
Abstract
The Clarion Clipperton Fracture Zone (CCZ) is a vast deep-sea region harboring a highly diverse benthic fauna, which will be affected by potential future deep-sea mining of metal-rich polymetallic nodules. Despite the need for conservation plans and monitoring strategies in this context, the majority of taxonomic groups remain scientifically undescribed. However, molecular rapid assessment methods such as DNA barcoding and Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS) provide the potential to accelerate specimen identification and biodiversity assessment significantly in the deep-sea areas. In this study, we successfully applied both methods to investigate the diversity of meiobenthic copepods in the eastern CCZ, including the first application of MALDI-TOF MS for the identification of these deep-sea organisms. Comparing several different species delimitation tools for both datasets, we found that biodiversity values were very similar, with Pielou’s evenness varying between 0.97 and 0.99 in all datasets. Still, direct comparisons of species clusters revealed differences between all techniques and methods, which are likely caused by the high number of rare species being represented by only one specimen, despite our extensive dataset of more than 2000 specimens. Hence, we regard our study as a first approach toward setting up a reference library for mass spectrometry data of the CCZ in combination with DNA barcodes. We conclude that proteome fingerprinting, as well as the more established DNA barcoding, can be seen as a valuable tool for rapid biodiversity assessments in the future, even when no reference information is available.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The deep sea is among the least investigated habitats on earth, harboring highly diverse benthic communities as well as mineral resources (Ramirez-Llodra et al. 2011). The Clarion Clipperton Fracture Zone (CCZ), located in the eastern equatorial Pacific (Fig. 1), has been targeted for the exploration of polymetallic nodules (Clark et al. 2013), which are a potential resource of copper, nickel, cobalt and rare earth elements (Wegorzewski and Kuhn 2014; Kuhn and Rühlemann 2021). There is general consensus that deep-sea mining will have adverse effects on the benthic communities (Gollner et al. 2017; Jones et al. 2017; Niner et al. 2018), and that recolonization of certain taxonomic groups will be slow (Miljutin et al. 2011). To preserve biodiversity in these deep-sea environments, it is not only important to define appropriate marine protected areas and preservation zones (Wedding et al. 2013; Jones et al. 2020; Uhlenkott et al. 2020), but also to develop tools for rapid biodiversity assessment to monitor the deep-sea fauna (Mohrbeck et al. 2015; Macheriotou et al. 2019). Biodiversity is generally high in the deep sea (George et al. 2014), but is often contrasted by low abundances of specimens (Smith et al. 2008; Ramirez-Llodra et al. 2010). Moreover, the actual biodiversity in the deep sea and specifically in nodule areas can currently only be predicted and is most likely underestimated (e.g. Christodoulou et al. 2019). Meiofauna studies conducted in the CCZ have so far focused mainly on the most abundant taxon Nematoda (Miljutina et al. 2010; Miljutin et al. 2015; Singh et al. 2016; Hauquier et al. 2019). Valid species descriptions of Copepoda from the CCZ only exist for some larger-sized species belonging to the orders Calanoida, e.g Aetideidae (Markhaseva et al. 2017), Siphonostomatoida (Mahatma et al. 2008) and Harpacticoida, e.g. Aegisthidae (Mercado-Salas et al. 2019). No large-scale evaluation of the copepod biodiversity in the CCZ has been published yet. Some new Kinorhynch species of the CCZ have been described in Sánchez et al. (2019) and Sánchez et al. (2022).

Bathymetric map of the study area positioned in the eastern Clarion Clipperton Fracture Zone (CCZ). The red dots represent multicore sampling sites
Until now, biodiversity assessment and species descriptions in the CCZ focused on morphology and DNA barcoding (Janssen et al. 2015; Herzog et al. 2018; Christodoulou et al. 2019; Mercado-Salas et al. 2019; Brix et al. 2020). This molecular method allows for species discrimination with little taxonomic expertise compared to pure morphological identification (Hebert et al. 2003; Tautz et al. 2003). Different algorithms exist for species delineation based on DNA barcoding, discriminating species either by distance-based or by tree-based approaches (Christodoulou et al. 2020; Paulus et al. 2022; Korfhage et al. 2022). Among the most commonly used methods is the distance-based Automatic Barcode Gap Discovery (ABGD), delimiting species according to higher within-species similarity compared to other species (Puillandre et al. 2012). The tree-based General Mixed Yule Coalescent method (GMYC) investigates a similar pattern, but is rather based on coalescence of branches in phylogenetic trees (Pons et al. 2006). The term reverse taxonomy describes the identification of specimens by analyzing their genetic information before carrying out a taxonomic investigation using morphological methods (Markmann and Tautz 2005). The first use of reverse-taxonomy in the CCZ using part of the mitochondrial cytochrome c oxidase 1 (CO1) gene focused on the macrofaunal taxa Isopoda and Polychaeta in two areas approx. 1300 km apart (Janssen et al. 2015). Other taxa that have been investigated using this molecular approach in addition to morphological examination include Tanaidacea (Jakiel et al. 2019), Ophiuroidea (Christodoulou et al. 2020) and Amphipoda (Mohrbeck et al. 2021; Jażdżewska et al. 2022).
In contrast to molecular barcoding, Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS) investigates the proteome fingerprint, i.e. the mass and amount of a subset of cell molecules comprised mainly of small proteins and peptides. Although MALDI-TOF MS is most commonly used in microbiology (Singhal et al. 2015), it also proved to be useful in distinguishing a large variety of different marine metazoan taxa such as isopods, cnidarians, mollusks and fish (Holst et al. 2019; Rossel et al. 2020; Wilke et al. 2020; Paulus et al. 2022; Korfhage et al. 2022; Kürzel et al. 2022). For copepods, it has successfully been used to discriminate a large pool of harpacticoid meiobenthic copepods (Rossel and Martínez Arbizu 2018a, 2019) as well as calanoid epipelagic (Laakmann et al. 2013), mesopelagic (Bode et al. 2017) and bentho-pelagic (Renz et al. 2021) copepods. This method is especially useful as a monitoring tool due to its relatively fast processing times and low costs per investigated specimen (for further details, see Rossel et al. 2019). An automatic discrimination of species based on the mass spectra derived from MALDI-TOF MS based on different clustering approaches has successfully been applied for biodiversity estimates of harpacticoid copepods (Rossel and Martínez Arbizu 2020) and calanoid copepods (Renz et al. 2021).
In addition to the fact that there is no need for comprehensive taxonomic expertise and that processing times are significantly faster compared to morphological investigations, another advantage of DNA barcoding and MALDI-TOF MS is that both methods can be used to identify operational taxonomic units (OTUs) instead of validated species. Morphological OTUs are often error-prone (Janssen et al. 2015) and identification catalogues are rarely available even for the larger megafauna organisms (Horton et al. 2021). Identification to the species level is thus rare for deep-sea meiofauna. Even the most abundant taxon Nematoda is usually only determined to the genus level in the CCZ (Miljutina et al. 2010; Singh et al. 2016; Hauquier et al. 2019). Despite difficulties for some taxa, molecular methods can aid to identify potential species and, hence, allow for a higher resolution of biodiversity assessments. Also, molecular identifiers are usually archived in repositories such as BOLD for DNA barcodes (Ratnasingham and Hebert 2007) or Dryad for mass spectrometry data. Hence, the identification can later be verified or the taxonomic resolution can be improved based on future species descriptions. Most importantly, identifications can more easily be matched between studies. As deep-sea mining in the polymetallic nodule fields of the CCZ is approaching at a rapid pace, the development of appropriate tools to monitor the (changing) environment is vital. Hence, in this study, we aim at (i) evaluating the applicability of proteome fingerprinting for biodiversity estimation and (ii) estimating species richness of meiobenthic harpacticoids in the CCZ using proteome fingerprinting and COI barcodes. To evaluate the confidence in biodiversity prediction using these molecular markers, we inter-compare different approaches on species delimitation for both methods, i.e. the distance-based ABGD and the tree-based GMYC method for the COI data as well as two different clustering approaches (partitioning around medoids and consensus clustering) for the proteomic spectra.
Material and methods
Sample collection and preparation
Sediment sampling in the CCZ was conducted using a multicorer during the cruises MANGAN 2018 (SO262: 05/04 to 29/05/2018, Rühlemann and Shipboard Scientific Party 2019) and MiningImpact2 (SO268/2: 30/03 to 22/05/2019, Haeckel and Linke 2021), both on the German research vessel SONNE (Fig. 1). The study area is located within the eastern part of the German contract area for the exploration of polymetallic nodules, which has been licensed by the German Federal Institute for Geosciences and Natural Resources (BGR) from the International Seabed Authority (ISA). In 2018, 16 biological sampling sites within four clusters of environmental grid layers were randomly chosen from a relatively small, ca. 5 × 5 km sampling area (see Fig. 1) using the R package vegan (Oksanen et al. 2017). The spatial layers of environmental variables used to distinguish as many different habitats within the study area as possible were based mainly on bathymetry and backscatter values (Uhlenkott et al. 2019). In addition, nine sites were manually selected to include the full range of bathymetric conditions and oceanographic characteristics that could, for example influence the potential direction(s) of spreading of a mining-related sediment plume (Uhlenkott et al. 2019). In 2019, meiofauna samples were obtained at eleven sites in close vicinity, about 5 km to the south of the sampling area of 2018 (Fig. 1). An overview of sampling stations is provided in Table 1.
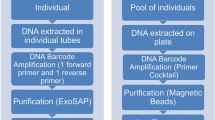
Meiofauna was sampled using multicores with an inner diameter of 94–96 mm. Bottom water was sieved over a 32-μm sieve and fixed with 99.8% ethanol denatured with methyl ethyl ketone together with, in 2018, the upper 3 cm and in 2019 the upper 5 cm of sediment in a Kautex wide-neck bottle (1000 ml). All samples were re-fixed with the same fixative after 24 h and stored at −20°C. To extract all meiofauna organisms from the sediment, samples were centrifuged according to the differential flotation method (Heip et al. 1985) with the colloidal gel Levasil®. Centrifuged samples were transferred into a Kautex wide-neck bottle (100 ml) and further stored at −20°C in the same fixative. All copepods were sorted out of the supernatant under a dissecting microscope. Prior to molecular processing, all individuals were photographed to document their basic morphology, and the ontogenetic stage was determined.
Further processing was conducted according to two different protocols. In the first approach conducted on 58% of all available specimens, the individual was cut into two pieces. The posterior part was used for DNA barcoding, while the anterior part was used for investigations with MALDI-TOF MS. In the second, enhanced protocol conducted on 42% of the specimens, the individuals were first prepared for MALDI-TOF MS and then washed with 10 μl molecular grade water before they were processed for DNA barcoding, to increase biomass used for the MALDI measurements. The change of protocol only influenced the success rate of MALDI-TOF MS but had no influence on the resulting DNA barcode or the mass spectrum. Furthermore, the exuviae could be retained for potential morphological investigations in the future.
DNA barcoding
DNA was extracted in 20 μl chelex (InstaGene Matrix, Bio-Rad) for 50 min at 56°C, followed by a denaturation of the enzymes for 10 min at 96°C. PCR was conducted directly using the extract as DNA template with Accu Start (2× PCR master mix, Quantabio) (Suppl. Tab. 1) in a 20-μl starting solution. A variety of primers and primer combinations was used (Table 2). The success of all reactions was checked on a 1%-agarose gel; all PCR-products that produced a band were sent to Macrogen Europe, Amsterdam, Netherlands, for sequencing on an Applied Biosystems 3730XL sequencer. A negative control was used in all PCR runs.
Resulting sequencing reads were assembled in Geneious R7 v. 7.0.6. and checked for contamination (e.g. bacteria, fungi, non-crustacean taxa) using the basic local alignment search tool (BLAST) (Altschul et al. 1997). Subsequently, sequences were aligned in SeaView (Gouy et al. 2010) using the MUSCLE algorithm (Edgar 2004). Alignments were checked and misalignments by the algorithm were corrected manually. Possible nuclear mitochondrial DNA segments (NUMTs) were discarded by excluding sequences containing stop codons.
MALDI-TOF MS
The tissue was transferred with 5-μl ethanol into a 0.2-ml microcentrifuge tube. After the ethanol had evaporated at room temperature, 2.5-μl α-cyano-4-hydroxycinnamic acid (HCCA) was added and the tissue was incubated for at least 5 min. Thereafter, the extract with the HCCA was transferred to a metallic target plate and measured in a Microflex LT/SH System (Bruker Daltonics) using the method MBTAuto. Peak evaluation was carried out in a mass peak range between 2 and 10 k Dalton (Da) using a centroid peak detection algorithm, a signal-to-noise threshold of 2 and a minimum intensity threshold of 600. To create a sum spectrum, 160 satisfactory shots were summed up.
Raw spectra were imported to R and further processed using the R-packages MALDIquantForeign (Gibb 2015) and MALDIquant (Gibb and Strimmer 2012). Spectra were square-root transformed, smoothed using the Savitzky Golay method (Savitzky and Golay 1964), baseline corrected using the Statistics-sensitive Non-linear Iterative Peak-clipping algorithm (SNIP) (Ryan et al. 1988) and spectra normalized using the Total Ion Current (TIC) method. Repeated measurements were averaged by using mean intensities.
Signal-to-noise (SNR) cut-off values and peak detection half window size (HWS) were optimized by comparing unsupervised (without a reference library) biodiversity estimation using partitioning around medoids (PAM) sensu Rossel and Martínez Arbizu (2020) to the results of the DNA-based method ABGD. SNR and HWS values were chosen based on the highest adjusted rand index (ARI) between the PAM clustering calculated using the R-package mclust (Scrucca et al. 2016) and ABGD. A higher SNR-value will discard mass peaks of low intensity that are considered to be noise. A high SNR will result in more discarded mass peaks and thus in less mass peaks in the final dataset. The highest mass peak within a certain HWS will be picked as the most relevant mass peak in this range. A smaller HWS will result in more mass peaks in the final dataset.
Due to the current lack of standard objective methods, quality control was mainly carried out by expert opinion. In that context, mass spectra with strong noise, low peak intensities or an exceedingly low number of peaks were discarded.
Assessment of species richness
We applied four different unsupervised species delimitation approaches, two based on DNA barcoding and two based on proteomic spectra. To assign DNA sequences to molecular operational taxonomic units (MOTUs), ABGD was applied on the whole dataset (Puillandre et al. 2012) using the default setting on the ABGD web application (https://bioinfo.mnhn.fr/abi/public/abgd/abgdweb.html). As another method for species delimitation, GMYC analysis (Pons et al. 2006) was used by creating an ultrametric tree in BEAST. This tree was then used for GMYC analysis in R using the package splits (R-Core-Team, 2018; Ezard et al. 2021).
Using mass spectra derived from MALDI-TOF MS, PAM clustering previously tested on harpacticoid copepods by Rossel and Martínez Arbizu (2020) was applied using Hellinger-transformed data (Legendre and Gallagher 2001). For dimensionality reduction, a principal component analysis (PCA) was carried out on the data using command “prcomp” in R. We applied the silhouette index (Rousseeuw 1987) as an internal validation measure for the optimal clustering result. The silhouette analysis uses the difference between normalized separation, i.e. minimum of pairwise distances between clusters, and compactness, i.e. maximum of pairwise distances within the clusters. For each data point, a silhouette width is calculated and the average of these widths then provides the validation criterion. The largest silhouette was chosen as the best estimation for proteomic operational taxonomic units (POTUs) (Fig. 2a).

a Plot of silhouette values obtained by PAM clustering. Each dot represents the silhouette value (y-axis) obtained from a certain number of clusters (x-axis; varying from 2 to 726). The highest value was obtained for 499 clusters and is marked in red. b Ambiguous clustering plot for the designation of the number of stable clusters from consensus clustering. Each dot represents the proportion of ambiguous clustering (y-axis) per number of clusters (x-axis). The optimal number of clusters is marked in red
To evaluate stability and reproducibility of POTU delimitation, a second clustering approach was applied using consensus clustering based on hierarchical clustering (HC) with single linkage using the R-package ConsensusClusterPlus (Wilkerson and Hayes 2010) as applied in a previous study by Renz et al. (2021). A consensus matrix was calculated based on 100 repetitions of HC using Euclidean distance of Hellinger-transformed peak intensities based on 80% of randomly chosen compounds. Outer clustering of the consensus matrix was repeated using HC with single linkage. The number of stable and reproducible clusters was inferred from the consensus analysis using the proportion of ambiguous clustering (PAC) as internal validation measure (Șenbabaoğlu et al. 2014). PAC is defined as the fraction of sample pairs with consensus values in the interval above 0 (i.e. sample pairs that are never in the same cluster) and below 1 (i.e. sample pairs that are always in the same cluster). In a truly stable clustering, a consensus matrix contains only 0 and 1, and the PAC would have a score of 0. Here, we used 0.1 as lower and 0.9 as upper limit. From this, we inferred the number (n) of stable clusters by visual inspection of the first distinct minimum, i.e. when the difference in PAC between n and n+1 clusters approaches 0 (Fig. 2b).
For every resulting copepod community, Shannon’s diversity (H’) (Shannon 1948) using the ‘diversity’ function from the R-package vegan (Oksanen et al. 2017) and Pielou’s evenness (Pielou 1966) were calculated as a measure of biodiversity. Finally, communities based on DNA and MALDI-TOF data were compared using a Mantel test (Mantel 1967) applied to distance matrices calculated from community data.
Rarefaction curves were computed using the ‘rarecurve’ function from the R-package vegan (step = 20, sample = 100).
Inter-comparison of the species delimitation approaches
To estimate variability between the species delimitation approaches, all resulting species clusters were compared by calculating the ARI using the R-package mclust (Scrucca et al. 2016). Except for direct comparisons between two species delimitation tools, the DNA-based method ABGD was used as baseline identification, as methods for delimitation of proteomic data as well as ABGD are similarity based, making these more comparable. Thus, the ARI was always calculated in relation to ABGD results. Additionally, the SNR and the HWS were varied to investigate ARI and biodiversity variability in relation to data processing.
Results
MOTUs (Molecular Operational Taxonomic Units)
DNA sequences were obtained for 1296 out of 2115 copepod specimens. Applying ABGD resulted in 718 MOTUs (H’=6.30, J=0.96) (Table 3). Intraspecific JC69 distances for these MOTUs ranged from 0 to 0.15 (mean=0.02), and interspecific distances from 0.11 to 0.42 (mean=0.20). GMYC produced 794 MOTUs (H’=6.44, J=0.96) (Table 3).
From the 2115 specimens used in our study, quality-controlled MALDI mass spectra were retained for 1445 specimens. However, both COI and MALDI information was available for a subset of 727 specimens, which were used for the comparison of methods. For these, ABGD resulted in 440 MOTUs (H’=5.84, J=0.96) and GMYC in 489 MOTUs (H’=5.60, J=0.97) (Table 3).
Quality control and processing of proteomic spectra
Using the dataset of 727 specimens for which both COI and MALDI information was available, we tested how data processing steps influence the accuracy of unsupervised biodiversity estimation using PAM clustering sensu Rossel and Martínez Arbizu (2020) based on the protein mass spectra in consistency with cluster formation based on the ABGD approach. We found that varying SNR and HWS had a major impact on estimation accuracy. However, the adjusted Rand index (ARI) of ABGD delimitation and PAM was found to span a narrow range from 0.48 to 0.56. The best ARI was found for SNR = 10 and HWS = 20.
POTUs (Proteomic Operational Taxonomic Units)
PAM clustering sensu Rossel and Martínez Arbizu (2020) of the 727 specimens with concurrent COI barcodes resulted in 499 POTUs (H’=6.06, J=0.98) (Table 3; Fig. 2a). Determination of POTUs using hierarchical clustering with consensus clustering (HC_CC) after Renz et al. (2021) resulted in 527 clusters (H’=6.09, J=0.97) (Fig. 2b).
Evaluation of clustering approaches
Consistency between the PAM-based POTU clusters and AGBD-based MOTU clusters described above was evaluated by adjusted rand (ARI = 0.57) (Table 3) and a detailed comparison of cluster composition. Over 70% of MOTU clusters with less than five specimens were correctly identified using proteome fingerprinting, while MOTUs containing more specimens were not retained completely. Figure 3 displays where wrong assignments occur frequently. Species clusters containing larger numbers of specimens are often split into smaller fractions, albeit retaining large numbers of conspecific specimens in the same clusters. Singletons often cluster non-specifically into other clusters. A Mantel test carried out on distance matrices from community tables resulting from PAM clustering and ABGD delimitation resulted in an r-value of 0.56 (p=0.001). Stability of clusters based on proteome patterns was evaluated using a consensus cluster approach based on HC after Renz et al. (2021). In total, 527 robust clusters (H’=6.09, J=0.97) were identified with an ARI of 0.60, with a high number of specimens being wrongly identified as singletons (n=215).

Alluvial plot of “relocation” of specimens, comparing ABGD species delimitation (middle) to the unsupervised delimitations based on MALDI-TOF MS data (left and right). Blue lines indicate that a specimen identified as a MALDI POTU is also correspondingly observed as ABGD MOTU. Red lines indicate relocation into a POTU with less than 50% of specimens found in its assigned ABGD MOTU. Magenta, turquoise and light blue indicate clustering with at least 50%, 66% or 80% of conspecific specimens, respectively
Even though occasional clustering of conspecific specimens of different ontogenetic stages was observed, this was not found to be a consistent pattern throughout all species. Such conspecific specimens were also frequently grouped into different clusters. These often differed by the average number of mass peaks (e.g. Fig. 4) and maximum intensities of the respective mass spectra.

ABGD MOTU 017 was found to be the species with the most specimens based on DNA barcoding, but was divided into three clusters by PAM clustering of proteomic data (a, c, d). Mass spectra of specimens displayed in a and c are visually similar, whereas those of d are quite different. The difference in average mass-peak number of these groups is displayed in b. a Cluster 78; c Cluster 65; d Cluster 472
Impact of data quality
The formation of clusters based on the intensity and the number of peaks indicates a large influence of mass spectra quality on the POTU delimitation. Since objective quality control tools are not available, a stricter visual quality control was carried out and the dataset was reduced to a total of 321 specimens (n = 235 species) to test if the accuracy of unsupervised delimitation methods can be enhanced using only high-quality mass spectra (Table 3). Again, SNR and HWS were varied to investigate the range of accuracy and to examine data-processing influence.
The ARI based on PAM ranged from 0.45 to 0.79 with the best ARI being detected at HWS = 10 and SNR = 19. This resulted in highly similar Shannon diversities (H’=5.37, J=0.98, nspec=243) in comparison to ABGD (H’=5.31, J=0.97). HC_CC resulted in a total number of 286 POTUs, with an ARI of 0.50. Shannon diversity (H’=5.59) and Pielou’s evenness (J=0.99) based on this method were a bit higher, especially because species with two specimens are frequently split into singleton clusters (Fig. 5, right side). In general, results from this high-quality dataset showed greater overlaps with barcoding delimitation (Fig. 5). A Mantel test carried out on distance matrices from community tables resulting from PAM clustering and ABGD delimitation resulted in a high r-value of 0.73 (p=0.001).

Alluvial plot of “relocation” of specimens for a reduced dataset after a strict visual quality control (see text for details). ABGD species delimitation (middle) is compared to the unsupervised delimitations based on MALDI-TOF MS data (left and right). Blue lines indicate that a specimen identified as a MALDI POTU is also correspondingly observed as ABGD MOTU. Red lines indicate relocation into a POTU with less than 50% of specimens found in its assigned ABGD MOTU. Magenta, turquoise and light blue indicate clustering with at least 50%, 66% or 80% of conspecific specimens, respectively
Implications of data processing for biodiversity assessments
Due to the lack of reference information (e.g. morphological identification or some molecular identifier) in studies where MALDI-TOF MS is applied as a standalone method, data processing (i.e. peak identification) cannot be optimized in a standardized way. Therefore, variability of evenness and biodiversity from PAM clustering were investigated varying the processing steps with the highest impact on results (SNR and HWS). Irrespective of data processing, results of PAM clustering are in high agreement with genetic results (Fig. 6). MALDI-TOF results from HC_CC overestimated diversity and evenness due to the high number of singletons compared to genetic tools.

Comparisons between adjusted rand index (ARI), Pielou’s evenness (J, in blue) and Shannon diversity (H’, in red) obtained by PAM clustering from datasets in which HWS and SNR were varied. Triangles display the corresponding value from HC_CC in the optimized dataset according to the comparison of PAM clustering and ABGD. Diamonds represent values obtained from GMYC. Lines are values obtained from ABGD species delimitation
Estimation of biodiversity
In total, a dataset of 1445 copepod specimens was analyzed by MALDI-TOF (Table 3). This dataset includes the 727 specimens with a concurrent barcode reference as described in the previous sections. For the remaining 718 specimens, a reference was not available. Using PAM clustering on this complete dataset, 815 POTUs were defined (H’=6.49; J=0.97; Table 3), while HC_CC resulted in 1023 POTUs and subsequently in H’=6.70 and J=0.97 (Table 3). This fits well with the high biodiversity and evenness values obtained for the previously analyzed datasets.
Furthermore, a dataset comprising 1296 DNA barcodes of benthic copepods from the sampling area was analyzed using ABGD and GMYC (Table 3). This dataset includes the 727 specimens for which a MALDI-TOF mass spectrum was also available. ABGD obtained 718 MOTUs with H’=6.30 and J=0.96. GMYC, on the other hand, produced 794 MOTUs with H’=6.44 and J=0.96.
Rarefaction curves based on the different methods applied in this study to the different datasets described above do not reach an asymptote and all emphasize that we are far from having discovered all species to be expected in the study area (Fig. 7).

Rarefaction curves generated for all tested datasets in this study. Numbers represent the size of the dataset for which the corresponding method applies (HC_CC; PAM; DNA_GMYC; DNA_ABGD). For each method, the number of defined species is shown in relation to the number of specimens analyzed
Discussion
The analyses carried out in this study show that unsupervised biodiversity assessment of deep-sea benthic copepods based on proteome fingerprinting is possible and is in general agreement with DNA-based methods. Hence, MALDI-TOF MS can be regarded as a potential tool for accelerated and standardized assessment of biodiversity, in addition to DNA barcoding which has already been applied in several studies on benthic biodiversity in the CCZ (Janssen et al. 2015; Jakiel et al. 2019; Christodoulou et al. 2020). So far, no assessment method applied in the CCZ was able to identify all collected specimens. However, it is important to mention that juvenile copepods were also identified in our approach. These are often not considered in morphological studies, although they make up one- to two-thirds of the specimens. In our study, a MALDI spectrum was successfully obtained for 68% of the specimens, whereas 61% of the specimens provided a DNA barcode. In previous studies from the CCZ, the success rate in obtaining COI-barcodes varied between 17 and 26% for benthic polychaetes (Janssen et al. 2019; Bonifácio et al. 2020), but was higher for Isopoda (42%, Janssen et al. 2019) and Ophiuroidea (57%, Christodoulou et al. 2020). One possibility to potentially handle these constraints in the comparative use of DNA barcoding and MALDI-TOF MS for biodiversity assessment would be to work with a pre-defined number of specimens. This method has also been applied to avoid unbalanced datasets in photographic surveys of benthic megafauna (Simon-Lledó et al. 2019). In this regard, the number of investigated specimens could be set in relation to the overall abundance of the analyzed taxa, as commonly applied in the investigation of nematodes (Hauquier et al. 2019).
The success rate of MALDI-TOF measurements on copepods (68%) was low compared to other studies, which usually reached 95 (Kaiser et al. 2018) to 100% (Renz et al. 2021). A high net efficiency identification rate due to low measurement failures coupled with relatively high accuracy has been discussed as a strong advantage of proteomic fingerprinting for species delimitation in copepods (Renz et al. 2021). The fact that we did not obtain mass spectra from 32% of our specimens in this study might be related to their small size and thus the small amount of biomass available for analyses. Additionally, some specimens were cut into separate parts for simultaneous DNA and MALDI analyses, which further reduced the amount of material available for the smaller specimens. Furthermore, juvenile stages were analyzed that induced additional biomass differences within one species. Subsequently, low biomasses may be a central cause for false partitioning of species into different clusters. This insufficient amount of biomass might also be an explanation for the variations in the mass-peak number for some specimens, which probably lead to larger Euclidean distances between specimens with different peak numbers and thus to their assignment to different clusters. This effect may have specifically influenced consensus clustering, where clustering based on subsets of markers resulted in higher dissimilarity between conspecific specimens and the prediction of a higher number of singleton clusters (Renz et al. 2021).
Previous studies on various animal groups such as insects, arachnids and crustaceans have already highlighted the influence of sample storage on protein mass spectra used for supervised identification (Mathis et al. 2015; Nebbak et al. 2017; Rossel and Martínez Arbizu, 2018b), in which identification success decreases with sample and data quality. This has also been emphasized by studies measuring the freshness of samples based on MALDI-TOF measurements (Ulrich et al. 2017). However, these studies have also shown that quality decrease does not always affect all specimens in a sample in the same way, resulting in good identification for some specimens while others could not be measured successfully anymore (Rossel and Martínez Arbizu 2018b). Although all samples were fixed in highly concentrated ethanol and stored at −20°C in this study, it cannot be excluded that reduced data quality might be related to degradation in some samples and specimens.
Even though results from different techniques as well as species delimitation methods are not completely congruent, performance as well as the resulting delimitation of taxonomic units is generally similar. Comparing the POTU delimitation of the high-quality proteome fingerprint dataset using the PAM algorithm to the MOTU delimitation using ABGD accounted for an ARI of 0.79. The comparison of GMYC and ABGD led to an ARI of only 0.84, although these delimitations are based on an identical dataset. Thus, with good-quality measurement as a prerequisite, proteome-based data are generally capable of providing an accurate picture of species richness, which is also supported on community level by the Mantel test. Also, even though high overlap of species defined by proteome fingerprinting and by COI DNA barcoding was shown various times (Bode et al. 2017; Rossel and Martínez Arbizu 2019; Renz et al. 2021; Yeom et al. 2021), none of the methods can claim to actually show natural species boundaries. Different methods can, even applied to the same data, return different species boundaries as is shown by the comparison of GMYC and ABGD. This is even more likely when delimitation methods rely on different kinds of data such as ABGD relying on DNA and PAM clustering relying on proteomic fingerprints. Furthermore, although the high overlap between genetic and proteomic data in the smallest dataset probably originates from better data quality, the difference in cluster sizes may also play a role, since the higher-quality dataset did not contain species equivalents with more than seven specimens.
Generally, the high number of rare MOTUs/POTUs and singletons poses difficulties on all species delimitation tools. The applied unsupervised methods depend on clustering approaches and, thus, on distances between and within clusters. If the cluster is only composed of a single specimen, no variability within the cluster can be derived and, hence, no distance within the cluster. Using MALDI-TOF MS data, no hard thresholds for within and between species variability were reported so far, in contrast to barcoding genes such as COI. Here, species were previously delimited solely based on percentage sequence divergence, e.g. in the program CD-HIT (Huang et al. 2010). A potential reason for the difficulty to define MALDI-TOF-based species delimitations is the higher variability of mass spectra signals, in turn originating from multiple factors such as ecological differences (Karger et al. 2019), quality disparities (Rossel and Martínez Arbizu 2018b) but also differences in data processing. Also, there is potential for more variability due to varying signal intensities and slight differences of masses in certain molecules in comparison to DNA, which can only exhibit four different possible stages at fixed positions along a sequence. Clearly, changing cut-off values such as the SNR and peak-picking half-window sizes will, even if only slightly, alter mass spectra and thus also distances between specimens. Hence, results of unsupervised delimitation tools such as the two clustering methods applied in this study (Rossel and Martínez Arbizu 2020; Renz et al. 2021) have to be treated with care. If proteome fingerprinting is applied as a stand-alone method for biodiversity assessment, optimization of the mass spectrometry data based on comparison with the DNA-based delimitation method ABGD for reference as applied in the study described here is not possible. However, our results suggest that even though data processing influences the outcome, the variability is not immensely high. Furthermore, general diversity as well as species evenness is relatively stable and similar compared to DNA-based results. These results are very promising and invite the inclusion of mass spectrometry data in reverse taxonomy approaches, which have been proposed as an acceleration and simplification of benthic-specimen and species identification in the CCZ (Janssen et al. 2015; Glover et al. 2016).
Economic interest in the polymetallic nodules of the CCZ is steadily increasing, due to their high content of metals such as manganese, nickel, copper and cobalt (Kuhn et al. 2017; Hein et al. 2020). Currently (2022), the International Seabed Authority has already issued 17 licenses for the exploration of polymetallic nodules in the CCZ. In this context, it is especially important to develop effective, fast and low-cost methods to initially investigate the baseline and potentially later to monitor the benthic fauna exposed to mining activities (Lins et al. 2021). MALDI-TOF MS could be especially attractive for quantitative assessments (specimen-by-specimen) of benthic communities as it does not require an extensive taxonomic knowledge and is considerably cheaper than DNA barcoding (Rossel et al. 2019). Furthermore, MALDI-TOF MS data can be used not only to distinguish species, but also the developmental stages within species (Rossel et al. 2022). Hence, it might be a useful, additional tool to monitor resettlement and dispersion strategies of individual taxa at impacted sites.
In this context, our unsupervised species delimitations can also be regarded as the first step towards the development of a mass-spectra reference library of meiobenthic Copepoda of the CCZ. However, considerations on the best form of data deposition of mass spectra are still ongoing, while DNA barcodes can easily be deposited and accessed via the BOLD database (Ratnasingham and Hebert 2007), which is also linked to GenBank (Benson et al. 2012), the most established database for sequence data. Should specimen identification based on MALDI-TOF MS be applied on a large scale, for example for monitoring purposes, a data repository for data from the CCZ will become mandatory to enable comparisons. Furthermore, the proteomic approach using MALDI-TOF MS requires well-adapted standardization. We were able to show that varying values such as SNR and HWS can have a major impact on species delimitation and biodiversity estimation. This optimization of the workflow, however, can only be carried out and adapted using morphological, genetic or other delimitation methods.
Conclusion
Unsupervised biodiversity assessments using MALDI-TOF MS can accelerate laborious morphological identifications in the context of biodiversity assessments of meiofauna in the CCZ and can be especially useful for the analysis of impacts associated with potential future deep-sea mining. We have shown that for benthic copepods, estimations of biodiversity, evenness and species numbers in comparison to those derived from DNA barcoding data are very similar, provided that the mass spectrometry data are of good quality. Still, variability in mass spectra quality is one of the main factors influencing the resulting species delimitation and needs to be further investigated and improved in the future. In our study area within the CCZ, with a high diversity of copepod species, we obtained a high number of singletons despite our large dataset of over 2000 specimens. In this context, the application of unsupervised methods needs to be undertaken with caution. To not only rely on unsupervised methods, future work needs to aim at collecting reference data for DNA-based as well as MALDI-TOF-based studies to allow species identification to the lowest possible taxonomic level.
During routine monitoring surveys where mere identifications of specimens are required, proteomic fingerprinting can be a valuable alternative to DNA barcoding. The low costs allow a far more extensive assessment compared to costly DNA barcoding. Also, there is no need to find a specific set of primers as the standard preparation was successfully tested for a variety of animal species, again saving costs and time. Our study shows that the assessment nevertheless has the capability to provide accurate species identifications once further optimized.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J et al (2012) GenBank. Nucleic Acids Res 41:D36–D42. https://doi.org/10.1093/nar/gks1195
Bode M, Laakmann S, Kaiser P, Hagen W, Auel H, Cornils A (2017) Unravelling diversity of deep-sea copepods using integrated morphological and molecular techniques. J Plankton Res 39:600–617. https://doi.org/10.1093/plankt/fbx031
Bonifácio P, Martínez Arbizu P, Menot L (2020) Alpha and beta diversity patterns of polychaete assemblages across the nodule province of the eastern Clarion-Clipperton Fracture Zone (equatorial Pacific). Biogeosciences 17:865–886. https://doi.org/10.5194/bg-17-865-2020
Brix S, Osborn KJ, Kaiser S, Truskey SB, Schnurr SM, Brenke N et al (2020) Adult life strategy affects distribution patterns in abyssal isopods – implications for conservation in Pacific nodule areas. Biogeosciences 17:6163–6184. https://doi.org/10.5194/bg-17-6163-2020
Cheng F, Wang M, Sun S, Li C, Zhang Y (2013) DNA barcoding of Antarctic marine zooplankton for species identification and recognition. Adv Polar Sci 24:119–127. https://doi.org/10.3724/SP.J.1085.2013.00119
Christodoulou M, O’Hara T, Hugall AF, Khodami S, Rodrigues CF, Hilario A et al (2020) Unexpected high abyssal ophiuroid diversity in polymetallic nodule fields of the northeast Pacific Ocean and implications for conservation. Biogeosciences 17:1845–1876. https://doi.org/10.5194/bg-17-1845-2020
Christodoulou M, O’Hara TD, Hugall AF, Martínez Arbizu P (2019) Dark ophiuroid biodiversity in a prospective abyssal mine field. Curr Biol 29:3909–3912.e3. https://doi.org/10.1016/j.cub.2019.09.012
Clark AL, Cook Clark J, Pintz S (2013) Towards the developement of a regulatory framework for polymetallic nodule exploitation in the area. ISA Technical Study Series 11, International Seabed Authority, Kingston, Jamaica.
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Ezard T, Fujisawa T, Tim B (2021) splits: SPecies’ LImits by Threshold Statistics. Available at: https://R-Forge.R-project.org/projects/splits/
Folmer O, Black MB, C, V. R. (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3:294–299
Geller J, Meyer C, Parker M, Hawk H (2013) Redesign of PCR primers for mitochondrial cytochrome c oxidase subunit I for marine invertebrates and application in all-taxa biotic surveys. Mol Ecol Resour 13:851–861. https://doi.org/10.5194/bg-17-1845-2020
George KH, Veit-Köhler G, Martínez Arbizu P, Seifried S, Rose A, Willen E et al (2014) Community structure and species diversity of Harpacticoida (Crustacea: Copepoda) at two sites in the deep sea of the Angola Basin (Southeast Atlantic). Org Divers Evol 14:57–73. https://doi.org/10.1007/s13127-013-0154-2
Gibb S (2015) MALDIquantForeign: import/export routines for MALDIquant. A package for R. https://CRAN.R-project.org/package=MALDIquantForeign
Gibb S, Strimmer K (2012) MALDIquant: quantitative analysis of mass spectrometry data. Bioinformatics 28:2270–2271. https://doi.org/10.1093/bioinformatics/bts447
Glover AG, Dahlgren TG, Wiklund H, Mohrbeck I, Smith CR (2016) An end-to-end DNA taxonomy methodology for benthic biodiversity survey in the Clarion-Clipperton Zone, central Pacific abyss. J Mar Sci Eng 4:2. https://doi.org/10.3390/jmse4010002
Gollner S, Kaiser S, Menzel L, Jones DOB, Brown A, Mestre NC et al (2017) Resilience of benthic deep-sea fauna to mining activities. Mar Environ Res 129:76–101. https://doi.org/10.1016/j.marenvres.2017.04.010
Gouy M, Guindon S, Gascuel O (2010) SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27:221–224. https://doi.org/10.1093/molbev/msp259
Haeckel M, Linke P (2021) RV SONNE Fahrtbericht / Cruise Report SO268: assessing the impacts of nodule mining on the deep-sea environment. Berichte aus dem GEOMAR Helmholtz Zenrtum für Ozeanforschung Kiel 59:1–802
Hauquier F, Macheriotou L, Bezerra TN, Egho G, Martínez Arbizu P, Vanreusel A (2019) Distribution of free-living marine nematodes in the Clarion–Clipperton Zone: implications for future deep-sea mining scenarios. Biogeosciences 16:3475–3489. https://doi.org/10.5194/bg-16-3475-2019
Hebert PDN, Ratnasingham S, deWaard JR (2003) Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc Biol Sci 270(Suppl 1):S96–S99. https://doi.org/10.1098/rsbl.2003.0025
Hein JR, Koschinsky A, Kuhn T (2020) Deep-ocean polymetallic nodules as a resource for critical materials. Nat Rev Earth Environ 1:158–169. https://doi.org/10.1038/s43017-020-0027-0
Heip C, Vincx M, Vranken G (1985) The ecology of marine nematodes. Oceanography and Marine Biology: an annual review (1985).
Herzog S, Amon DJ, Smith CR, Janussen D (2018) Two new species of Sympagella (Porifera: Hexactinellida: Rossellidae) collected from the Clarion-Clipperton Zone, East Pacific. Zootaxa 4466:152–163. https://doi.org/10.11646/zootaxa.4466.1.12
Holst S, Heins A, Laakmann S (2019) Morphological and molecular diagnostic species characters of Staurozoa (Cnidaria) collected on the coast of Helgoland (German Bight, North Sea). Mar Biodivers 49:1775–1797. https://doi.org/10.1007/s12526-019-00943-1
Horton T, Marsh L, Bett BJ, Gates AR, Jones DOB, Benoist NMA et al (2021) Recommendations for the standardisation of open taxonomic nomenclature for image-based identifications. Front Mar Sci 8:620702. https://doi.org/10.3389/fmars.2021.620702
Huang Y, Niu B, Gao Y, Fu L, Li W (2010) CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26:680–682. https://doi.org/10.1093/bioinformatics/btq003
Jakiel A, Palero F, Błażewicz M (2019) Deep ocean seascape and pseudotanaidae (crustacea: tanaidacea) diversity at the clarion-clipperton fracture Zone. Sci Rep 9:17305. https://doi.org/10.1038/s41598-019-51434-z
Janssen A, Kaiser S, Meißner K, Brenke N, Menot L, Martínez Arbizu P (2015) A reverse taxonomic approach to assess macrofaunal distribution patterns in abyssal Pacific polymetallic nodule fields. PLoS ONE 10:e0117790. https://doi.org/10.1371/journal.pone.0117790
Janssen A, Stuckas H, Vink A, Martínez Arbizu P (2019) Biogeography and population structure of predominant macrofaunal taxa (Annelida and Isopoda) in abyssal polymetallic nodule fields: implications for conservation and management. Mar Biodivers 49:2641–2658. https://doi.org/10.1007/s12526-019-00997-1
Jażdżewska AM, Brandt A, Martínez Arbizu P, Vink A (2022) Exploring the diversity of the deep sea—four new species of the amphipod genus Oedicerina described using morphological and molecular methods. Zool J Linnean Soc 194:181–225. https://doi.org/10.1093/zoolinnean/zlab032
Jones DO, Ardron JA, Colaço A, Durden JM (2020) Environmental considerations for impact and preservation reference zones for deep-sea polymetallic nodule mining. Mar Policy 118: 103312. https://doi.org/10.1016/j.marpol.2018.10.025
Jones DO, Kaiser S, Sweetman AK, Smith CR, Menot L, Vink A et al (2017) Biological responses to disturbance from simulated deep-sea polymetallic nodule mining. PLoS One 12:e0171750. https://doi.org/10.1371/journal.pone.0171750
Kaiser P, Bode M, Cornils A, Hagen W, Martínez Arbizu P, Auel H et al (2018) High-resolution community analysis of deep-sea copepods using MALDI-TOF protein fingerprinting. Deep Sea Res Part Oceanogr Res Pap 138:122–130. https://doi.org/10.1016/j.dsr.2018.06.005
Karger A, Bettin B, Gethmann JM, Klaus C (2019) Whole animal matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry of ticks – Are spectra of Ixodes ricinus nymphs influenced by environmental, spatial, and temporal factors? PLoS ONE 14:e0210590. https://doi.org/10.1371/journal.pone.0210590
Korfhage SA, Rossel S, Brix S, McFadden CS, Ólafsdóttir SH, Martínez Arbizu P (2022) Species delimitation of Hexacorallia and Octocorallia around Iceland using nuclear and mitochondrial DNA and proteome fingerprinting. Front Mar Sci 9:838201. https://doi.org/10.3389/fmars.2022.838201
Kuhn T, Rühlemann C (2021) Exploration of polymetallic nodules and resource assessment: a case study from the German contract area in the Clarion-Clipperton Zone of the Tropical Northeast Pacific. Minerals 11:618. https://doi.org/10.3390/min11060618
Kuhn T, Wegorzewski A, Rühlemann C, Vink A (2017) Composition, formation, and occurrence of polymetallic nodules. In: Sharma R (ed) Deep-Sea Mining: Resource Potential, Technical and Environmental Considerations. Springer International Publishing, Cham, pp 23–63. https://doi.org/10.1007/978-3-319-52557-0_2
Kürzel K, Kaiser S, Lörz A-N, Rossel S, Paulus E, Peters J et al (2022) Correct species identification and its implications for conservation using Haploniscidae (Crustacea, Isopoda) in Icelandic waters as a proxy. Front Mar Sci 8:795196. https://doi.org/10.3389/fmars.2021.795196
Laakmann S, Gerdts G, Erler R, Knebelsberger T, Martínez Arbizu P, Raupach MJ (2013) Comparison of molecular species identification for North Sea calanoid copepods (Crustacea) using proteome fingerprints and DNA sequences. Mol Ecol Resour 13:862–876. https://doi.org/10.1111/1755-0998.12139
Legendre P, Gallagher ED (2001) Ecologically meaningful transformations for ordination of species data. Oecologia 129:271–280. https://doi.org/10.1007/s004420100716
Lins L, Zeppilli D, Menot L, Michel LN, Bonifácio P, Brandt M et al (2021) Toward a reliable assessment of potential ecological impacts of deep-sea polymetallic nodule mining on abyssal infauna. Limnol Oceanogr Methods 19:626–650. https://doi.org/10.1002/lom3.10448
Macheriotou L, Guilini K, Bezarra TN, Tytgat B, Nguyen DT, Phuong Nguyen TX et al (2019) Metabarcoding free-living marine nematodes using curated 18S and CO1 reference sequence databases for species-level taxonomic assignments. Ecol Evol 9:1211–1226. https://doi.org/10.1002/ece3.4814
Mahatma R, Martínez Arbizu P, Ivanenko VN (2008) A new genus and species of Brychiopontiidae Humes, 1974 (Crustacea: Copepoda: Siphonostomatoida) associated with an abyssal holothurian in the Northeast Pacific nodule province *. Zootaxa 1866:290–302. https://doi.org/10.5281/zenodo.183905
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Markhaseva EL, Mohrbeck I, Renz J (2017) Description of Pseudeuchaeta vulgaris n. sp.(Copepoda: Calanoida), a new aetideid species from the deep Pacific Ocean with notes on the biogeography of benthopelagic aetideid calanoids. Mar Biodivers 47:289–297. https://doi.org/10.1007/s12526-016-0527-9
Markmann M, Tautz D (2005) Reverse taxonomy: an approach towards determining the diversity of meiobenthic organisms based on ribosomal RNA signature sequences. Philos Trans R Soc Lond Ser B Biol Sci 360:1917–1924. https://doi.org/10.1098/rstb.2005.1723
Mathis A, Depaquit J, Dvovrák V, Tuten H, Bañuls A-L, Halada P et al (2015) Identification of phlebotomine sand flies using one MALDI-TOF MS reference database and two mass spectrometer systems. Parasit Vectors 8:266. https://doi.org/10.1186/s13071-015-0878-2
Mercado-Salas NF, Khodami S, Martínez Arbizu P (2019) Convergent evolution of mouthparts morphology between Siphonostomatoida and a new genus of deep-sea Aegisthidae Giesbrecht, 1893 (Copepoda: Harpacticoida). Mar Biodivers 49:1635–1655. https://doi.org/10.1007/s12526-018-0932-3
Miljutin D, Miljutina M, Messié M (2015) Changes in abundance and community structure of nematodes from the abyssal polymetallic nodule field, Tropical Northeast Pacific. Deep Sea Res Part Oceanogr Res Pap 106:126–135. https://doi.org/10.1016/j.dsr.2015.10.009
Miljutin DM, Miljutina MA, Arbizu PM, Galéron J (2011) Deep-sea nematode assemblage has not recovered 26 years after experimental mining of polymetallic nodules (Clarion-Clipperton Fracture Zone, Tropical Eastern Pacific). Deep Sea Res Part Oceanogr Res Pap 58:885–897. https://doi.org/10.1016/j.dsr.2011.06.003
Miljutina MA, Miljutin DM, Mahatma R, Galéron J (2010) Deep-sea nematode assemblages of the Clarion-Clipperton Nodule Province (tropical north-eastern Pacific). Mar Biodivers 40:1–15. https://doi.org/10.1007/s12526-009-0029-0
Mohrbeck I, Horton T, Jażdżewska AM, Martínez Arbizu P (2021) DNA barcoding and cryptic diversity of deep-sea scavenging amphipods in the Clarion-Clipperton Zone (Eastern Equatorial Pacific). Mar Biodivers 51:26. https://doi.org/10.1007/s12526-021-01170-3
Mohrbeck I, Raupach MJ, Arbizu PM, Knebelsberger T, Laakmann S (2015) High-throughput sequencing—the key to rapid biodiversity assessment of marine metazoa? PLoS One 10:e0140342. https://doi.org/10.1371/journal.pone.0140342
Nebbak A, El Hamzaoui B, Berenger J-M, Bitam I, Raoult D, Almeras L et al (2017) Comparative analysis of storage conditions and homogenization methods for tick and flea species for identification by MALDI-TOF MS. Med Vet Entomol 31:438–448. https://doi.org/10.1111/mve.12250
Niner HJ, Ardron JA, Escobar EG, Gianni M, Jaeckel A, Jones DO et al (2018) Deep-sea mining with no net loss of biodiversity—an impossible aim. Front Mar Sci 5:53. https://doi.org/10.3389/fmars.2018.00053
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H (2017) Vegan: community ecology package. R package version 2.4-5. https://CRAN.R-project.org/package=vegan
Paulus E, Brix S, Siebert A, Martínez Arbizu P, Rossel S, Peters J, Svavarsson J, Schwentner M (2022) Recent speciation and hybridization in Icelandic deep-sea isopods: An integrative approach using genomics and proteomics. Mol Ecol 31(1):313–330. https://doi.org/10.1111/mec.16234
Pielou EC (1966) The measurement of diversity in different types of biological collections. J Theor Biol 13:131–144. https://doi.org/10.1016/0022-5193(66)90013-0
Pons J, Barraclough TG, Gomez-Zurita J, Cardoso A, Duran DP, Hazell S et al (2006) Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst Biol 55:595–609. https://doi.org/10.1080/10635150600852011
Puillandre N, Lambert A, Brouillet S, Achaz G (2012) ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol Ecol 21:1864–1877. https://doi.org/10.1111/j.1365-294X.2011.05239.x
Ramirez-Llodra E, Brandt A, Danovaro R, De Mol B, Escobar E, German C et al (2010) Deep, diverse and definitely different: unique attributes of the world’s largest ecosystem. Biogeosciences 7:2851–2899. https://doi.org/10.5194/bg-7-2851-2010
Ramirez-Llodra E, Tyler PA, Baker MC, Bergstad OA, Clark MR, Escobar E et al (2011) Man and the last great wilderness: human impact on the deep sea. PLoS One 6:e22588. https://doi.org/10.1371/journal.pone.0022588
Ratnasingham S, Hebert PD (2007) BOLD: the Barcode of life data system (http://www.barcodinglife.org). Mol Ecol Resour 7:355–364. https://doi.org/10.1111/j.1471-8286.2007.01678.x
R-Core-Team (2018) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Renz J, Markhaseva EL, Laakmann S, Rossel S, Martínez Arbizu P, Peters J (2021) Proteomic fingerprinting facilitates biodiversity assessments in understudied ecosystems: a case study on integrated taxonomy of deep sea copepods. Mol Ecol Resour 21:1936–1951. https://doi.org/10.1111/1755-0998.13405
Rossel S, Barco A, Kloppmann M, Martínez Arbizu P, Huwer B, Knebelsberger T (2020) Rapid species level identification of fish eggs by proteome fingerprinting using MALDI-TOF MS. J Proteome 231:103993. https://doi.org/10.1016/j.jprot.2020.103993
Rossel S, Kaiser P, Bode-Dalby M, Renz J, Laakmann S, Auel H, Hagen W, Arbizu PM, & Peters J (2022) Proteomic fingerprinting enables quantitative biodiversity assessments of species and ontogenetic stages in Calanus congeners (Copepoda, Crustacea) from the Arctic Ocean. Molecular Ecology Resources, 00, 1–14. https://doi.org/10.1111/1755-0998.13714
Rossel S, Khodami S, Martínez Arbizu P (2019) Comparison of rapid biodiversity assessment of meiobenthos using MALDI-TOF MS and Metabarcoding. Front Mar Sci 6:659. https://doi.org/10.3389/fmars.2019.00659
Rossel S, Martínez Arbizu P (2018a) Automatic specimen identification of Harpacticoids (Crustacea: Copepoda) using Random Forest and MALDI-TOF mass spectra, including a post hoc test for false positive discovery. Methods Ecol Evol 9:1421–1434. https://doi.org/10.1111/2041-210X.13000
Rossel S, Martínez Arbizu P (2018b) Effects of sample fixation on specimen identification in biodiversity assemblies based on proteomic data (MALDI-TOF). Front Mar Sci 5:149. https://doi.org/10.3389/fmars.2018.00149
Rossel S, Martínez Arbizu P (2019) Revealing higher than expected diversity of Harpacticoida (Crustacea: Copepoda) in the North Sea using MALDI-TOF MS and molecular barcoding. Sci Rep 9:9182. https://doi.org/10.1038/s41598-019-45718-7
Rossel S, Martínez Arbizu P (2020) Unsupervised biodiversity estimation using proteomic fingerprints from MALDI-TOF MS data. Limnol Oceanogr Methods 18:183–195. https://doi.org/10.1002/lom3.10358
Rousseeuw PJ (1987) Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J Comput Appl Math 20:53–65. https://doi.org/10.1016/0377-0427(87)90125-7
Rühlemann C, Shipboard Scientific Party (2019) Geology, Biodiversity and Environment of the German license area for the exploration of polymetallic nodules in the equatorial NE Pacific. Cruise Report of R/V SONNE Cruise MANGAN 2018, BGR, Hannover, 318 pp
Ryan C, Clayton E, Griffin W, Sie S, Cousens D (1988) SNIP, a statistics-sensitive background treatment for the quantitative analysis of PIXE spectra in geoscience applications. Nucl Instrum Methods Phys Res Sect B Beam Interact Mater At 34:396–402. https://doi.org/10.1016/0168-583X(88)90063-8
Sánchez N, González-Casarrubios A, Cepeda D et al. (2022). Diversity and distribution of Kinorhyncha in abyssal polymetallic nodule areas of the Clarion-Clipperton Fracture Zone and the Peru Basin, East Pacific Ocean, with the description of three new species and notes on their intraspecific variation. Mar Biodivers 52:52 https://doi.org/10.1007/s12526-022-01279-z
Sánchez N, Pardos F, Martínez Arbizu P (2019) Deep-sea Kinorhyncha diversity of the polymetallic nodule fields at the Clarion-Clipperton Fracture Zone (CCZ). Zool Anz 282:88–105. https://doi.org/10.1016/j.jcz.2019.05.007
Savitzky A, Golay MJ (1964) Smoothing and differentiation of data by simplified least squares procedures. Anal Chem 36:1627–1639. https://doi.org/10.1021/ac60214a047
Scrucca L, Fop M, Murphy TB, Raftery AE (2016) mclust 5: clustering, classification and density estimation using Gaussian finite mixture models. R J 8:289–317.
Șenbabaoğlu Y, Michailidis G, Li JZ (2014) Critical limitations of consensus clustering in class discovery. Sci Rep 4:6207. https://doi.org/10.1038/srep06207
Shannon CE (1948) A mathematical theory of communication. Bell Syst Tech J 27:379–423. https://doi.org/10.1002/j.1538-7305.1948.tb01338.x
Simon-Lledó E, Bett BJ, Huvenne VAI, Schoening T, Benoist NMA, Jones DOB (2019) Ecology of a polymetallic nodule occurrence gradient: Implications for deep-sea mining. Limnol Oceanogr 64:1883–1894. https://doi.org/10.1002/lno.11157
Singh R, Miljutin DM, Vanreusel A, Radziejewska T, Miljutina MM, Tchesunov A et al (2016) Nematode communities inhabiting the soft deep-sea sediment in polymetallic nodule fields: do they differ from those in the nodule-free abyssal areas? Mar Biol Res 12:345–359. https://doi.org/10.6084/m9.figshare.3370666
Singhal N, Kumar M, Kanaujia PK, Virdi JS (2015) MALDI-TOF mass spectrometry: an emerging technology for microbial identification and diagnosis. Front Microbiol 6:791. https://doi.org/10.3389/fmicb.2015.00791
Smith CR, De Leo FC, Bernardino AF, Sweetman AK, Arbizu PM (2008) Abyssal food limitation, ecosystem structure and climate change. Trends Ecol Evol 23:518–528. https://doi.org/10.1016/j.tree.2008.05.002
Tautz D, Arctander P, Minelli A, Thomas RH, Vogler AP (2003) A plea for DNA taxonomy. Trends Ecol Evol 18:70–74. https://doi.org/10.1016/S0169-5347(02)00041-1
Uhlenkott K, Edullantes C, Ercan T, Gatzemeier N, Khodami S, Martínez Arbizu P et al (2019) Benthic biodiversity. In: Cruise report: Mangan 2018. Bundesanstalt für Geowissenschaften und Rohstoffe (BGR), Hannover, Germany.
Uhlenkott K, Vink A, Kuhn T, Martínez Arbizu P (2020) Predicting meiofauna abundance to define preservation and impact zones in a deep-sea mining context using random forest modelling. J Appl Ecol 57:1210–1221. https://doi.org/10.1111/1365-2664.13621
Ulrich S, Beindorf P, Biermaier B, Schwaiger K, Gareis M, Gottschalk C (2017) A novel approach for the determination of freshness and identity of trouts by MALDI-TOF mass spectrometry. Food Control 80:281–289. https://doi.org/10.1016/j.foodcont.2017.05.005
Wedding LM, Friedlander A, Kittinger J, Watling L, Gaines S, Bennett M et al (2013) From principles to practice: a spatial approach to systematic conservation planning in the deep sea. Proc R Soc B Biol Sci 280:20131684. https://doi.org/10.1098/rspb.2013.1684
Wegorzewski AV, Kuhn T (2014) The influence of suboxic diagenesis on the formation of manganese nodules in the Clarion Clipperton nodule belt of the Pacific Ocean. Mar Geol 357:123–138. https://doi.org/10.1016/j.margeo.2014.07.004
Wilke T, Renz J, Hauffe T, Delicado D, Peters J (2020) Proteomic fingerprinting discriminates cryptic gastropod species. Malacologia 63:131–137. https://doi.org/10.4002/040.063.0113
Wilkerson MD, Hayes DN (2010) ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26:1572–1573. https://doi.org/10.1093/bioinformatics/btq170
Yeom J, Park N, Jeong R, Lee W (2021) Integrative description of cryptic Tigriopus species from Korea using MALDI-TOF MS and DNA barcoding. Front Mar Sci 8:495. https://doi.org/10.3389/fmars.2021.648197
Acknowledgements
We thank Captain Lutz Mallon and the crew of the research vessel “SONNE” for their help and expertise during the scientific cruises SO262 and SO268. Also, we want to thank Dr. Carsten Rühlemann for his valuable comments on our manuscript. Furthermore we would like to thank the anonymous reviewers for their comments that helped to increase the quality of the publication. This is publication 90 of the Senckenberg am Meer Molecular Laboratory and publication 16 of the Proteomics Laboratory.
Funding
Open Access funding enabled and organized by Projekt DEAL. This research was funded by the German Federal Ministry of Education and Research (BMBF) (grant numbers 03F0812E and 03F0707E). This work was supported by the DFG initiative 1991 “Taxon-omics” (grant nos. RE2808/3-1 and RE2808/3-2).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval
No animal testing was performed during this study.
Sampling and field studies
All necessary permits for sampling and observational field studies have been obtained by the authors from the competent authorities and are mentioned in the acknowledgements, if applicable. The study is compliant with CBD and Nagoya protocols.
Data availability
Genetic data is available in the BOLD project: PNCM - Copepoda obtained in the German trial area for a mining test in the CCZ and investigated with MALDI-TOF MS and DNA barcoding and on GenBank (accession numbers: OL437472 - OL438767). Mass spectrometry data is stored at DataDryad (DOI: https://doi.org/10.5061/dryad.qfttdz0m3).
Author Contribution
Pedro Martínez Arbizu, Annemiek Vink, Katja Uhlenkott and Sven Rossel carried out sediment sampling. Sven Rossel and Katja Uhlenkott carried out lab work and collected the data. Sven Rossel, Janna Peters, Katja Uhlenkott and Pedro Martínez Arbizu analyzed the data. Katja Uhlenkott, Sven Rossel and Janna Peters drafted the manuscript. All authors significantly revised the draft. All authors conceived and designed the research.
Additional information
Communicated by N. Sánchez
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is a contribution to the Topical Collection Biodiversity in Abyssal Polymetallic Nodule Areas
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rossel, S., Uhlenkott, K., Peters, J. et al. Evaluating species richness using proteomic fingerprinting and DNA barcoding—a case study on meiobenthic copepods from the Clarion Clipperton Fracture Zone. Mar. Biodivers. 52, 67 (2022). https://doi.org/10.1007/s12526-022-01307-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12526-022-01307-y