
Abstract
Introduction
Epithelial–mesenchymal transition (EMT) is an important biological process in tumor invasion and metastasis, and thus a potential indicator of the progression and drug resistance of breast cancer. This study comprehensively analyzed EMT-related genes in triple-negative breast cancer (TNBC) to develop an EMT-related prognostic gene signature.
Methods
With the application of The Cancer Genome Atlas (TCGA) database, Molecular Taxonomy of Breast Cancer International Consortium (METABRIC), and the Genotype-Tissue Expression (GTEx) database, we identified EMT-related signature genes (EMGs) by Cox univariate regression and LASSO regression analysis. Risk scores were calculated and used to divide patients with TNBC into high-risk group and low-risk groups by the median value. Kaplan–Meier (K–M) and receiver operating characteristic (ROC) curve analyses were applied for model validation. Independent prognostic predictors were used to develop nomograms. Then, we assessed the risk model in terms of the immune microenvironment, genetic alteration and DNA methylation effects on prognosis, the probability of response to immunotherapy and chemotherapy, and small molecule drugs predicted by The Connectivity Map (Cmap) database.
Results
Thirteen EMT-related genes with independent prognostic value were identified and used to stratify the patients with TNBC into high- and low-risk groups. The survival analysis revealed that patients in the high-risk group had significantly poorer overall survival than patients in the low-risk group. Populations of immune cells, including CD4 memory resting T cells, CD4 memory activated T cells, and activated dendritic cells, significantly differed between the high- and low-risk groups. Moreover, some therapeutic drugs to which the high-risk group might show sensitivity were identified.
Conclusions
Our research identified the significant impact of EMGs on prognosis in TNBC, providing new strategies for personalizing TNBC treatment and improving clinical outcomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Epithelial–mesenchymal transition (EMT) is an important biological process in tumor invasion and metastasis, and thus may be a potential indicator of the progression and drug resistance of breast cancer. |
This study established a prognostic risk model for triple-negative breast cancer (TNBC) based on 13 EMT-related signature genes (EMGs). |
High-risk patients with TNBC had shorter overall survival compared to low-risk patients and were more likely to experience immunosuppression and thus less likely to benefit from immune checkpoint inhibitor (ICI) therapy or chemotherapy, which aids the development of individualized therapy for patients with TNBC. |
Introduction
Triple-negative breast cancer (TNBC) is a type of breast cancer with negative expression of estrogen (ER), progesterone (PR), and human epidermal growth factor receptor 2 (HER2; also known as ERBB2) [1]. It mostly occurs in premenopausal young women under 40 years old, who account for approximately 15–20% of all patients with TNBC [2]. It often occurs in the form of high-grade invasive ductal carcinoma, and although it does not represent a high proportion of breast cancer cases, TNBC has significantly higher rates of early recurrence, distant metastasis, and mortality than other breast cancer subtypes [3, 4]. Moreover, disease-free survival and prognosis are poor in patients with TNBC, and since TNBC is not sensitive to endocrine therapy or molecular targeted therapy, chemotherapy remains the main systemic treatment, but the efficacy is poor [5]. With the emergence of drug resistance in TNBC, there is an urgent need to develop new treatment regimens and targets [6].
Epithelial–mesenchymal transition (EMT) is a cellular program associated with embryonic development, cancer progression, metastasis, tumor recurrence, and cancer cell drug resistance [7]. During transformation, cell–cell or extracellular matrix interactions remodel, allowing epithelial cells to separate from each other or the basement membrane, activating transcription to promote mesenchymal formation [8]. One known feature of TNBC is the predominance of cells with mesenchymal properties. These cells are present in poorly differentiated cancer cells through EMT, suggesting that EMT is involved in developing TNBC [9].
This study used the method of Li et al. [10] and Wang et al. [11]. We developed EMT-related signature genes (EMGs) related to prognosis based on The Cancer Genome Atlas (TCGA) database, which was validated by the METABRIC database. On the basis of the EMGs, high-risk patients with TNBC had shorter overall survival (OS) compared to low-risk patients and were more likely to experience immunosuppression and thus less likely to benefit from immune checkpoint inhibitor (ICI) therapy or chemotherapy, which is consistent with the EMT features. This study highlights the functional role of EMGs and uncovers a potential prognostic biomarker for individualized treatment for TNBC.
Methods
Data Acquisition and Preprocessing
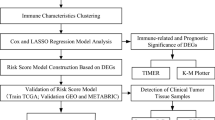
Figure 1 presents the study flowchart. The RNA sequencing (RNA-seq) data and clinical information of 1246 TNBC samples were obtained from the TCGA database (https://gdc-portal.nci.nih.gov/). Combined with clinical data, after we selected patients with TNBC (ER−, PR−, and HER2−) and removed incomplete data, 136 samples were subjected to subsequent analysis. The RNA-seq data and clinical information of an additional 294 tumor samples were downloaded from the METABRIC database (https://www.cbioportal.org/study?id=brca_metabric). Gene expression was analyzed in 91 normal tissues collected from the Genome Tissue Expression (GTEx) database (https://gtexportal.org/home/datasets). Gene expression was quantified and normalized by fragments per kilobase of transcript per million mapped reads (FPKM) and log2(TPM + 1)-based transformation. The clinical features of the TCGA patients are shown in Supplementary Table S1.

Overview of data processing steps
The gene sets related to EMT are collected from the following three datasets: 1184 EMT-RDGs were extracted from the EMT gene database (http://dbemt.bioinfo-minzhao.org/download.cgi), 200 EMT-RDGs are picked up from the HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION gene set, and 153 EMT-RDGs are selected from GO_EPITHELIAL_TO_MESENCHYMAL_TRANSITION in the Molecular Signatures Database v7.4 (MSigDB, http://www.gsea-msigdb.org/gsea/msigdb/index.jsp).
This study was based on publicly available data and patients were not directly involved in the entire research process; therefore, the need for ethical committee approval and informed consent was waived.
Identification of Key Prognostic Genes and Establishment of the Model
After 153 duplicated genes were removed, a total of 1384 EMT-RDGs were collected for our subsequent analysis. The differential gene expression analyses of the data in TNBC and normal tissues were performed using the “DESeq2” R package (|log2 (fold change)| > 0.32, p < 0.05 were considered as significant). Then, univariate Cox proportional hazard regression analysis was performed to determine the EMT-RDGs among these differentially expressed genes (DEGs) which were associated with the OS of patients, and only EMT-RDGs with p value < 0.05 were selected for subsequent analyses. To avoid overfitting the prognostic signature and narrowing the genes for prediction of the OS, LASSO Cox regression was used to build the EMGs. Finally, a prognostic formula for principal component analysis (PCA) was developed with 13 EMGs and their corresponding coefficients as follows: risk score = ∑(coefficient × expression of signature genes). On the basis of the median risk score, the patients were classified as high-risk group and low-risk group. To evaluate the differences in survival rate between the two groups, the Kaplan–Meier (K–M) survival curve was generated using the R package “survival”. Furthermore, the R package “survivalROC” was used to generate time-dependent receiver operating characteristic (ROC) curves to evaluate the predictive power of the prognostic model.
Validation of the EMGs for TNBC
To verify the prognostic value of DEGs, we used the METABRIC dataset, which contained 294 TNBC samples, as validation cohort. Each patient’s risk score was calculated using the same formula. Univariate and multivariate Cox regression analyses were performed to analyze the independent prognosis of the 13-gene prognostic model. All independent prognostic factors were used to construct a nomogram to predict the survival of patients with TNBC at 3, 5, and 8 years.
Cancer Cell Line Encyclopedia (CCLE) Analysis of EMGs
In addition, we further searched for the expression levels of EMGs in cell lines via the CCLE database (https://portals.broadinstitute.org/ccle). The CCLE is a high-throughput web-based tool with large numbers of human cancer cell lines (n = 1457) and unique datasets (n = 136,488) [12]. An RNA sequencing method was selected to search for expression of EMGs in 60 breast cancer cell lines.
Tumor Immune Microenvironment Analysis
The immune score, stromal score, ESTIMATE score, immune cell infiltration, immune signature, and immune-related gene expression of the high and low-risk groups were compared to characterize the tumor immune microenvironment of the prognostic signature. The enrichment level of 22 immune signatures in each TNBC sample was quantified by single-sample gene set enrichment analysis (ssGSEA) in the R package “GSVA”. Heat maps and violin plots were drawn to observe the difference in immune cell infiltration between the high- and low-risk groups.
Genetic Alteration and DNA Methylation Effects on Prognosis
Genetic alterations, including gene mutations and copy number alterations, are the potential factors impacting expression. We evaluated the genetic alterations of EMGs with Oncoprinter from cBioportal and the impacts of EMGs on patient with TNBC survival. DNA methylation is another risk factor that affects the expression of EMGs. The influence of DNA methylation on EMG expression was assessed by DNMIVE, and the impact of a single-methylation CpG site on the OS of patients with TNBC was analyzed by MethSurv [13].
Evaluation of the Sensitivity of Patients with TNBC to Chemotherapeutic Agents
The “pRRophetic” package in R was used to predict the half-maximal inhibitory concentration (IC50) of chemotherapy drugs in the high- and low-risk groups of patients with TNBC and to infer the sensitivity of the different patients. By constructing the ridge regression model based on Genomics of Drug Sensitivity in Cancer (GDSC) (http://www.cancerrxgene.org/), cell line expression spectrum, and TCGA gene expression profiles, the package could apply a pRRophetic algorithm to predict drug IC50 [14].
Prediction of Potential Small Molecule Agents in Patients with TNBC
CMap database (https://clue.io/) was used to predict potential drugs [15]. The full range of up- and downregulated genes between high-risk group and low-risk group was submitted to the CMap database to predict potential drug targets for the treatment of TNBC. Enrichment scores were calculated from − 1 to 1. Enrichment scores between − 1 and 0 indicated that the drug might reverse gene expression (a candidate for TNBC). In contrast, enrichment scores between 0 and 1 indicated that the drug might induce gene expression. p values < 0.05 were considered statistically significant.
Statistical Analysis
All statistical analysis was performed using the R software 4.1.1. The Wilcoxon rank-sum test was used to compare the differences between groups of continuous data. The relationships between EMGs and risk score and immune infiltration were determined by Spearman’s correlation analysis. The Cox proportional hazard model was performed to estimate the β regression coefficient, hazard ratios, p value, and their corresponding 95% confidence interval for each selected risk predictor. On the basis of the multivariate Cox analysis, a nomogram was constructed with the “rms” package in R. The C-index and calibration curve with the bootstrap method were used to evaluate the prediction performance of the nomogram. A p value < 0.05 was considered statistically significant.
Results
Identification of EMT-Related DEGs
On the basis of the screening criteria above, a total of 136 TNBC samples RNA-seq data and matched clinical information were downloaded from the TCGA database, and the data for 91 normal samples data were obtained from the GTEx database. The differential expression analysis of 136 cancer samples and 91 normal samples was performed using the “Deseq2” package and identified a total of 9416 DEGs (|log2 (fold change)| > 0.32, p < 0.05). There were 5874 upregulated and 3542 downregulated DEGs, of which 725 genes (430 upregulated genes, 295 downregulated genes) were associated with EMT-RDGs (Venn diagram is shown in Supplementary Fig. S1).
Establishment of a Risk Signature with 13 EMGs
To identify EMT-RDGs associated with OS, a univariate Cox proportional hazard regression analysis was initially performed on 725 EMT-RDGs in the TCGA database. The result showed that 27 EMT-RDGs were significantly associated with the OS (Fig. 2a), which were subjected to LASSO Cox regression analysis. Of these genes, 12 genes were considered risk factors (HR > 1) and that their overexpression might reduce survival, while overexpression of the remaining 13 (HR < 1) might improve the survival of patients. LASSO analyses were used subsequently to remove EMGs that may be closely related to other EMGs (Fig. 2b, c). Finally, 13 EMGs were screened to predict OS in patients from the TCGA-TNBC dataset, and the risk score was calculated as follows: HOOK1 expression × (− 0.139) + PEG10 expression × (− 0.348) + BOP1 expression × 0.097 + GLIPR2 expression × (− 0.01) + P2RY2 expression × 0.007 + CCND2 expression × (− 0.219) + IRS2 expression × (− 0.206) + ITGB3 expression × 0.267 + CCL25 expression × (− 0.156) + CEACAM5 expression × (0.031) + CEACAM1 expression × (− 0.315) + PMEPA1 expression × (0.263) + HMOX1 expression × (0.014). The 13 EMGs are shown in Supplementary Table S2. According to the median risk score (cutoff = − 0.1605), 68 of the 136 TNBC samples were classified as high risk, and the remaining 68 were classified as low risk (the risk score, survival status, and gene expression heatmap of EMGs are presented in Fig. 2d). As the shown in the time-dependent ROC curve, the area under curve (AUC) values for 3, 5, and 8 years were 0.944, 0.878, and 0.866 (Fig. 2e). K–M survival curves revealed that patients in the high-risk group had significantly poorer OS than patients in the low-risk group (Fig. 2f). Additionally, analysis of the CCLE dataset (https://www.broadinstitute.org/ccle) also showed differential expression of EMGs in breast cell lines (Fig. 3).

Establishment and evaluation of the prognostic model. a The forest plots illustrate the univariate Cox analysis of 27 genes significantly associated with OS. b Coefficients were calculated for each lambda. Each line represents a gene confidence value. c LASSO regression analysis of EMGs based on univariate Cox regression analysis. The horizontal axis represents the log value of the independent variable λ, whilst the vertical axis represents the partial likelihood deviance of the log value of each independent variable λ. d The risk curve of the risk model shows that the risk increases from left to right. According to the median value, the samples were divided into high-risk (red line) and low-risk (blue line) groups. Heatmap showing expression of the six genes screened from EMGs in TNBC. e The ROC curve evaluates the effectiveness of the risk model. f K–M survival curve of the risk score, with the ordinate indicating the survival rate and the abscissa indicating the total survival time. The red curve represents the high-risk group, the blue curve represents the low-risk group, and there was a significant difference between the groups (> 0.0001)

Expression of EMGs in different breast cancer cell lines. The heatmap showed the expression levels of 13 EMGs in breast cancer cell lines. The upper blocks in red indicate overexpression, whereas the bottom in blue indicate underexpression
Validation of EMT-Related Gene Signature in the METABRIC Database
The METABRIC cohort was used to confirm the external validity and accuracy of the prognostic model. According to the formula of the training set (TCGA-TNBC), the patients were classified as high risk (n = 142) or low risk (n = 152). The OS of patients in the high-risk group was significantly lower than that in the low-risk group, which was consistent with the results of the TCGA cohort (p = 0.0042). As shown in Fig. 3c, the AUC of the ROCs was 0.521 (3-year ROC), 0.596 (5-year ROC), and 0.625 (8-year ROC), suggesting that the EMT gene prediction model has a good prognostic prediction. The detailed result can be found in the Supplementary Fig. S2.
Independent Prognostic Value of the 13-Gene Prognostic Model
The univariate analysis and multivariate analyses identified biomarkers that could independently predict the survival of patients in both the training and validation cohorts (Fig. 4a, b). The pathological stage was also confirmed as an independent predictive factor in both cohorts. Two predictive nomograms in the TCGA and METABRIC cohorts (Fig. 4c, d) were constructed to further improve predictive ability. Calibration curves of the two nomograms between predicted and observed 3-year, 5-year, and 8-year outcomes in both cohorts (TCGA, Fig. 4e; METABRIC, Fig. 4f) were also plotted. Moreover, the decision curve analyses indicated that our nomogram had a high potential for clinical decision-making at 3, 5, and 8 years in both cohorts (TCGA, Fig. 4g–i; METABRIC, Fig. 4j–l).


Estimating the risk model’s independent prognostic value. Univariate and multivariate Cox regression analyses of OS in the TCGA (a) and METABRIC (b) cohorts. Risk scores were significantly associated with OS prediction. Nomograms for predicting survival proportion of patients at 3, 5, and 8 years in TCGA (c) and METABRIC (d) cohorts. e–f Calibration curves of both cohorts. g–l DCA (decision curve analysis) of the nomograms based on OS in TCGA (g–i) and METABRIC (j–l) cohorts for 3-year, 5-year, and 8-year risk
Landscape of the Tumor Immune Microenvironment in TNBC
The “estimate” R package based on the ssGSEA algorithm was used to explore the immune infiltration in both groups. The results demonstrated no difference in immune (Fig. 5a), stromal (Fig. 5b), and ESTIMATE scores (Fig. 5c) between high-risk groups and low-risk groups. But there was a trend of higher immune scores in the low-risk group and a trend of higher stromal scores in the high-risk group. CD4 memory resting T cells, CD4 memory activated T cells, and dendritic cells activated were significantly higher in the low-risk group than in the high-risk group. Moreover, the results of the CIBERSORT algorithm also demonstrated that CD4 memory resting T cells, CD4 memory activated T cells, and dendritic cells activated were significantly higher in the low-risk group, and the high-risk group was associated with more macrophages MO, monocytes and eosinophils (Fig. 5d). Furthermore, in the TCGA cohort, the risk score negatively correlated with resting dendritic cells (R = − 0.21, p = 0.015), CD4 memory activated T cells (R = − 0.23, p = 0.0077), and CD8 T cells (R = − 0.22, p = 0.0085) but positively correlated with M0 macrophages (R = 0.24, p = 0.0053) and activated mast cells (R = 0.22, p = 0.01) (Fig. 5f).

Analysis of immune infiltration patterns in breast cancer samples from TCGA dataset. “ns” represents no significant difference in the infiltration of immune cells between the two samples; *p < 0.05. Comparison of ImmuneScore (a), StromalScore (b), and ESTIMATEScore (c) between the patient groups. d Boxplots depicting the CIBERSORT scores of 22 immune cells of the high-risk patients compared to low-risk patients. e Heatmap of the expression of immune cells. f The relationship between the risk score and immune infiltration
The immune-related pathways, including the transforming growth factor-β (TGFβ) family member pathway, interferon receptor, interferons, natural killer cell cytotoxicity, TCR signaling pathway, and TNF family members, had higher activity in the low-risk group than in the high-risk group (Fig. 6a). Meanwhile, the low-risk patients had significantly higher expression levels of TNFRSF9 (tumor necrosis receptor superfamily), TIGIT (T cell immunoreceptor with Ig and ITIM domains), IDO1 (indoleamine 2,3-dioxygenase 1), and LAG3 (lymphocyte activation gene 3) (p < 0.01) (Fig. 6b). These results indicate that patients with a low EMT-related signature score might respond better to ICI treatment.

Analysis of immune-related gene immune pathways and checkpoints. a Boxplots depicting the immune-related pathways of the high-risk patients compared to the low-risk patients. b The gene expression of PDCD1, TNFRSF9, CTLA4, TIGIT, IDO1, CD274, and LAG3 in the high- and low-risk groups. c Heatmap of the differences in immune-related gene mRNA expression between the high-risk and low-risk patients. d Heatmap of the correlation between 13-gene mRNA expression with the ssGSEA scores of 21 immune cells. Adjusted p values: ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001
Furthermore, heatmap was used to show differences in immune-related gene mRNA expression between high- and low-risk patients in the TCGA cohort and analyze the correlation between the 13 genes of the signature and the ssGSEA scores of 21 immune cells (Fig. 6c, d).
Immune Signature Predicts Immunotherapy Benefits
The overall expression of the common immune checkpoints in the low-risk group was higher than that in the high-risk group, indicating low immune cell infiltration in high-risk patients. The difference in cytolytic activity (Fig. 8a) between the two groups was estimated by quantifying the granzyme A (GZMA) expression, which was attributed to the CYT score. Moreover, difference in mRNAsi (Fig. 8b), a novel stemness index for evaluating the dedifferentiation potential of tumor cells, was compared between the two groups. The low-risk group also had a higher T cell inflammation score (Fig. 8c).
DNA Methylation Might Indicate a Diverse Prognosis, but Not Genetic Alterations
We obtained the genetic alteration data of EMGs in the TCGA cohort from cBioportal. BOP1 had the highest frequency of genetic alteration (15%), while only 0.4% of patients had genetic alterations in HMOX1 (Fig. 7a). Patients with genetic alterations in the EMGs did not show different OS rates compared with those without the alterations (p = 0.995) (Fig. 7b). Regarding DNA methylation, we first evaluated the promoter methylation and gene expression levels. The result showed that the promoter methylation of PEG10, GLIPR2, P2RY2, CCND2, IRS2, ITGB3, CEACAM5, CEACAM1, and PMEPA1 had negative correlations with mRNA expression and only found that the promoter methylation of HMOX1 was positively associated with mRNA expression (Fig. 7c). Additionally, we revealed the impact of single CpG on TNBC prognosis, which is displayed in Supplementary Table S3.

EMG genetic alteration and DNA methylation effects on TNBC. a Distribution of genetic alteration of eight EMGs; b EMG genetic alteration did not impact the OS of patients with TNBC; c The DNA methylation of ten EMGs impacted its expression
Identification of Potential Drugs for Patients with TNBC
The public pharmacogenomics database Genomics of Drug Sensitivity in Cancer (https://www.cancerrxge-ne.org) was used to predict the chemotherapy response of prognostic subtypes with 15 common chemotherapy drugs (AZD8055, bicalutamide, docetaxel, GDC0941, GSK269962A, imatinib, lapatinib, metformin, MG.132, MK.2206, NVP.BEZ235, pazopanib, PF.4708671, PHA.665752, temsirolimus), showing a significant difference in the estimated IC50 between high-risk and low-risk groups (Fig. 8d). The p values of the 15 drugs are shown in Supplementary Table S4. Patients in the low-risk group showed higher sensitivity to these drugs than the high-risk group did.

a–c Box plots of the cytolytic score, mRNAsi, and T cell inflammation scores. d The chemotherapy response estimation of 15 potential therapeutic drugs for TNBC
CMAP Analysis to Predict Drug Treatment
Further, the CMap database (https://portals.broadinstitute.org/cmap/) was used to screen for small molecule drugs. The top 15 drugs/molecules with positive correlations and the top 15 drugs/molecules with negative correlations were obtained from CMap. Neratinib is a recommended drug for patients with TNBC. Other drugs/molecules, including lerisetron, aspirin, sodium butyrate, amonafide, and fluspirilene, may serve as potential drugs for patients with TNBC. The results are presented in Supplementary Table S5.
Discussion
TNBC is one of the most commonly malignant tumors in the world and displays poorer prognosis compared to other breast cancer subtypes [14]. Currently, the treatment of TNBC mainly includes adjuvant chemotherapy plus surgical resection for an early stage and adjuvant chemotherapy for an advanced stage. However, as a result of the lack of targetable receptors and tumor heterogeneity, TNBC represents a clinically challenging endeavor [16]. Accurate prognosis is a critical factor of precision medicine in stratifying risks and developing an optimal management plan. Therefore, it is essential to establish a novel biomarker to predict the prognosis and provide reliable treatment targets of TNBC.
It is known that EMT is a cellular process which has been associated with a variety of tumor functions, including tumor initiation, tumor stemness, tumor cell migration, and drug resistance [17]. TNBC is one of the most studied malignant tumors, and a growing body of research has revealed that it is closely associated with EMT. The study by Grasset et al. confirmed that the chemoresistance and frequent early relapse in patients with TNBC may be associated with the presence of different EMT cell states in the same tumor [18]. Several signaling pathways are also related to the EMT process in TNBC, like Notch signaling, Hedgehog signaling, Wnt/β-catenin signaling, and PI3K/AKT signaling pathways [19]. Shome and Ghosh indicated that the synergistic action of targeting both the EGFR and Wnt/β-catenin signaling pathways may overcome the EMT process in TNBC cells to treat this disease [20]. In addition, many efforts have been made to further explore the specific effect of the EMT process on the prognosis of breast cancer. Groza et al. observed five EMT signature genes closely related to invasive breast carcinoma prognosis and developed a prognostic model [21]. And a prognostic model based on EMT-related lncRNAs in TNBC was also developed in another study [22]. However, it is a pity that few studies have further explored the clinical significance of mRNA in the prognosis of TNBC and performed the prediction of targeted drugs.
In this study, we constructed and validated 13 EMGs to predict the prognosis of patients with TNBC using TCGA and METABRIC datasets (BOP1, GLIPR2, CCL25, CCND2, HMOX1, CEACAM5, CEACAM1, P2RY2, PMEPA1, ITGB3, IRS2, PEG10, and HOOK1). Of these genes, CCND2 expression inhibited cancer cell growth and migration ability, and CCL25, also known as thymus-expressed chemokine (TECK), is the ligand for CCR9. Previously, Hung et al. [23] showed that CCL25 was not expressed in TNBC tumors by IHC; however, our study showed that although the expression of CCL25 was low, CCL25 was expressed in TNBC tumors, primarily in cancer cells [24]. Transmembrane prostate androgen-inducible protein (PMEPA1) is highly expressed in TNBC and can promote the formation of TNBC cell colonies, tumor spheroids, and cell migration [25]. Experimental studies have shown that P2RY2 regulates the migration and invasion of breast cancer cells, which may be related to EMT pathways. In addition, the expression of P2RY2 was negatively correlated with the differentiation grade of breast tumors, and TNBC is a subtype of breast cancer with a lower degree of differentiation, so the expression of P2RY2 may be higher, which is also consistent with our results [26]. Thus, these genes could be used to develop an EMT-related gene signature to predict the prognosis of TNBC.
Survival analyses demonstrated that 13 genes were significantly associated with the prognosis of patients with TNBC, and a poorer prognosis was observed in high-risk patients. A nomogram based on independent prognostic factors, including the signature and pathological stage, was constructed to improve the accuracy of clinical decisions and develop stratified treatment, showing favorable predictive performance for prognosis in patients with TNBC. Decision curve analysis (DCA) determined that the prognostic signature had better predictive potential than the pathological stage alone [27].
Immune cells play dual roles in the tumor microenviroment (TME), either tumor-promoting or tumor-suppressing activities [28]. The value of EMT-related genes for predicting the prognosis of patients with TNBC and its influence on the TME of TNBC can be helpful for the diagnosis and treatment of TNBC and ultimately improve patient prognosis. In this study, the high-risk group had a lower infiltration of immune-promoting cells such as CD4+ T memory cells, NK T cells, and dendritic cells than the low-risk group. This indicated that the EMT process might protect tumors from the intrinsic antitumor immune response by creating an immunosuppressive microenvironment. It has previously been shown that perturbation of EMT reprograms TNBC immuno-subtype immune microenvironment [29]. CD4 and CD8 T cells play critical roles in immunosurveillance against cancer [30], while myeloid dendritic cells confer an increase in immune activation by antigen presentation and activation of those T cells [28, 31]. The lack of these cells could contribute to the poor prognosis of these high-risk group patients. The same result was found by Chae et al. in non-small cell lung cancer, confirming the prognostic value of the risk score [32]. Low-risk patients had a higher proportion of antitumor immune cells, immune signature, and CYT score, while high-risk patients had a higher score of stromal components induced by the TGFβ pathway. In previous studies, Sulaiman et al. reported the complexity of the TGFβ family pathway and discussed how the dysregulation of the TGFβ pathway promotes oncogenic attributes in TNBC, negatively affecting patient prognosis [33].
Further analysis showed that the 13 EMG signatures predicted the responses to several common chemotherapeutic agents. In our study, the low-risk group was more sensitive to chemotherapy drugs such as bicalutamide, docetaxel, imatinib, and lapatinib than the high-risk group, suggesting that EMT in the tumor microenvironment can influence the response to immune checkpoint inhibitor therapy which is in agreement with a study by Taki et al. [34]. The present study indicated that bicalutamide inhibited the proliferation and invasion of TNBC cells by targeting the AR signaling pathway and downregulating MMP-2/-9 protein expression [35]. Docetaxel, a semisynthetic paclitaxel analogue, causes cell cycle arrest by interfering with microtubule function. TGFβ-induced EMT contributes to the reduced response of TNBC to bicalutamide and docetaxel [36]. The co-treatment with clinically validated inhibitors of c-ABL (imatinib) and EGFR (lapatinib) results in synergistic growth inhibition in TNBC cells via suppression of albumin-induced EMT, but the mechanism remains to be determined [37, 38]. We found that low-risk patients may benefit more from immunotherapy and chemotherapy than high-risk patients. This implies that more studies are needed to develop new treatment strategies or multidrug combinations to improve the prognosis of high-risk patients.
This study had some limitations. First, it was a retrospective study, and independent prospective cohorts are needed to validate the prognostic model developed. Second, this risk model was based on the public databases TCGA and METABRIC, which have incomplete data, such as personal clinical history and treatment intervention. Third, further experimental studies are needed to elucidate the underlying mechanism of the EMT gene signature in TNBC.
Conclusions
A novel prognostic model based on 13 EMT-related genes was constructed and validated to predict the overall survival of patients with TNBC. Furthermore, according to the EMT analysis and evaluation of chemotherapy efficacy, the features indirectly demonstrated that low-risk patients were more likely to benefit from immune therapy and chemotherapy. This study provides a new reference for further exploration of the mechanisms of EMT and tumor immunity, as well as insights to guide personalized treatment of patients with TNBC.
References
Ai D, Yao J, Yang F, et al. TRPS1: a highly sensitive and specific marker for breast carcinoma, especially for triple-negative breast cancer. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2021;34(4):710-9.
Urueña C, Sandoval TA, Lasso P, et al. Evaluation of chemotherapy and P2Et extract combination in ex-vivo derived tumor mammospheres from breast cancer patients. Scientific reports. 2020;10(1):19639.
Wolff AC, Hammond ME, Hicks DG, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31(31):3997-4013.
Vagia E, Mahalingam D, Cristofanilli M. The landscape of targeted therapies in TNBC. Cancers (Basel). 2020;12(4):916.
Chaudhary LN, Wilkinson KH, Kong A. Triple-negative breast cancer: who should receive neoadjuvant chemotherapy? Surg Oncol Clin N Am. 2018;27(1):141–53.
Jhan JR, Andrechek ER. Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics. 2017;18(17):1595–609.
Zhang N, Ng AS, Cai S, Li Q, Yang L, Kerr D. Novel therapeutic strategies: targeting epithelial-mesenchymal transition in colorectal cancer. Lancet Oncol. 2021;22(8):e358-e68.
Dongre A, Weinberg RA. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20(2):69–84.
Vijay GV, Zhao N, Den Hollander P, et al. GSK3β regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res. 2019;21(1):37.
Li F, Song QZ, Zhang YF, et al. Identifying the EMT-related signature to stratify prognosis and evaluate the tumor microenvironment in lung adenocarcinoma. Front Genet. 2022;13:1008416.
Wang N, Gu Y, Chi J, et al. Screening of DNA damage repair genes involved in the prognosis of triple-negative breast cancer patients based on bioinformatics. Front Genet. 2021;12:721873.
Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603-7.
Modhukur V, Iljasenko T, Metsalu T, Lokk K, Laisk-Podar T, Vilo J. MethSurv: a web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics. 2018;10(3):277-88.
Won KA, Spruck C. Triple-negative breast cancer therapy: current and future perspectives (Review). Int J Oncol. 2020;57(6):1245–61.
Lamb J, Crawford ED, Peck D, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science (New York, NY). 2006;313(5795):1929-35.
Yan C, Liu Q, Jia R. Construction and validation of a prognostic risk model for triple-negative breast cancer based on autophagy-related genes. Front Oncol. 2022;12: 829045.
Xue W, Sun C, Yuan H, et al. Establishment and analysis of an individualized emt-related gene signature for the prognosis of breast cancer in female patients. Disease markers. 2022;2022:1289445.
Grasset EM, Dunworth M, Sharma G, et al. Triple-negative breast cancer metastasis involves complex epithelial-mesenchymal transition dynamics and requires vimentin. Science translational medicine. 2022;14(656):eabn7571.
Zhang H, Wang J, Yin Y, Meng Q, Lyu Y. The role of EMT-related lncRNA in the process of triple-negative breast cancer metastasis. Biosci Rep. 2021;41(2):BSR20203121.
Shome R, Ghosh SS. Tweaking EMT and MDR dynamics to constrain triple-negative breast cancer invasiveness by EGFR and Wnt/β-catenin signaling regulation. Cell Oncol (Dordr). 2021;44(2):405–22.
Groza IM, Braicu C, Jurj A, et al. Cancer-associated stemness and epithelial-to-mesenchymal transition signatures related to breast invasive carcinoma prognostic. Cancers (Basel). 2020;12(10):3053.
Guo J, Yi X, Ji Z, et al. Development of a prognostic model based on the identification of EMT-related lncRNAs in triple-negative breast cancer. J Oncol. 2021;2021:9219961.
Hung CS, Wang SC, Yen YT, Lee TH, Wen WC, Lin RK. Hypermethylation of CCND2 in lung and breast cancer is a potential biomarker and drug target. Int J Mol Sci. 2018;19(10):3096.
Wang X, Su W, Tang D, et al. An immune-related gene prognostic index for triple-negative breast cancer integrates multiple aspects of tumor-immune microenvironment. Cancers (Basel). 2021;13(21):5342.
Haque MA, Abdelaziz M, Puteri MU, et al. PMEPA1/TMEPAI is a unique tumorigenic activator of AKT promoting proteasomal degradation of PHLPP1 in triple-negative breast cancer cells. Cancers (Basel). 2021;13(19):4934.
Qiu Y, Liu Y, Li WH, Zhang HQ, Tian XX, Fang WG. P2Y2 receptor promotes the migration and invasion of breast cancer cells via EMT-related genes Snail and E-cadherin. Oncol Rep. 2018;39(1):138-50.
He F, Chen Z, Deng W, et al. Development and validation of a novel ferroptosis-related gene signature for predicting prognosis and immune microenvironment in head and neck squamous cell carcinoma. Int Immunopharmacol. 2021;98:107789.
Li Q, Pan Y, Cao Z, Zhao S. Comprehensive analysis of prognostic value and immune infiltration of chromobox family members in colorectal cancer. Front Oncol. 2020;10:582667.
Kim IS, Gao Y, Welte T, et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol. 2019;21(9):1113-26.
Yamamoto Y, Iwahori K, Funaki S, et al. Immunotherapeutic potential of CD4 and CD8 single-positive T cells in thymic epithelial tumors. Sci Rep. 2020;10(1):4064.
Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL. Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol. 2015;194(7):2985-91.
Chae YK, Chang S, Ko T, et al. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci Rep. 2018;8(1):2918.
Sulaiman A, McGarry S, Chilumula SC, Kandunuri R, Vinod V. Clinically translatable approaches of inhibiting TGF-β to target cancer stem cells in TNBC. Biomedicines. 2021;9(10):1386.
Taki M, Abiko K, Ukita M, et al. Tumor Immune Microenvironment during Epithelial-Mesenchymal Transition. Clin Cancer Res. 2021;27(17):4669-79.
Kong Y, Qu F, Yuan X, Yan X, Yu W. Effect of Bicalutamide on the proliferation and invasion of human triple negative breast cancer MDA-MB-231 cells. Medicine (Baltimore). 2020;99(17):e19822.
Wardhani BW, Puteri MU, Watanabe Y, Louisa M, Setiabudy R, Kato MTGF-β-induced TMEPAI attenuates the response of triple-negative breast cancer cells to doxorubicin and paclitaxel. J Exp Pharmacol. 2020;12:17-26.
Lee JY, Chang JW, Yang WS, et al. Albumin-induced epithelial-mesenchymal transition and ER stress are regulated through a common ROS-c-Src kinase-mTOR pathway: effect of imatinib mesylate. Am J Physiol Renal Physiol. 2011;300(5):F1214-22.
Desai K, Aiyappa R, Prabhu JS, et al. HR+HER2- breast cancers with growth factor receptor-mediated EMT have a poor prognosis and lapatinib downregulates EMT in MCF-7 cells. Tumour Biol. 2017;39(3):1010428317695028.
Acknowledgements
We sincerely acknowledge TCGA, METABRIC, and GTEx database for providing transcriptomic and clinical data. And we also thank all individuals who participated in this study and donated samples.
Funding
No funding or sponsorship was received for this study. Liang Yao funded the journal’s Rapid Service and Open Access fees.
Author Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Liang Yao, Bo Zhang and Rong Zhao. Analysis and interpretation of data: Qi Wang, Ya-Jing Zhang, Liu Yang and Zhou-Jun Yuan. The first draft of the manuscript was written by Bo Zhang, Rong Zhao, and Jun Yang. Revising the article critically: Qian-Jun Wang and Liang Yao. All authors read and approved the final manuscript.
Disclosures
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. Bo Zhang, Jun Yang, Qian-Jun Wang, and Liang Yao were affiliated with the Department of Breast Oncology, Shanxi Provincial Cancer Hospital at the time of the study and are now affiliated with the Department of Breast Oncology, Shanxi Hospital Affiliated to Cancer Hospital, Chinese Academy of Medical Sciences, and the Department of Breast Oncology, Cancer Hospital Affiliated to Shanxi Medical University.
Compliance with Ethics Guidelines
This study was based on based on publicly available data and patients were not directly involved in the entire research process; therefore, the need for ethical committee approval and informed consent was waived.
Data Availability
All data generated or analyzed during this study are included in this published article/as supplementary information files.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Zhang, B., Zhao, R., Wang, Q. et al. An EMT-Related Gene Signature to Predict the Prognosis of Triple-Negative Breast Cancer. Adv Ther 40, 4339–4357 (2023). https://doi.org/10.1007/s12325-023-02577-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-023-02577-z