ exit. The NCX1 and ATP-dependent Ca2+ pump are the two mechanisms that regulate Ca2+ exit via plasma membrane, and the dominant role of NCX1 has been well known. Especially during normal diastole of cardiomyocytes, NCX1 contributes to an approximately 20–30% reduction of [Ca2+]i by expelling Ca2+ from the cytoplasm [1–3].
The canine NCX1 protein has a molecular mass of 110 kDa and consists of 970 amino acids. In 1999, two research groups suggested that mammalian NCX1 protein comprises nine trans-membrane segments (TMS) and a large hydrophobic loop between 5 and 6 TMS, with the NH2- and COOH-terminals located on the external and internal sides, respectively [4, 5]. In 2013, two groups published ten TMS topology models of mammalian NCX1. The major difference between them is in the orientation of the three C-terminal TMS, but not in the large intracellular loop containing about 550 amino acids between TMS 5 and 6 [6, 7]. The large cytoplasmic domain is involved in the regulation of NCX1 by cytoplasmic factors including exchanger inhibitory peptide (XIP), Na+, Ca2+, and protein kinase C (PKC) [8, 9].
The pharmacological agents for NCX1 regulation are classified into two groups: stimulators and inhibitors. The pharmacology of NCX1 inhibitors has been reported by several researchers [9–11]. On the other hand, there are next to no reports on NCX1 stimulants among cardioprotective agents. This review is about the properties of NCX1 stimulators among cardioprotective drugs including nicorandil, pinacidil, flecainide, sodium nitroprusside (SNP), and sildenafil, which we have investigated to date, as well as atrial natriuretic peptide (ANP) and dofetilide.
NCX1 and NO/cGMP/PKG signaling pathway
In the cytoplasm, nitrate oxide synthase (NOS) produces NO, which activates a soluble guanylate cyclase (GC) and increases intracellular guanosine 3′,5′-cyclic monophosphate (cGMP) and thereby activates cGMP dependent protein kinase (PKG). Several studies have indicated that NCX1 function may be stimulated through the NO/cGMP/PKG signaling pathway in in vitro [12–14]. Horie et al. (1991) reported in single cardiac cells that nicorandil, which has nitrate-like activity and a KATP channel opening activity, decreases the resting level of intracellular Ca2+ ([Ca2+]i) via cGMP-mediated activation of plasma membrane transporters [15]. In addition, Baczkó et al. (2004) reported that, in rat cardiac myocytes, another KATP channel opener (pinacidil) prevents hypoxia/reoxygenation-induced Ca2+ overload [16]. This effect was due to hyperpolarization of the diastolic membrane potential, which may facilitate Ca2+ exit by NCX1. From these two reports, we suspected a relationship between KATP channel openers and NCX1. Therefore, we investigated the effects of KATP channel openers on NCX1 function and possible involvement of the NO/cGMP/PKG signaling pathway.
Nicorandil
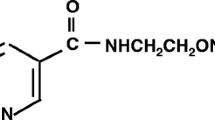
Nicorandil (N-(2-hydroxyethyl)-nicotinamide nitrate) is widely used as an anti-angina drug with nitrate-like activity and KATP channel opening activity. This agent has cardioprotective effects by shortening action potential duration (APD) and hyperpolarizing membrane potential during cardiac ischemia/reperfusion injury. Nicorandil has multiple additional effects including anti-fibrotic activity, anti-apoptotic activity, and reactive oxygen species (ROS) prevention [17]. Regarding ion channels, nicorandil enhances Ca2+-dependent K+ current in rat smooth muscles [18], and cAMP-dependent Cl− current in guinea-pig cardiomyocytes via increasing intracellular cGMP [19]. In addition, nicorandil acutely increases cGMP levels by activating soluble GC via NO-dependent or -independent pathways in smooth muscles and cardiac cells [20–23].
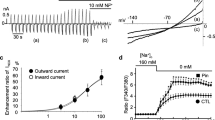
In patch-clamp and fluorescent Ca2+ indicator (Fura-2/AM) studies, we examined the acute effect of nicorandil on cardiac Na+/Ca2+ exchange current (INCX1) in single guinea-pig ventricular cells. Nicorandil enhanced INCX1 in a concentration-dependent manner, with EC50 values of 8.3 and 6.6 μM for the outward and inward INCX, respectively, and Hill coefficients of approximately 1 [24] (Fig. 1a–c). Since nicorandil has nitrate-like activity, we first focused on the NO/cGMP/PKG signaling pathway. We observed that 8-Br-cGMP at 100 μM significantly enhanced INCX compared to control in single guinea-pig ventricular cells [24]. The nicorandil-induced INCX was significantly inhibited by ODQ, a soluble GC inhibitor, at 10 μM [24] (Fig. 1e). Interestingly, the nicorandil-induced INCX increase was hardly prevented by L-NAME, an NO synthase (NOS) inhibitor, at 10 μM [24] (Fig. 1d). Liou et al. (2011) reported that nicorandil increased NO and eNOS phosphorylation in cardiac fibroblasts and these effects were time dependent [25]. It took more than 30 min for nicorandil to significantly increase both eNOS phosphorylation and NO generation [25]. Since the perfusion time (< 5 min) of nicorandil was shorter than 30 min in our experiment, the nicorandil-induced INCX increase must be NO independent in single guinea-pig ventricular cells. Similar results were obtained by Minamiyama et al. (2007), who reported that nicorandil elevated cGMP levels without NO generation in rat liver, aorta, and human coronary smooth muscle cells in vitro [23]. Although we did not examine the effect of PKG on INCX, our results suggest that nicorandil-induced INCX1 increase is mediated by the PKG signaling pathway through an increase in intracellular cGMP.

Effect of nicorandil on INCX1 (modified from [24] with permission). a Chart recording on INCX1. The ramp pulse was initially depolarized from a holding potential of − 60 to +60 mV, then hyperpolarized to − 120 mV, and then depolarized back to − 60 mV at a rate of 680 mV/s. A ramp pulse was given every 10 s. INCX was inhibited completely by KB-R7943, a potent inhibitor of INCX, at 50 μM. b I–V curves on INCX1. c Concentration–response curves of the pinacidil on INCX. d and e Summarized data of L-NAME and ODQ on INCX1. Cont control, Nico nicorandil, KB-R KB-R7943
Furthermore, to clarify the site of action of nicorandil on NCX1, we used the fibroblast cell line, CCL39 stably expressing a canine heart NCX1 isoform and its mutant. In this NCX1 mutant, amino acids (Δ)247–671 are deleted, which is a large portion of the long intracellular loop between TMS 5 and 6, and which includes the XIP region, Ca2+ binding domain, phosphorylation sites, and various modulating sites [8, 9, 26]. We examined the effects of nicorandil on INCX in cells expressing wild-type NCX1, its mutants, and in isolated guinea-pig cardiac ventricular myocytes [24] (Fig. 2a, c). The enhancement ratios of INCX1 by nicorandil were similar between the wild-type NCX1 expressing cells and the guinea-pig cardiac ventricular myocytes [24] (Fig. 2c). On the other hand, nicorandil did not increase INCX1 in the mutant expressing cells [24] (Fig. 2b, c). These results indicated that the large intracellular loop between TMS 5 and 6 may be responsible for the site of action of nicorandil on NCX1.

Effect of nicorandil on INCX in wild type and mutant NCX1 expressing CCL39 cells (modified from [24] with permission). a and b Chart recordings of nicorandil on INCX1 in NCX1 wild type and mutant Δ247–671, and I–V curves. Cont control, Nico nicorandil, KB-R, KB-R7943. c Summarized data. Control: Myocyte or CCL39WT or CCL39Mutant without nicorandil
Pinacidil
Pinacidil, which was initially developed as an antihypertensive drug, is a non-selective KATP channel opener without nitrate-like activity. The non-selective KATP channel openers have properties that open both plasma membrane KATP (pmKATP) and mitochondria KATP (mitoKATP) channels.
In the patch-clamp study and Fura-2/AM study, we examined the effect of pinacidil on INCX1 in single guinea-pig cardiac ventricular myocytes. Pinacidil enhanced INCX1 in a concentration-dependent manner with EC50 values of 23.5 and 23.0 μM for the outward and inward INCX1, respectively, and Hill coefficients of approximately 1 [27] (Fig. 3a).

Effect of pinacidil on INCX1 (modified from [27] with permission). a Concentration–response curves of the pinacidil (Pin) on INCX1. b I–V curves and summary data of L-NAME (top), ODQ (middle) and KT5823 (bottom) on Pin-induced INCX1. c Summary data of MPG on Pin-induced INCX1
KATP channels are regulated by the NO/cGMP/PKG signaling pathway in rat and rabbit hearts [28, 29]. On the other hand, two groups suggested a possible link between KATP channels and NO generation in the rabbit mesenteric artery and rat heart [30, 31]. We examined the relationship between the INCX1 increasing effect of pinacidil and the NO/cGMP/PKG signaling pathway. In our study, L-NAME, ODQ, and KT-5823, a PKG inhibitor, completely blocked the pinacidil-induced INCX1 increase [27] (Fig. 3b). Glibenclamide, a non-selective KATP channel blocker, completely blocked the pinacidil-induced INCX1, but 5-HD, a selective mitoKATP channel inhibitor, did not [27] (Fig. 4a). These results suggest that the pinacidil-induced INCX1 increase may be due to pmKATP channel opening, but not due to mitoKATP channel opening.

a Summary data of glibenclamide, 5-HD and MPG on pinacidil-induced INCX1 increase (modified from [27] with permission). b Summary data of pinacidil on NO in isolated cardiac ventricular myocytes. The effect of glibenclamide and L-NAME on pinacidil-induced NO (modified from [27] with permission). c Summary data of sildenafil on pinacidil-induced INCX increase (modified from [27] with permission). CTL control, Glib glibenclamide, Pin pinacidil, Sil sildenafil
The next question that arises is how pinacidil generates NO. We tested NO production by pinacidil using a fluorometric assay kit in single cardiomyocytes. Pinacidil increased NO about twice as much as the control and pinacidil-induced NO was significantly inhibited by glibenclamide and L-NAME [27] (Fig. 4b). These results suggest that pinacidil may generate NO directly, or indirectly by pmKATP channel opening, and increase INCX1 by phosphorylation via PKG as a result of activation of the NO/cGMP/PKG signaling pathway.
Reactive oxygen species (ROS) enhance NCX1 function in cardiac ventricular myocytes [32–34]. Krenz et al. (2002) have reported that KATP channel opening contributed to generation of ROS in vascular smooth muscles [35]. There may be a positive feedback relationship for ROS release by interaction between pmKATP channel and mitoKATP channel opening in cardiomyocytes. However, in our study, 30 μM pinacidil-induced INCX1 was not inhibited by 1 mM N-2-(mercaptopropionyl) glycine (MPG), an ROS scavenger, or by 5-HD [27] (Figs. 3c, 4a right). There are two reports that affirm these results. Pinacidil generated ROS in a concentration-dependent manner in the rabbit heart, but it took more than 30 min for pinacidil to significantly increase ROS [36]. Holmuhamedov et al. (1998) indicated that in isolated cardiac mitochondria pinacidil acutely decreased mitochondrial membrane potential by mitoKATP channel opening in a concentration-dependent manner at a concentration of 100 μM or higher [37]. These reports suggest that 30 μM pinacidil application for 2–3 min may not generate ROS by mitoKATP channel opening. Therefore, pinacidil-induced INCX1 is not caused by ROS and/or mitoKATP channel opening.
There are multiple isoforms of phosphodiesterases (PDEs) in cardiomyocytes that can hydrolyze cAMP and/or cGMP. Sildenafil, a PDE5 inhibitor, has specific properties such as inhibiting hydrolyzation of cGMP and increasing intracellular cGMP accumulation [38]. In our patch-clamp study, sildenafil at 10 μM further increased 10 μM pinacidil-induced INCX1 [27] (Fig. 4c). In the case of a low or high concentration of pinacidil, the signaling pathway that enhances NCX1 function may be different.
KATP channel openers and NO production
How is KATP channel opening involved in the production of NO? KATP channels in vascular smooth muscle cells regulate the membrane potential. Opening of smooth muscle KATP channels by KATP channel openers causes membrane hyperpolarization. The opening of KATP channels in the endothelium may elevate intracellular Ca2+ concentration, which stimulates the secretion of vasoactive factors via NOS activation [39]. NO release was dependent on the activation of endothelial KATP channels in the pig endocardial artery [40]. Recently, it was reported in endothelial colony-forming cells that the KATP channel openers nicorandil and iptakalim led to Ca2+ influx and activation of CaMKII, with increased phosphorylation levels of CaMKII, eNOS, and Akt, while these phosphorylations were abolished by glibenclamide [41]. These results suggest that intracellular Ca2+ increase may contribute to the opening of KATP channels, but this concept is still controversial. Katakam et al. (2015) indicated for the first time that diazoxide, a mitoKATP channel opener, depolarized mitochondria and increased [Ca2+]i in cultured neurons [42]. Diazoxide thus increased nNOS phosphorylation and increased NO production. Our study suggests that nicorandil, a hybrid KATP channel opener, increases INCX1 though the cGMP/PKG signaling pathway. Since the pinacidil-induced NO increase was inhibited by glibenclamide and L-NAME in a fluorometric assay, pinacidil may directly or indirectly generate NO [27] (Fig. 4b). From these results, we proposed that pinacidil, which does not possess nitrate-like activity, increases INCX1 though the NO/cGMP/PKG signaling pathway.
However, how does KATP channel opening induce the pinacidil activation of NOS? The opening of KATP channels in the endothelium may elevate intracellular Ca2+ concentration, which stimulates NOS activation [39]. NCX contributes to Ca2+ homeostasis in endothelial cells. In vascular endothelial cells, consistent with a pivotal role of NCX in Ca2+-dependent activation of eNOS, NCX protein was detected in caveolin-rich membrane fractions containing both eNOS and caveolin-1. This suggests that a functional interaction between endothelial NCX and eNOS may take place in caveolae [43]. Therefore, KATP channels, NOS, and NCX may be colocalized in caveolae of the plasma membrane. It is known that NCX1 increased or up-regulated in animal and clinical studies in heart failure (HF). Cardiomyocytes as well as endothelial cells and smooth muscle cells contain caveolin-1, -2, and -3 [44]. The muscle-specific isoform, caveolin-3, increased in HF [45]. NCX1 co-precipitated with caveolin-3 [44]. Myocardial NO signaling may be elevated in HF. The increase in caveolin-3 and sarcolemmal caveolae is associated with augmented nitric oxide signaling in canine pacing-induced HF [45]. Endothelial NOS (eNOS) and neuronal NOS (nNOS) are constitutively expressed in cardiomyocytes and endothelial cells, and inducible NOS (iNOS) is also expressed in normal cardiomyocytes [46, 47]. Various ion channels in cardiomyocytes are colocalized with different types of NOS, as reviewed by Gonzales et al. (2009) [48]. There may be a close relationship between KATP channel opening and NOS activation in cardiomyocytes. If there are microdomains such as caveolae where KATP channels, NOS, GC, and PKG are colocalized just below the cardiac cell membrane [43, 48], NOS may be activated by the opening of KATP channels and induce NO production. We found two reports that support this hypothesis. One notes that KATP channel opening by levosimendan may activate nNOS and thereby generate NO in the hippocampus and temporal cortex [49]. Another report indicates that vasodilatation caused by KATP channel opening by minoxidil was inhibited by L-NAME in rat renal vascular smooth muscles [50]. Assuming that the KATP channel opening may mechanically or redox chemically activate NOS, which may be colocalized with the KATP channel, the activation of NOS generates NO and activates the cGMP/PKG signaling pathway in cardiomyocytes. In endothelial colony-forming cells, the KATP channel openers nicorandil and iptakalim led to Ca2+ influx, and activated CaMKII with increased phosphorylation levels of CaMKII, eNOS, and Akt, while their phosphorylation was abolished by glibenclamide [41]. KATP channel opening decreases [Ca2+]i in cardiomyocytes. Therefore, the mechanism of NOS activation in cardiomyocytes by KATP channel opening may be different from that of endothelial cells. However, we have not been able to find any report to date on the molecular link between KATP channel opening and NOS activation. On the contrary, we found reports that KATP channel openers such as pinacidil, diazoxide, cromakalim, and minoxidil did not increase cGMP in rat intact aorta smooth muscle [51, 52]. Whether all KATP channel openers activate NOS and increase INCX1 needs to be investigated.
SNP and ANP
Vasodilatating NO donors such as sodium nitroprusside (SNP) and alpha-human atrial natriuretic peptide (α-hANP) are widely used for the treatment of congestive heart failure. SNP is an atrial and venous dilator that decreases cardiac preload and afterload. ANP activates GC and increases cGMP as a second messenger. ANP has various effects, including as an anti-inflammatory, and inhibitory effects on the rennin–angiotensin system and sympathetic tone [53].
SNP and 8-Br-cGMP, a membrane-permeable analog of cGMP, stimulated NCX1 activity by stimulating soluble GC via an NO-dependent or NO-independent pathway in vascular smooth muscle cells, C6 glioma cells, and astrocytes [12, 14, 54]. We examined the effects of SNP and 8-Br-cGMP on INCX1 in guinea-pig cardiomyocytes by the patch-clamp method. SNP at 1 mM and 8-Br-cGMP at 100 μM stimulated INCX1 [24, 27] (Fig. 5a). Nashida et al. (2011) reported that SNP decreases intracellular Ca2+ concentration by activation of the Ca2+ exit mode of NCX1 through the NO/cGMP/PKG signaling pathway [55]. Furukawa et al. (1991) reported that α-human ANP at 100 nM increases the Ca2+ efflux via NCX1 (ionomycin-induced 45Ca2+ efflux) in rat aorta vascular smooth muscle cells and suggested that the NCX1 function increase by ANP may be dependent on the cGMP/PKG signaling pathway [12].

Antiarrhythmic drugs that activate NCX1 function
Flecainide
Flecainide is a class Ic antiarrhythmic agent in Vaughan Williams classification and is used primarily in the treatment of supraventricular arrhythmias [56]. The acute effects of flecainide are inhibition of peak Na+ channels (peak INa), late Na+ channels (late INa), L-type Ca2+ channels (ICa-L), two voltage-gated K+ channels, i.e., delayed rectifier K+ channels at the rapid component (IKr) and the transient outward K+ channels (Ito), and the human Ether-à-go-go-Related Gene (hERG) potassium channel [57].
Flecainide at 5 μM decreased the amplitude of DADs in dog Purkinje fibers [58]. We examined the effect of flecainide on INCX1 in single guinea-pig cardiac ventricular cells. Flecainide at 30–100 μM stimulated INCX1 by 30–60% in a concentration-dependent manner by the patch-clamp method [59] (Fig. 5b). Sikkel et al. (2013) reported that flecainide at 5 μM significantly stimulated NCX1-mediated Ca2+ efflux in isolated rat cardiomyocytes and suggested that this effect contributed to reducing [Na+]i [60].
Cardioprotective drugs that inhibit INa, ICa, and IK such as amiodarone, bepridil, aprindine, and dronedarone inhibited INCX1 in a concentration-dependent manner in isolated guinea-pig cardiomyocytes, as reviewed previously [10]. In addition, ranolazine and carvedilol, which inhibited INCX1, also suppressed INa, ICa, and IK [61, 62]. However, strangely, only flecainide, which suppressed INa, ICa, and IK, activated INCX1 in our study. Further studies are required to elucidate the molecular mechanisms of flecainide that activated the NCX1 function.
Dofetilide
Dofetilide, a Class III antiarrhythmic drug in Vaughan Williams classification, prolongs APD by inhibiting delayed outward rectifying K+ current and has a positive inotropic effect in guinea-pig cardiomyocytes [63]. Dofetilide increased the amplitude of DADs induced by cardiac glycoside acetyl-strophanthidin in isolated cardiac Purkinje fibers using microelectrode techniques [64]. Dofetilide dose-dependently increased INCX1 with EC50 values of 0.149 μM and 0.249 μM for the inward and outward components, respectively, in rat cardiac ventricular myocytes [65]. However, there has been no report on the molecular mechanisms of activation of INCX1 by dofetilide.
NCX1 stimulators that protect or inhibit delayed afterdepolarizations (DADs)
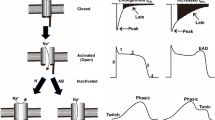
An augmented NCX1 function may play an important role in cardiac arrhythmogenesis. The cardiac arrhythmia is induced by concomitant triggers such as extrasystole, intracellular Ca2+ overload, and spontaneous Ca2+ release. The activated Ca2+ efflux mode of NCX1 may cause DADs, and ventricular arrhythmias [66, 67]. Therefore, NCX inhibitors may have antiarrhythmic actions by inhibiting intracellular Ca2+ overload in cardiomyocytes, or by directly inhibiting the inward INCX1 [68, 69]. In our study, 30 μM carvedilol, which suppressed INCX1, also inhibited ouabain-induced DADs with 0.1 Hz pulse stimuli in isolated guinea-pig ventricular myocytes [62] (Fig. 6a). DADs are almost entirely due to the inward INCX1, not Ca2+-activated Cl− current or Ca2+-activated non-selective cation current [70]. Though nicorandil, pinacidil, and flecainide protected against or attenuated both spontaneous and triggered activities such as ouabain- or acetylstrophantidin-induced DADs in in vitro and in vivo studies [71–76], these three drugs enhanced INCX1 in our patch-clamp experiment using guinea-pig cardiac ventricular myocytes. Furthermore, in this study nicorandil also protected against ouabain-induced DADs in single guinea-pig cardiac ventricular myocytes [24] (Fig. 7b, c). Why do nicorandil, pinacidil, and flecainide, which increase INCX1, prevent or suppress DADs?

Effect of carvedilol on DADs ([62] with permission). a (Left) Control condition. (Middle) DADs were induced by ouabain and electrical stimulation. (Right) The inhibitory effect of carvedilol on DADs. b Summarized data of carvedilol on DAD amplitude. c Summarized data of carvedilol on resting membrane potential (RMP)

Effect of nicorandil on action potential and DADs ([24] with permission). a The effect of nicorandil on action potential in a ventricular myocyte. b (Left) Control condition. (Middle) DADs were induced by ouabain. (Right) The inhibitory effect of nicorandil on DADs. c Summarized data of nicorandil on DADs
Both nicorandil and pinacidil have a KATP channel opening effect. Pharmacological properties in common to these two drugs are shortening APD and hyperpolarizing membrane potential by ATP-sensitive K+ (KATP) channel opening. In our study, nicorandil inhibited ICa and shortened APD via KATP channel opening [24] (Fig. 7a). The INCX1 increase by nicorandil and pinacidil may be as a result of phosphorylation by PKG via the cGMP/PKG signaling pathway. While the Na+/K+ pump in the plasma membrane is also activated by cGMP, the cGMP-mediated increase in NCX1 function decreases [Ca2+]i in cardiac cells [13]. Furthermore, both the functional densities of NCX1 and the Na+/K+ pump were 3- to 3.5-fold more in the transverse tubule plasma membrane than that in the external plasma membrane in rat cardiac ventricular myocytes [77]. In rat vascular smooth muscle cells, the Na+/K+ pump may affect the gap junction conductivity by changing [Ca2+]i of the microdomain via modulation of NCX1 activity [78]. The functional interaction between NCX1 and the Na+/K+ pump may be pivotal for the contraction of cardiac muscle. Especially in the microdomain of the plasma membrane in the heart, NCX1 and the Na+/K+ pump may closely interact to regulate [Na+]i and [Ca2+]i. Both nicorandil and pinacidil may decrease resting [Ca2+]i via the activation of both NCX1 and the Na+/K+ pump by the cGMP/PKG signaling cascade in cardiomyocytes. The cardioprotective effects of nicorandil and pinacidil against DADs may be mainly due to shortening APD in addition to the enhancement of Ca2+ efflux by NCX1.
[Na+]i is a key modulator of intracellular Ca2+ cycling in the heart. An enhancement of late INa increases the intracellular Na+ concentration and thereby increases Ca2+ influx via the outward mode of NCX1 during the plateau phase of the action potential (AP). Late INa-mediated [Na+]i loading may increase diastolic [Ca2+]i, Ca2+ extrusion by the inward mode of NCX1, and DADs formation [79, 80]. The inhibitory effect of flecainide on cardiac Nav1.5 channels increased the triggering threshold by inhibiting both peak INa and late INa. Therefore, flecainide indirectly reduced [Ca2+]i by the Ca2+ efflux mode of NCX1 as well as the incidence of DADs [81]. On the other hand, in one report, flecainide at 6 μM failed to abolish isoproterenol-induced DADs but suppressed isoproterenol-induced triggered activity in mice [82]. Further studies are required to clarify whether or not flecainide inhibits DADs.
Up-regulation of NCX1 gene expression and NCX1 inhibitor
Xu et al. (2009) reported that chronic administration of KB-R7943, an NCX inhibitor, up-regulated NCX1 gene expression in both isolated cardiomyocytes and intact mouse heart [83]. In response to chronic NCX1 inhibition, p-38 forms NCX1-p38 complex [83]. Furthermore, NCX1-p38 complex results in NCX1 up-regulation via activation of p-38 signaling pathway [83]. During hypertrophy and heart failure, up-regulation of NCX1 can be considered as a compensatory adaptation to improve contractile function. However, this compensation invites an increased risk of arrhythmia, such as DADs.
Summary
The KATP channel openers nicorandil and pinacidil, and ANP and SNP, as well as the Na+ channel blocker flecainide and the K+ channel blocker dofetilide, increased NCX1 function (Table 1). The effects of nicorandil and ANP on NCX1 may be mediated by a PKG signaling pathway through an increase in intracellular cGMP (Fig. 8). The effect of pinacidil on NCX1 is mediated by a PKG signaling pathway and pmKATP channel opening (Fig. 8). Little is known about the coexistence and functional cooperation mechanism among NCX1, NOSs and KATP channels in caveolae on the membrane in cardiomyocytes. The effect of SNP on increasing NCX1 may be dependent on the NO/cGMP/PKG signaling pathway (Fig. 8). On the other hand, the molecular mechanisms of flecainide and dofetilide, which activated the NCX1 function, have not been reported. The up-regulation of NCX1 during hypertrophy and heart failure can be considered a compensatory adaptation to improve contractile function. However, this compensation increases risk of arrhythmia. Therefore, further studies are also required to elucidate the role of NCX1 gene expression for myocardial protection.

Scheme of a possible signaling pathway for NCX1 activation by nicorandil, pinacidil, ANP, SNP, and sildenafil. Nicorandil and ANP increase cGMP, SNP generates NO, sildenafil accumulates cGMP, and four agents subsequently activate PKG. Pinacidil opens the pmKATP channel, which generates NO and activates guanylate cyclase, increases cGMP, and subsequently activates PKG. PKG directly or indirectly phosphorylates and stimulates NCX1
References
Blaustein MP, Lederer WJ (1999) Sodium/calcium exchange: its physiological implications. Physiol Rev 79:763–854
Bers DM (2000) Calcium fluxes involved in control of cardiac myocyte contraction. Cir Res 87:275–281
Sarai N, Kobayashi T, Matsuoka S, Noma A (2006) A simulation study to rescue the Na+/Ca2+ exchanger knockout mice. J Physiol Sci 56:211–217
Iwamoto T, Nakamura TY, Pan Y, Uehara A, Imanaga I, Shigekawa M (1999) Unique topology of the internal repeats in the cardiac Na+/Ca2+ exchanger. FEBS Lett 446:264–268
Nicoll DA, Ottolia M, Lu L, Lu Y, Philipson KD (1999) A new topological model of the cardiac sarcolemmal Na+-Ca2+ exchanger. J Biol Chem 274:910–917
Ren X, Philipson KD (2013) The topology of the cardiac Na+/Ca2+ exchanger, NCX1. J Mol Cell Cardiol 57:68–71
Szerencsei RT, Kinjo TG, Schnetkamp PPM (2013) The topology of the C-terminal sections of the NCX1 Na+/Ca2+ exchanger and the NCKX2 Na+/Ca2+-K+ exchanger. Channels 7:109–114
Iwamoto T, Pan Y, Wakabayashi S, Imagawa T, Yamanaka HI, Shigekawa M (1996) Phosphorylation-dependent regulation of cardiac Na+/Ca2+ exchanger via protein kinase C. J Biol Chem 271:13609–13615
Shigekawa M, Iwamoto T (2001) Cardiac Na+/Ca2+ exchange: molecular and pharmacological aspects. Cir Res 88:864–876
Watanabe Y, Koide Y, Kimura J (2006) Topics on the Na+/Ca2+ exchanger: pharmacological characterization on Na+/Ca2+ exchanger inhibitors. J Pharmacol Sci 102:7–16
Iwamoto T, Watanabe Y, Kita S, Blaustein MP (2007) Na+/Ca2+ exchange inhibitors: a new class of calcium regulators. Cardiovasc Hematol Disord Drug Targets 7:188–198
Furukawa K, Ohshima N, Tawada-Iwata Y, Shigekawa M (1991) Cyclic GMP stimulates Na+/Ca2+ exchange in vascular smooth muscle cells in primary culture. J Biol Chem 266:12337–12341
Nishimura J (2006) Topics on the Na+/Ca2+ exchanger: involvement of Na+/Ca2+ exchanger in the vasodilator-induced vasorelaxation. J Pharmacol Sci 102:27–31
Kitao T, Takuma K, Kawasaki T, Inoue Y, Ikehara A, Nashida T, Ago Y, Matsuda T (2010) The Na+/Ca2+ exchanger-mediated Ca2+ influx triggers nitric oxide-induced cytotoxicity in cultured astrocytes. Neurochem Int 57:58–66
Horie M, Suzuki H, Hayashi S, Zang W-J, Komori M, Okada Y, Fujita J, Kawai C (1991) Nicorandil reduced the basal level of cytosolic free calcium in single guinea pig ventricular myocytes. Cell Struc Funct 16:433–440
Baczkó I, Giles WR, Light PE (2004) Pharmacological activation of plasma-membrane KATP channels reduces reoxygenation-induced Ca2+ overload in cardiac myocytes via modulation of the diastolic membrane potential. Br J Pharmacol 141:1059–1067
Aizawa K, Takahashi Y, Higashijima N, Serizawa K, Yogo K, Ishizuka N, Endo K, Fukuyama N, Hirano K, Ishida H (2015) Nicorandil prevents sirolimus-induced production of reactive oxygen species, endothelial dysfunction, and thrombus formation. J Pharmacol Sci 127:284–291
Kajioka S, Oike M, Kitamura K (1990) Nicorandil opens a calcium-dependent potassium channel in smooth muscle cells of the rat portal vein. J Pharmacol Exp Ther 254:905–913
Nishimura N, Reien Y, Matsumoto A, Ogura T, Miyata Y, Suzuki K, Nakazato Y, Daida H, Nakaya H (2010) Effects of nicorandil on the cAMP-dependent Cl- current in guinea-pig ventricular cells. J Pharmacol Sci 112:415–423
Kukovetz WR, Holzmann S, Braida C, Pöch G (1991) Dual mechanism of the relaxing effect of nicorandil by stimulation of cyclic GMP formation and by hyperpolarization. J Cardiovasc Pharmacol 17:627–633
Meisheri KD, Cipkus-Dubray LA, Hosner JM, Khan SA (1991) Nicorandil-induced vasorelaxation: functional evidence for K+ channel-dependent and cyclic GMP-dependent components in a single vascular preparation. J Cardiovasc Pharmacol 17:903–912
Ishizuka N, Saito K, Akima M, Matsubara S, Saito M (2000) Hypotensive interaction of sildenafil and nicorandil in rats through the cGMP pathway but not by KATP channel activation. Jpn J Pharmacol 84:316–324
Minamiyama Y, Takemura S, Hai S, Suehiro S, Okada S, Funae Y (2007) Nicorandil elevates tissue cGMP levels in a nitric-oxide-independent manner. J Pharmacol Sci 103:33–39
Wei JZ, Watanabe Y, Takeuchi K, Yamashita K, Tashiro M, Kita S, Iwamoto T, Watanabe H, Kimura J (2016) Nicorandil stimulates a Na+/Ca2+ exchanger by activating guanylate cyclase in guinea pig cardiac myocytes. Pflugers Arch 468:693–703
Liou JY, Hong HJ, Sung LC, Chao HH, Chen PY, Cheng TH, Chan P, Liu JC (2011) Nicorandil inhibits angiotensin-II-induced proliferation of cultured rat cardiac fibroblasts. Pharmacology 87:144–151
Pan Y, Iwamoto T, Uehara A, Nakamura TY, Imanaga I, Shigekawa M (2000) Physiological functions of the regulatory domains of cardiac Na+/Ca2+ exchanger NCX1. Am J Physiol 279:C393–C402
Iguchi K, Saotome M, Yamashita K, Hasan P, Sasaki M, Maekawa Y, Watanabe Y (2019) Pinacidil, a KATP channel opener, stimulates cardiac Na+/Ca2+ exchanger function through the NO/cGMP/PKG signaling pathway in guinea-pig cardiac ventricular myocytes. Naunyn Schmiedeberg’s Arch Pharmacol 392:949–959
Han J, Kim N, Joo H, Kim E, Earm Y (2002) ATP-sensitive K+ channel activation by nitric oxide and protein kinase G in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol 283:H1545–H1554
Cuong DV, Kim N, Youm JB, Joo H, Warda M, Lee JW, Park WS, Kim T, Kang S, Kim H, Han J (2006) Nitric oxide-cGMP-protein kinase G signaling pathway induces anoxic preconditioning through activation of ATP-sensitive K+ channels in rat hearts. Am J Physiol Heart Circ Physiol 290:H1808–H1817
Murphy ME, Brayden JE (1995) Nitric oxide hyperpolarizes rabbit mesenteric arteries via ATP-sensitive potassium channels. J Physiol 486:47–58
Mączewski M, Beręsewicz A (1997) Inhibition of nitric oxide synthesis and ischemia/reperfusion attenuate coronary vasodilator response to pinacidil in isolated rat heart. J Physiol Pharmacol 48:737–749
Goldhaber JI (1996) Free radicals enhance Na+/Ca2+ exchange in ventricular myocytes. Am J Physiol Heart Circ Physiol 271:H823–H833
Eigel BN, Gursahani H, Hardley RW (2004) ROS are required for rapid reactivation of Na+/Ca2+ exchanger in hypoxic reoxygenated guinea pig ventricular myocytes. Am J Physiol Heart Circ Physiol 286:H955–H963
Hinata M, Matsuoka I, Iwamoto T, Watanabe Y, Kimura J (2007) Mechanism of Na+/Ca2+ exchanger activation by hydrogen peroxide in guinea-pig ventricular myocytes. J Pharmacol Sci 103:283–292
Krenz M, Oldenburg O, Wimpee H, Cohen MV, Garlid KD, Critz SD, Downey JM, Benoit JN (2002) Opening of ATP-sensitive potassium channels causes generation of free radicals in vascular smooth muscle cells. Basic Res Cardiol 97:365–373
Han J, Kim N, Park J, Seog D-H, Joo H, Kim E (2002) Opening of mitochondrial ATP-sensitive potassium channels evokes oxygen radical generation in rabbit heart slices. J Biochem 131:721–727
Holmuhamedov EL, Jovanović S, Dzeja PP, Jovanović A, Terzic A (1998) Mitochondrial ATP-sensitive K+ channels modulate cardiac mitochondria function. Am J Physiol 275:H1567–H1576
Lugnier C (2011) PDE inhibitors: a new approach to treat metabolic syndrome? Curr Opin Pharmacol 11:698–706
Gendron ME, Thorin E, Perrault LP (2004) Loss of endothelial KATP channel-dependent, NO-mediated dilation of endocardial resistance coronary arteries in pigs with left ventricular hypertrophy. Br J Pharmacol 143:285–291
Foster MN, Coetzee WA (2016) KATP channels in the cardiovascular system. Physiol Rev 96:177–252
Wu Y, He M-Y, Ye J-K, Ma S-Y, Huang W, Wei Y-Y, Kong H, Wang H, Zeng X-N, Xie W-P (2017) Activation of ATP-sensitive potassium channels facilitates the function of human endothelial colony-forming cells via Ca2+/Akt/eNOS pathway. J Cell Mol Med 21:609–620
Katakam PVG, Dutta S, Sure VN, Grovenburg SM, Gordon AO, Peterson NR, Rutkai I, Busija DW (2016) Depolarization of mitochondria in neurons promotes activation of nitric oxide synthase and generation of nitric oxide. Am J Physiol Heart Circ Physiol 310:H1097–H1106
Teubl M, Groschner K, Kohlwein SD, Mayer B, Schmidt K (1999) Na+/Ca2+ exchange facilitates Ca2+-dependent activation of endothelial nitric-oxide synthase. J Biol Chem 274:29529–29535
Bossuyt J, Taylor BE, James-Kracke M, Hale CC (2002) Evidence for cardiac sodium-calcium exchanger association with caveolin-3. FEBS Lett 511:113–117
Hare JM, Lofthouse RA, Juang GJ, Colman L, Ricker KM, Kim B, Senzaki H, Cao S, Tunin RS, Kass DA (2000) Contribution of caveolin protein abundance to augmented nitric oxide signaling in conscious dogs with pacing-induced heart failure. Circ Res 86:1085–1092
Michel T, Feron O (1997) Nitric oxide synthase: which, where, why, and how? J Clin Invest 100:2146–2157
Brahmajothi MV, Campbell DL (1999) Heterogeneous basal expression of nitric oxide synthase and superoxide dismutase isoform in mammalian heart: implications for mechanisms governing indirect and direct nitric oxide-related effects. Cir Res 85:575–587
Gonzalez DR, Treuer A, Sun Q-A, Stamler JS, Hare JM (2009) S-nitrosylation of cardiac ion channels. J Cardiovasc Pharmacol 54:188–195
Gooshe M, Tabaeizadeh M, Aleyasin AR, Mojahedi P, Ghasemi K, Yousefi F, Vafaei A, Amini-Khoei H, Amiri S, Dehpour AR (2017) Levosimendan exerts anticonvulsant properties against PTZ-induced seizures in mice through activation of nNOS/NO pathway: role for KATP channel. Life Sci 168:38–46
El-Gowelli HM, El-Gowilli SM, Elsalakawy LK, El-Mas MM (2013) Nitric oxide synthase/K+ channel cascade triggers the adenosine A(2B) receptor-sensitive renal vasodilation in female rats. Eur J Pharmacol 702:116–125
Southerton JS, Weston AH, Bray KM, Newgreen DT, Taylor SG (1988) The potassium channel opening action of pinacidil: studies using biochemical, ion flux and microelectrode techniques. Naunyn Schmiedeberg’s Arch Pharmacol 338:310–318
Newgreen DT, Bray KM, McHarg AD, Weston AH, Duty S, Brown BS, Kay PB, Edwards G, Longmore J, Southerton JS (1990) The action of diazoxide and minoxidil sulphate on rat blood vessels: a comparison with cromakalim. Br J Pharmacol 100:605–613
Ozawa T, Shinke T, Shite J, Takaoka H, Inoue N, Matsumoto H, Watanabe S, Yoshikawa R, Otake H, Matsumoto D, Ogasawara D, Yokoyama M, Hirata K (2015) Effects of human atrial natriuretic peptide on myocardial performance and energetics in heart failure due to previous myocardial infarction. J Cardiol 66:232–238
Asano S, Matsuda T, Takuma K, Kim HS, Sato T, Nishikawa T, Baba A (1995) Nitroprusside and cyclic GMP stimulate Na+-Ca2+ exchange activity in neuronal preparations and cultured astrocytes. J Neurochem 64:437–441
Nashida T, Takuma K, Fukuda S, Kawasaki T, Takahashi T, Baba A, Ago Y, Matsuda T (2011) The specific Na+/Ca2+ exchange inhibitor SEA0400 prevents nitric oxide-induced cytotoxicity in SH-SY5Y cells. Neurochem Int 59:51–58
Falk RH, Fogel RI (1994) Flecainide. J Cardiovasc Electrophysiol 5:964–981
Melgari D, Zhang Y, Harchi AEI, Dempsey CE, Hancox J (2015) Molecular basis of hERG potassium channel blockade by the class Ic antiarrhythmic flecainide. J Mol Cell Cardiol 86:42–53
Krassói L, Varró A, Papp JG (1996) Effect of restacorin on the early and delayed after depolarization in dog Purkinje fibres. Acta Physiol Hung 84:299–300
Kuroda Y, Yuasa S, Watanabe Y, Ito S, Egashira T, Seki T, Hattori T, Ohno S, Kodaira M, Suzuki T, Hashimoto H, Okata S, Tanaka A, Aizawa Y, Murata M, Aiba T, Makita N, Furukawa T, Shimizu W, Kodama I, Ogawa S, Kokubun N, Horigome H, Horie M, Kamiya K, Fukuda K (2017) Flecainide ameliorates arrhythmogenicity through NCX flux in Andersen-Tawil syndrome-iPS cell-derived cardiomyocytes. Biochem Biophys Rep 9:245–256
Sikkel MB, Collins TP, Rowlands C, Shah M, O’Gara P, Williams AJ, Harding SE, Lyon AR, MacLeod KT (2013) Flecainide reduces Ca2+ spark and wave frequency via inhibition of the sarcolemmal sodium current. Cardiovasc Res 98:286–296
Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, Cordeiro JM, Thomas G (2004) Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation 110:904–910
Tashiro M, Watanabe Y, Yamakawa T, Yamashita K, Kita S, Iwamoto T, Kimura J (2017) Suppressive effect of carvedilol on Na+/Ca2+ exchange current in isolated guinea-pig cardiac ventricular myocytes. Pharmacology 99:40–47
Tande PM, Bjørnstad H, Yang T, Refsum H (1990) Rate-dependent class III antiarrhythmic action, negative chronotropy, and positive inotropy of a novel IK blocking drug, UK-68,798: potent in guinea pig but no effect in rat myocardium. J Cardiovasc Pharmacol 16:401–410
Xie JY, Yuan CS, Zhou Z, January CT (2000) Enhancement of delayed afterdepolarizations and triggered activity by class III antiarrhythmic drugs: multiple effects of E-4031 and dofetilide. Methods Find Exp Clin Pharmacol 22:67–76
Zhang X-P, Wu B-W, Yang C-H, Wang J, Niu S-C, Zhang M-S (2009) Dofetilide enhances the contractility of rat ventricular myocytes via augmentation of Na+-Ca2+ exchange. Cardiovasc Drugs Ther 23:207–214
Sipido KR, Volders PG, de Groot SH, Verdonck F, Van de Werf F, Wellens HJ, Vos MA (2000) Enhanced Ca2+ release and Na/Ca exchange activity in hypertrophied canine ventricular myocytes: potential link between contractile adaptation and arrhythmogenesis. Circulation 102:2137–2144
Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM (2001) Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Cir Res 88:1159–1167
Hobai IA, O’Rourke B (2004) The potential of Na+/Ca2+ exchange blockers in the treatment of cardiac diseases. Expert Opin Investig Drugs 13:653–664
Antoons G, Sipido KR (2008) Targeting calcium handling in arrhythmia. Europace 10:1364–1405
Schlotthauer K, Bers DM (2000) Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization: underlying mechanism and threshold for triggered action potentials. Circ Res 87:774–780
Imanishi S, Arita M, Aomine M, Kiyosue T (1984) Antiarrhythmic effects of nicorandil on canine cardiac Purkinje fibers. J Cardiovasc Pharmacol 6:772–779
Lathrop DA, Nànàsi PP, Varrò A (1990) In vitro cardiac models of dog Purkinje fibre triggered and spontaneous electrical activity: effects of nicorandil. Br J Pharmacol 99:119–123
Spinelli W, Sorota S, Siegal M, Hoffman BF (1991) Antiarrhythmic actions of the ATP-regulated K+ current activated by pinacidil. Circ Res 68:1127–1137
Carlsson L, Abrahamsson C, Drews L, Duker G (1992) Antiarrhythmic effects of potassium channel openers in rhythm abnormalities related to delayed repolarization. Circulation 85:1491–1500
Krassói L, Varró A, Papp JG (1996) Effect of restacorin on the early and delayed after depolarization in dog Purkinje fibres. Acta Physiol Hung 84:299–300
Watanabe I, Okamura Y, Ohkubo K, Nagashima K, Mano H, Sonoda K, Kofune M, Kunimoto S, Kasamaki Y, Hirayama A (2011) Effect of ATP-sensitive K+ channel opener nicorandil in a canine model of proarrhythmia. Int Heart J 52:318–322
Despa S, Brette F, Orchard CH, Bers DM (2003) Na/Ca exchange Na/K-ATPase function are equally concentrated in transverse tubules of rat ventricular myocytes. Biophys J 85:3388–3396
Matchkov VV, Gustafsson H, Rahman A, Boedtkjer DMB, Gorintin S, Hansen AK, Bouzinova EV, Praetorius HA, Aalkjaer C, Nilsson H (2007) Interaction between Na+/K+-pump and Na+/Ca2+-exchanger modulates intercellular communication. Circ Res 100:1026–1035
Undrovinas NA, Maltsev VA, Belardinelli L, Sabbah HN, Undrovinas A (2010) Late sodium current contributes to diastolic cell Ca2+ accumulation in chronic heart failure. J Physiol Sci 60:245–257
Shryock JC, Song Y, Rajamani S, Antzelevitch C, Belardinelli L (2013) The arrhythmogenic consequences of increasing late INa in the cardiomyocyte. Cardiovasc Res 99:600–611
Salvage SC, Chandrasekharan KH, Jeevaratnam K, Dulhunty AF, Thompson AJ, Jackson AP, Huang CL-H (2018) Multiple targets for flecainide action: implications for cardiac arrhythmogenesis. Br J Pharmacol 175:1260–1278
Liu N, Denegri M, Ruan Y, Avelino-Cruz JE, Perissi A, Negri S, Napolitano C, Coetzee WA, Boyden PA, Priori SG (2011) Short communication: flecainide exerts an antiarrhythmic effect a mouse model of catecholaminergic polymorphic ventricular tachycardia by increasing the threshold for triggered activity. Cir Res 109:291–295
Xu L, Kappler C, Mani S, Shepherd N, Renaud L, Snider P, Conway SJ, Menick DR (2009) Chronic administration of KB-R7943 induces up-regulation of cardiac NCX1. J Biol Chem 284:27265–27272
Acknowledgements
I thank Dr. Junko Kimura for helpful and critical comments on the manuscript. This study was supported by Grants-in-Aid for Scientific Research (17K11047) from the Japan Society for Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author of this manuscript has no conflict of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Watanabe, Y. Cardiac Na+/Ca2+ exchange stimulators among cardioprotective drugs. J Physiol Sci 69, 837–849 (2019). https://doi.org/10.1007/s12576-019-00721-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12576-019-00721-5