
Abstract
Recent years have brought a significant amount of new results in the field of atherosclerosis. A better understanding of the role of different lipoprotein particles in the formation of atherosclerotic plaques is now possible. Recent cardiovascular clinical trials have also shed more light upon the efficacy and safety of novel compounds targeting the main pathways of atherosclerosis and its cardiovascular complications.
In this review, we first provide a background consisting of the current understanding of the pathophysiology and treatment of atherosclerotic disease, followed by our future perspectives on several novel classes of drugs that target atherosclerosis. The focus of this update is on the pathophysiology and medical interventions of low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), triglycerides (TG) and lipoprotein(a) (Lp(a)).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Atherosclerosis is a chronic condition in which arteries harden through build-up of plaques. Main classical risk factors for atherosclerosis include dyslipoproteinaemia, diabetes, cigarette smoking, hypertension and genetic abnormalities. In this review, we present an update on the pathophysiology of atherosclerosis and related current and possible future medical interventions with a focus on low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), triglycerides (TG) and lipoprotein(a) (Lp(a)).
Pathophysiology of atherosclerosis
Hypercholesterolaemia is considered one of the main triggers of atherosclerosis. The increase in plasma cholesterol levels results in changes of the arterial endothelial permeability that allow the migration of lipids, especially LDL-C particles, into the arterial wall. Circulating monocytes adhere to the endothelial cells that express adhesion molecules, such as vascular adhesion molecule-1 (VCAM-1) and selectins, and, consequently, migrate via diapedesis in the subendothelial space [1]. Once in the subendothelial space, the monocytes acquire macrophage characteristics and convert into foamy macrophages. LDL particles in the subendothelial space are oxidised and become strong chemoattractants. These processes only enhance the accumulation of massive intracellular cholesterol through the expression of scavenger receptors (A, B1, CD36, CD68, for phosphatidylserine and oxidised LDL) by macrophages, which bind native and modified lipoproteins and anionic phospholipids. The end result is a cascade of vascular modifications [1] described in Table 1. Clinical sequelae of atherosclerosis are vessel narrowing with symptoms (angina pectoris) and acute coronary syndromes due to plaque instability.
The majority of coronary thrombi are caused by plaque rupture (55–65%), followed by erosions (30–35%), and least frequently from calcified nodules (2–7%) [1]. Rupture-prone plaques typically contain a large, soft, lipid-rich necrotic core with a thin (≤65 µm) and inflamed fibrous cap. Other common features include expansive remodelling, large plaque size (>30% of plaque area), plaque haemorrhage, neovascularisation, adventitial inflammation, and ‘spotty’ calcifications. Vulnerable plaques contain monocytes, macrophages, and T‑cells. T‑cells promote the vulnerability of plaques through their effects on macrophages [2].
LDL-C, TG and HDL-C emerged as strong independent predictors of atherosclerotic disease after the analysis of the data from the Framingham study. While the role of other parameters is being investigated, TC, LDL-C and HDL-C remain to date the cornerstone in risk estimation for future atherosclerotic events. Low HDL-C has been shown to be a strong independent predictor of premature atherosclerosis [3] and is included in most of the risk estimation scores. Very high levels of HDL-C, however, have consistently not been found to be associated with atheroprotection. The mechanism by which HDL-C protects against atherosclerosis is still under debate and accumulating evidence strongly suggests that the proportion of dysfunctional HDL versus functional HDL rather than the levels may be of importance.
Hypertriglyceridaemia (HTG) has been shown to be an independent risk factor for cardiovascular disease (CVD). Moreover, high TG levels are often associated with low HDL-C and high levels of small dense LDL particles. The burden of HTG is high, with about one-third of adult individuals having TG levels >1.7 mmol/l (150 mg/dL) [3].
Lp(a) is a specialised form of LDL and consists of an LDL-like particle and the specific apolipoprotein (apo) A. Elevated Lp(a) is an additional independent risk marker and genetic data made it likely to be causal in the pathophysiology of atherosclerotic vascular disease and aortic stenosis [4].
Lipoprotein modification treatment
Current view
Medication to adequately control lipoprotein levels needs to be initiated when risk reduction through lifestyle modifications such as dietary changes, stimulation of physical activity and smoking cessation is not sufficient. In secondary prevention, medical therapy is almost invariably needed in addition to lifestyle optimisation.
LDL-C-lowering therapy
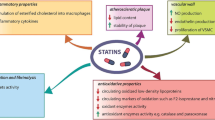
HMG-CoA reductase inhibitors (statins)
3-hydroxy-3-methyl–glutaryl-coenzyme A (HMG-CoA) reductase inhibitors (usually addressed as ‘statins’) induce an increased expression of LDL receptors (LDL-R) on the surface of the hepatocytes, which determines an increase in the uptake of LDL-C from the blood and a decreased plasma concentration of LDL-C and other apo B‑containing lipoproteins, including TG-rich particles [3].
Since the 1990s, statin therapy has shown its effect on cardiovascular outcome in several major landmark trials, summarised in Table 2.
Independent of baseline LDL-C level and baseline cardiovascular (CV) risk, meta-analyses concerning up to 27 statin CV outcome trials, showed a 22% risk reduction in CV events per 1 mmol/l reduction in LDL-C ([5–7]; Fig. 1).

Relation between proportional reduction in incidence of major coronary events and major vascular events and mean absolute LDL cholesterol reduction at 1 year. Square represents a single trial plotted against mean absolute LDL cholesterol reduction at 1 year, with vertical lines above and below corresponding to one SE of unweighted event rate reduction. Trials are plotted in order of magnitude of difference in LDL cholesterol difference at 1 year. For each outcome, regression line (which is forced to pass through the origin) represents weighted event rate reduction per mmol/l LDL cholesterol reduction. (Figure published with permission of the Lancet (owned by Elsevier))
It is currently known that both the baseline burden of atherosclerotic plaque and the degree of progression on serial evaluation significantly associate with risk of CV events [8, 9]. The difference in change in percent atheroma volume (PAV) between patients with and without an event can be as low as approximately 0.55% [10].
Not reaching the cholesterol treatment goals and non-compliance are two important causes for statin therapy failure. Although the LDL-C levels obtained in clinical trials are often low, the clinical reality seems different. Vonbank et al. [11] showed that in 2 cohorts of high-risk CV patients, one from 1999–2000 and the other one from 2005–2007, only 1.3% and 48.5% of patients, respectively, had the LDL-C < 1.8 mmol/l at 2‑year follow-up. The fear of possible side effects of statin therapy is an important reason for non-compliance and remains an underestimated problem in clinical practice. One study in high-dose statin patients reported that muscular pain prevented even moderate exertion during everyday activities in 38% of patients, while 4% of patients were confined to bed or unable to work [12]. Jukema et al. reviewed available data and concluded that statin use is associated with a small increase in type 2 diabetes mellitus incidence, but no convincing evidence was found for other major adverse effects such as cognitive decline or cancer [13].
Statins are therefore, in general, very efficient drugs that in an overwhelming amount of well conducted clinical trials showed consistent clinical event reductions with a very good safety profile. Nevertheless, side effects of importance may occur making the compound, as in any drug class, sometimes unsuitable for some individual patients.
Cholesterol absorption inhibitors
By inhibiting cholesterol absorption, ezetimibe reduces LDL-C. In clinical studies, ezetimibe as monotherapy reduced LDL-C by 15–22% and when combined with a statin it induced an incremental reduction in LDL-C levels of 15–20% [3]. No frequent major adverse effects have been reported [3]. Results from studies like PRECISE-IVUS [14] and IMPROVE-IT [15] support the use of ezetimibe as second-line therapy in association with statins when the therapeutic goal is not achieved at the maximum tolerated statin dose, in statin-intolerant patients, or in patients with contraindication to statins [3].
Bile acid sequestrants
At the highest dose, cholestyramine, colestipol or the recently developed colesevelam can produce a reduction in LDL-C of 18–25% [3]. The use of cholestyramine and colestipol is limited by gastrointestinal adverse effects and major drug interactions with other frequently prescribed drugs. Colesevelam appears to be better tolerated and to have less interaction with other drugs and can be combined with statins. Relatively little hard evidence is available from large clinical trials for this class of drugs.
Proprotein convertase subtilisin/kexin type-9 inhibitors
Inhibitors of proprotein convertase subtilisin/kexin type-9 (PCSK-9) offer the prospect of achieving even lower LDL-C levels than statins in combination with ezetimibe. PCSK-9 binds to LDL-R at the liver and stimulates the absorption and degradation of these receptors. Through inhibition of PCSK-9, the degradation of LDL-R is prevented thereby improving the absorption by the liver of LDL-C particles, which consequently leads to lower LDL-C plasma concentrations.
In 2015, reports were published from two phase 3 trials that measured the efficacy and safety of evolocumab and alirocumab, two monoclonal antibodies that inhibit PCSK-9 [16, 17]. In these trials, the PCSK-9 therapy significantly lowered LDL-C by ≈ 50% and in a preliminary (not powered) analysis reduced the incidence of CV events (Table 3). Other promising results were published from the GLAGOV [18] trial and demonstrated a significant percentage atheroma volume decrease with evolocumab (Table 3). Both evolocumab and alirocumab have been recently approved by the European Medicine Agency and the US Food and Drug Administration for the treatment of elevated plasma LDL-C. The PCSK-9 therapy is suitable in a wide range of patients provided that they express LDL-R, including those with heterozygous and homozygous familial hypercholesterolaemia with residual LDL-R expression [3]. Relatively high costs of the compounds and yet the lack of hard outcomes in large randomised controlled trials (RCTs) still limit their use in clinical practice.
The first results of two large RCTs investigating the long-term efficacy and safety of evolocumab (FOURIER trial) and alirocumab (ODYSSEY Outcomes trial) are underway and necessary [19, 20]. Recently, the development of another monoclonal PCSK-9 inhibitor, bococizumab, was stopped due to auto-antibodies formation against the compound that significantly reduced the LDL-C-lowering efficacy (The SPIRE program) [21].
TG-lowering therapy
Statins
Statins reduce the plasma concentration of TG-rich particles by inhibiting HMG-CoA reductase. Although recent evidence positions HTG as a CV risk factor, the benefits of lowering elevated TG levels are still modest.
Statins are the first-choice therapy in patients with HTG since they reduce both the CV risk and, in high doses, have a stronger effect on elevated TG levels (up to 27% reduction) [3, 22].
Fibrates
Fibrates are agonists of peroxisome proliferator-activated receptor-α (PPAR-α), acting via transcription factors regulating various steps in lipid and lipoprotein metabolism. Fibrates have good efficacy in lowering fasting TG as well as post-prandial TGs and TG-rich lipoprotein remnant particles, with lowering TG levels up to more than 50% [23]. However, results from 5 prospective RCTs and 5 meta-analyses failed to demonstrate superior CV outcomes with fibrates, especially when used on top of statins [3].
n-3 fatty acids
n-3 fatty acids (eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)) can lower TG possibly through interaction with PPARs. Although the underlying mechanism is poorly understood n‑3 fatty acids can reduce TG levels with up to 45%. A meta-analysis of 20 studies and 63,000 patients found no overall effect of omega-3 fatty acids on composite CV events. n‑3 fatty acids appear to be safe and not interact with other therapies [24].
Currently, there are two ongoing phase 3 randomised placebo-controlled clinical trials evaluating the effect of EPA on CV outcomes in 21,000 subjects with elevated serum TG [25, 26]. If TG are not controlled by statins or fibrates n‑3 fatty acids may be added to decrease TG further, as these combinations are safe and well tolerated [3].
HDL-C increasing therapy
Even though lifestyle changes may increase HDL-C levels to a certain degree, many patients will also require medication should a robust HDL-C increase be considered necessary. To date, there is no convincing evidence that artificially raising HDL-C leads to an improved CV outcome. However, if HDL-C increasing therapy is considered then the following options are available.
Cholesteryl ester transfer protein (CETP) inhibitors
The inhibition of CETP by small molecule inhibitors represents currently the most efficient pharmacological approach to influence low HDL-C, with an effect of ≥100% increase in HDL-C and frequently a reduction of LDL-C levels as well. Despite the impressive HDL-C increase, no effect has been seen yet on CV endpoints, as all the CETP-inhibitors studies [27–29] have failed to demonstrate this thus far.
Torcetrapib was discontinued following a higher mortality in the torcetrapib arm of the ILLUMINATE trial [27], the results of the dalcetrapib trial (Dal-OUTCOMES) showed no clinical impact in acute coronary patients and the ACCELERATE trial of evacetrapib in acute coronary patients on statins was terminated prematurely due to lack of efficacy signals [28, 29].
Of the CETP inhibitors initially developed, only anacetrapib is still active. In mice models it has been reported that anacetrapib attenuates atherosclerosis not by increasing HDL-C but rather by decreasing LDL-C by CETP inhibition and by a CETP independent reduction of plasma PCSK-9 level [30].
The REVEAL study, a very large phase 3 RCT with anacetrapib, is still underway and its results are expected in 2017 [31]. This trial will further elucidate whether the additional beneficial effects of anacetrapib on top of a statin can be translated into clinical benefit.
Statins
Statins produce elevations in HDL-C levels between 5–10% [32]. It is difficult to extract the amount of effect that HDL-C increase might have in the overall observed CV risk reduction with statins.
Fibrates
Fibrates increase HDL-C in a similar proportion with statins, namely between 5% in long-term trials (especially if type 2 DM patients are included) and up to 15% in short-term studies [23, 33]. The FIELD study failed to demonstrate that fenofibrate could significantly lower the CV risk [23].
Future perspectives
LDL-C-lowering therapy
PCSK-9 inhibition (non-monoclonal antibody)
A recent approach in decreasing PCSK-9 levels is the administration of small interfering RNA (siRNA) molecules directed against PCSK-9. The siRNA molecules enable the RNA-induced silencing complex, which cleaves messenger RNA (mRNA) molecules encoding PCSK-9 specifically. The cleaved mRNA is degraded and thus unavailable for protein translation, which results in decreased levels of the PCSK-9 protein. The phase 2 ORION trial showed that one subcutaneous injection of 300 mg inclisiran determined a mean LDL-C reduction of 51% after 6 months [34]. Inclisiran was well tolerated with no relevant safety concerns. These results support the start of the phase 3 program. The next step might be the development of a vaccine targeting PCSK-9. Crossey et al. provided in mice and macaques the proof-of-principle evidence that a vaccine targeting PCSK-9 peptide can effectively lower lipid levels and works synergistically with statins [35].
Bempedoic acid
Bempedoic acid is a first-in-class adenosine triphosphate (ATP) Citrate Lyase inhibitor. The mechanism of action involves the inhibition of cholesterol biosynthesis and the up-regulation of LDL-R, which in turn decreases plasma LDL-C levels. A phase 3 clinical trial (CLEAR Harmony) is currently conducted in patients with high CV risk and elevated LDL-C that is not adequately controlled under their current therapy. Almost 2000 subjects will be randomised for bempedoic acid or placebo and will be followed for 52 weeks [36]. In continuation of this trial, the CLEAR Outcomes trial will be conducted. This will be an event-driven study of 12,600 patients on either bempedoic acid or placebo with the primary efficacy endpoint of major adverse CV events. The results of this trial will be expected not earlier than 2022.
Peroxisome proliferator-activated receptor delta (PPARδ)
PPARδ is a nuclear receptor that regulates genes involved in lipid storage and transport. MBX-8025 is a selective agonist for PPARδ.
The recently presented partial results from a proof-of-concept phase II trial in patients with homozygous familial hypercholesterolaemia showed that the range of responses to MBX-8025 was broad, but that MBX-8025 could provide a clinically meaningful reduction in LDL-C for a subset of patients [37].
Other lipoprotein modification targets
Apo A-I mimetics
Apo A-I is the primary functional component of HLD-C and supports the rapid removal of cholesterol from plaque. The MILANO-PILOT study was a proof-of-concept study in which the impact on coronary plaque by MDCO-216 was measured in 120 acute coronary syndrome (ACS) patients using IVUS [38]. MDCO-216 is a complex of dimeric recombinant apolipoprotein A-I Milano and a phospholipid (POPC), and mimics pre-beta HDL. In this study, MDCO-216 did not produce a significant effect on coronary progression. Based on these results further development of the compound was halted. CER-001 is a different engineered pre-beta HDL compound and is currently being tested in a phase 2 clinical trial (CARAT) assessing the nominal change from baseline to follow-up (at 12 weeks) in the PAV in the target coronary artery of ACS patients. Results will be available in early 2017 [39]. CSL112 is a plasma-derived apolipoprotein A-I (apo A-I) and was tested in a phase II trial for safety and tolerability. CSL112 was well tolerated and did not significantly alter liver or kidney functions [40]. Assessment of the efficacy of CSL112 will be performed in an adequately powered phase 3 clinical trial.
Angiopoietin-like 3 (ANGPTL3)
ANGPTL3 is a protein and main regulator of lipoprotein metabolism. Its function is linked to the inhibition of lipoprotein lipase (LPL) activity. Earlier studies have identified that subjects with ANGPTL3 deficiency have reduced cholesterol and TG levels. Recently, a phase 1/2 study evaluated the safety, tolerability, pharmacokinetics and pharmacodynamics of ANGPTL3-LRx (an antisense inhibitor of ANGPTL3) in healthy volunteers with elevated TG and subjects with familial hypercholesterolaemia. There were no short-term safety concerns and ANGPTL3-LRx induced significant mean reductions in TGs (66%), LDL-C (35%) and total cholesterol (36%). Final results are expected in 2017 [41].
Lipoprotein(a) (Lp(a))
PCSK-9 inhibitors and nicotinic acid reduce Lp(a) by approximately 30% [16, 17, 42], however, an effect on CV events targeting Lp(a) has not been convincingly shown. A phase 2 clinical trial showed that IONIS-APO(a)Rx, an oligonucleotide targeting Lp(a), induced a lowering of Lp(a) levels of up to 71.6% [43]. A phase 1/2a first-in-man trial showed that IONIS-APO(a)-LRx, a ligand-conjugated antisense oligonucleotide designed to be highly and selectively taken up by hepatocytes, induced a lowering of Lp(a) levels of up to 92%. Both antisense oligonucleotides were short-term safe and well tolerated [43].
Plasma Lp(a) is currently not recommended for risk screening in the general population, but measurement should be considered in people with high CV risk or a strong family history of premature atherothrombotic disease [3].
Table 4 provides an overview of the most important ongoing lipoprotein modifying trials and their expected or recently published results.
Conclusions
Lowering LDL-C by statin therapy remains, to date, the cornerstone for the medical prevention and treatment of atherosclerotic disease since it is efficient and generally safe. In high-risk patients with statin intolerance or in high-risk patients who do not obtain the desired LDL-C level with intensive statin treatment, cholesterol absorption inhibitors, especially ezetimibe, should be considered. Bile acid sequestrants, fibrates and niacin are not recommended. Upcoming PCSK-9 inhibitors, whether in the form of monoclonal antibodies or new approaches, appear as potent agents for dyslipoproteinaemia. However, their long-term efficacy and safety still needs to be proven and costs may limit their practical use. HDL-C modulation through CETP inhibition and apo A-I mimetics did not yet provide evidence for better CV outcomes; the REVEAL and CARAT trials will shed light on the future of these drug classes. New classes of molecules targeting ANGPTL3 and Lp(a) have shown promising efficacy and good short-term safety profiles in several early phase trials and these results warrant further development.
References
Sakakura K, Nakano M, Otsuka F, Ladich E, Kolodgie FD, Virmani R. Pathophysiology of atherosclerosis plaque progression. Heart Lung Circ. 2013;22(6):399–411.
Yla-Herttuala S, Bentzon JF, Daemen M, et al. Stabilization of atherosclerotic plaques: an update. Eur Heart J. 2013;34(42):3251–8.
Catapano AL, Graham I, De Backer G, et al. ESC/EAS Guidelines for the Management of Dyslipidaemias: The Task Force for the Management of Dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Developed with the special contribution of the European Assocciation for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J. 2016;37(39):2999. doi:10.1093/eurheartj/ehw272.
Kronenberg F, Utermann G. Lipoprotein(a): resurrected by genetics. J Int Med. 2013;273(1):6–30.
Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet (lond Engl). 2010;376(9753):1670–81.
Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366(9493):1267–78.
Mihaylova B, Emberson J, Blackwell L, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380(9841):581–90.
Nicholls SJ, Hsu A, Wolski K, et al. Intravascular ultrasound-derived measures of coronary atherosclerotic plaque burden and clinical outcome. J Am Coll Cardiol. 2010;55(21):2399–407.
Jukema JW, Bruschke AV, van Boven AJ, et al. Effects of lipid lowering by pravastatin on progression and regression of coronary artery disease in symptomatic men with normal to moderately elevated serum cholesterol levels. The Regression Growth Evaluation Statin Study (REGRESS). Circulation. 1995;91(10):2528–40.
Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350(15):1495–504.
Vonbank A, Saely CH, Rein P, Sturn D, Drexel H. Current cholesterol guidelines and clinical reality: a comparison of two cohorts of coronary artery disease patients. Swiss Med Wkly. 2013;143:w13828.
Bruckert E, Hayem G, Dejager S, Yau C, Begaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients the PRIMO study. Cardiovasc Drugs Ther. 2005;19(6):403–14.
Jukema JW, Cannon CP, de Craen AJ, Westendorp RG, Trompet S. The controversies of statin therapy: weighing the evidence. J Am Coll Cardiol. 2012;60(10):875–81.
Tsujita K, Sugiyama S, Sumida H, et al. Impact of dual lipid-lowering strategy with ezetimibe and atorvastatin on coronary plaque regression in patients with percutaneous coronary intervention: the multicenter randomized controlled PRECISE-IVUS trial. J Am Coll Cardiol. 2015;66(5):495–507.
Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe added to Statin therapy after acute coronary syndromes. N Engl J Med. 2015;372(25):2387–97.
Sabatine MS, Giugliano RP, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1500–9.
Robinson JG, Farnier M, Krempf M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372(16):1489–99.
Nicholls SJ, Puri R, Anderson T, et al. Effect of Evolocumab on progression of coronary disease in statin-treated patients: the GLAGOV randomized clinical trial. JAMA. 2016;316:2373. doi:10.1001/jama.2016.16951.
Schwartz GG, Bessac L, Berdan LG, et al. Effect of alirocumab, a monoclonal antibody to PCSK9, on long-term cardiovascular outcomes following acute coronary syndromes: rationale and design of the ODYSSEY outcomes trial. Am Heart J. 2014;168(5):682–9.
Sabatine MS, Giugliano RP, Keech A, et al. Rationale and design of the Further cardiovascular OUtcomes Research with PCSK9 Inhibition in subjects with Elevated Risk trial. Am Heart J. 2016;173:94–101.
Ridker PM, Amarenco P, Brunell R, et al. Evaluating bococizumab, a monoclonal antibody to PCSK9, on lipid levels and clinical events in broad patient groups with and without prior cardiovascular events: Rationale and design of the Studies of PCSK9 Inhibition and the Reduction of vascular Events (SPIRE) Lipid Lowering and SPIRE Cardiovascular Outcomes Trials. Am Heart J. 2016;178:135–44.
Adams SP, Sekhon SS, Wright JM. Lipid-lowering efficacy of rosuvastatin. Cochrane Database Syst Rev. 2014; . doi:10.1002/14651858.CD010254.pub2.
Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366(9500):1849–61.
Kotwal S, Jun M, Sullivan D, Perkovic V, Neal B. Omega 3 Fatty acids and cardiovascular outcomes: systematic review and meta-analysis. Circ Cardiovasc Qual Outcomes. 2012;5(6):808–18.
Outcomes Study to Assess STatin Residual Risk Reduction With EpaNova in HiGh CV Risk PatienTs With Hypertriglyceridemia (STRENGTH). Identifier: NCT02104817. https://clinicaltrials.gov/ct2/show/NCT02104817?term=NCT02104817&rank=1. Accessed 12 Dec 2016.
A Study of AMR101 to Evaluate Its Ability to Reduce Cardiovascular Events in High Risk Patients With Hypertriglyceridemia and on Statin. The Primary Objective is to Evaluate the Effect of 4 g/Day AMR101 for Preventing the Occurrence of a First Major Cardiovascular Event (REDUCE-IT). Identifier: NCT01492361. https://clinicaltrials.gov/ct2/show/NCT01492361?term=NCT01492361&rank=1. Accessed 12 Dec 2016.
Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357(21):2109–22.
Schwartz GG, Olsson AG, Abt M, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367(22):2089–99.
Nicholls SJ, Lincoff AM, Barter PJ, et al. Assessment of the clinical effects of cholesteryl ester transfer protein inhibition with evacetrapib in patients at high-risk for vascular outcomes: rationale and design of the ACCELERATE trial. Am Heart J. 2015;170(6):1061–9.
van der Tuin SJ, Kuhnast S, Berbee JF, et al. Anacetrapib reduces (V)LDL cholesterol by inhibition of CETP activity and reduction of plasma PCSK9. J Lip Res. 2015;56(11):2085–93.
REVEAL: Randomized EValuation of the Effects of Anacetrapib Through Lipid-modification (REVEAL). Identifier: NCT01252953. https://clinicaltrials.gov/ct2/show/study/NCT01252953?term=anacetrapib&rank=9. Accessed 19 Dec 2016.
Barter PJ, Brandrup-Wognsen G, Palmer MK, Nicholls SJ. Effect of statins on HDL-C: a complex process unrelated to changes in LDL-C: analysis of the VOYAGER Database. J Lip Res. 2010;51(6):1546–53.
Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362(17):1563–74.
Trial to Evaluate the Effect of ALN-PCSSC Treatment on Low Density Lipoprotein Cholesterol (LDL-C) (ORION). Identifier: NCT02597127. https://clinicaltrials.gov/ct2/show/NCT02597127?term=NCT02597127&rank=1. Accessed 12 Dec 2016.
Crossey E, Amar MJ, Sampson M, et al. A cholesterol-lowering VLP vaccine that targets PCSK9. Vaccine. 2015;33(43):5747–55.
Evaluation of Long-Term Safety and Tolerability of ETC-1002 in High-Risk Patients With Hyperlipidemia and High CV Risk. Identifier: NCT02666664. https://clinicaltrials.gov/ct2/show/NCT02666664?term=NCT02666664&rank=1. Accessed 12 Dec 2016.
Study to Evaluate the Effects of MBX-8025 in Patients With HoFH. Identifier: NCT02472535. https://clinicaltrials.gov/ct2/show/NCT02472535?term=NCT02472535&rank=1. Last Accessed 12 Dec 2016.
MDCO-216 Infusions Leading to Changes in Atherosclerosis: A Novel Therapy in Development to Improve Cardiovascular Outcomes – Proof of Concept Intravascular Ultrasound (IVUS), Lipids, and Other Surrogate Biomarkers Trial (PILOT). Identifier: NCT02678923. https://clinicaltrials.gov/ct2/show/NCT02678923?term=NCT02678923&rank=1. Accessed 12 Dec 2016.
CER-001 Atherosclerosis Regression ACS Trial (CARAT). Identifier: NCT02484378. https://clinicaltrials.gov/ct2/show/NCT02484378?term=CARAT&rank=1. Accessed 12 Dec 2016.
Gibson CM, Korjian S, Tricoci P, et al. Safety and tolerability of CSL112, a reconstituted, infusible, plasma-derived apolipoprotein A‑I, after acute myocardial infarction: the AEGIS-I trial (ApoA-I event reducing in ischemic syndromes I). Circulation. 2016;134:1918. doi:10.1161/circulationaha.116.025687.
Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of IONIS ANGPTL3-LRx in Healthy Volunteers With Elevated Triglycerides and Subjects With Familial Hypercholesterolemia. Identifier: NCT02709850. https://clinicaltrials.gov/ct2/show/NCT02709850?term=NCT02709850&rank=1. Accessed 12 Dex 2016.
Seed M, O’Connor B, Perombelon N, O’Donnell M, Reaveley D, Knight BL. The effect of nicotinic acid and acipimox on lipoprotein(a) concentration and turnover. Atherosclerosis. 1993;101(1):61–8.
Viney NJ, van Capelleveen JC, Geary RS, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet. 2016;388(10057):2239–53.
The Scandinavian Simvastatin Survival. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344(8934):1383–9.
Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333(20):1301–7.
Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335(14):1001–9.
Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295(13):1556–65.
Nicholls SJ, Ballantyne CM, Barter PJ, et al. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365(22):2078–87.
The Lipid Metabolism-Atherogenesis Branch, National Heart, Lung, and Blood Institute. The lipid research clinics coronary primary prevention trial results. I. Reduction in incidence of coronary heart disease. JAMA. 1984;251(3):351–64.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
J.W. Jukema/his department has received research grants from and/or was speaker (with or without lecture fees) on a.o. (CME accredited) meetings sponsored by Amgen, Astra-Zeneca, Lilly, Merck-Schering-Plough, Pfizer, Sanofi Aventis, The Medicine Company, the Netherlands Heart Foundation, CardioVascular Research the Netherlands (CVON), the Interuniversity Cardiology Institute of the Netherlands and the European Community Framework KP7 Programme. S.C. Bergheanu and M.C. Bodde declare that they have no competing interests.
Additional information
S.C. Bergheanu and M.C. Bodde contributed equally to the manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bergheanu, S.C., Bodde, M.C. & Jukema, J.W. Pathophysiology and treatment of atherosclerosis. Neth Heart J 25, 231–242 (2017). https://doi.org/10.1007/s12471-017-0959-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12471-017-0959-2