
Abstract
Introduction
The effects of continuous infusions of ciprofol on its pharmacodynamic and pharmacokinetic properties and safety profiles in healthy Chinese subjects were evaluated.
Methods
In this open-label, randomized, two-way cross-over study, subjects received initial doses of continuous ciprofol/propofol as an infusion for 30 min in part 1 (n = 8) and a bolus dose in part 2 (n = 8) followed by maintenance infusions for a total of 4 h in part 1 and 12 h in part 2. Each subject participated in both parts with a washout time of at least 40 h.
Results
The safety and tolerability parameters of ciprofol were similar to those of propofol, and all treatment-emergent adverse events were mild. The incidences of injection pain and respiratory depression in subjects given ciprofol were lower than those receiving propofol. The pharmacokinetic parameters Cmax, tmax, t1/2, λz and MRT for ciprofol and propofol were similar, while CL, Vd and Vss were statistically significantly different. Pharmacodynamic parameters including the Richmond Agitation Sedation Scale and bispectral index profiles of ciprofol were similar to those of propofol.
Conclusion
Ciprofol has potential for clinical application for continuous intravenous infusion to maintain sedation for 12 h with the same safety, tolerability and efficacy as propofol.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Ciprofol (HSK 3486) is a novel 2,6-disubstituted phenol derivative, which exhibits tighter binding to the γ-aminobutyric acid type A (GABAA) receptor than propofol |
Previous phase 1 trials with ciprofol as a single and as an initial bolus dose, followed by a maintenance infusion dose for 30 min, have been conducted |
In the present study, continuous infusions of ciprofol for 4 and 12 h have been performed with propofol as the positive control |
What was learned from the study? |
Cmax, tmax, t1/2, λz and MRT as well as RASS and BIS profiles were similar for ciprofol and propofol with only mild treatment-emergent adverse events |
CL, Vd and Vss were statistically significantly different and the incidence of injection pain and respiratory depression was lower in the ciprofol group compared to the propofol group |
Ciprofol for continuous intravenous infusion to maintain sedation for 12 h was not inferior to propofol regarding tolerability and efficacy |
Introduction
Propofol is an intravenous anesthetic agent that is widely used for the induction and maintenance of anesthesia and sedation of patients in worldwide clinical practice [1,2,3]. Because propofol is formulated as an oil-in-water emulsion, due to its aqueous insolubility [4], up to 60% of patients experience injection pain. One-third of the pain levels have been described as severe or excruciating [5,6,7,8]. Injection pain induced by propofol is a well-known adverse event (AE). Numerous clinical maneuvers have been tried to suppress injection pain but with limited success [9,10,11]. Studies on the use of propofol for the induction and long-term maintenance of sedation reported that it caused respiratory depression [12,13,14,15].
Ciprofol (HSK3486), a novel 2,6-disubstituted phenol derivatives, is a close analog of propofol which exhibits tighter binding to the γ-aminobutyric acid type A (GABAA) receptor [16], has relatively lower lipophilicity and has a more suitable steric bulk than propofol. Phase 1 single-dose escalation studies have been conducted in healthy Chinese subjects (0.4–0.9 mg/kg) to evaluate the safety and pharmacokinetic-pharmacodynamic (PK/PD) properties of ciprofol after a single intravenous dose for the induction of anesthesia for colonoscopy procedures [17]. Another phase 1c study (NCT04029766) was conducted in healthy males in Australian to evaluate the safety and PK/PD properties of ciprofol after an initial bolus dose, followed by a maintenance infusion dose for 30 min. These studies showed that ciprofol was safe and well tolerated by subjects. The PD properties of ciprofol doses used for induction and maintenance of anesthesia were similar to those of propofol, with the advantage that ciprofol produced a lower incidence of injection pain [17,18,19]. The effects of ciprofol on respiration, blood pressure and heart rate were not inferior to those elicited by propofol [17, 19].
However, continuous infusion of ciprofol for sedation has not been studied in healthy subjects. Therefore, the present study was divided into two parts, with subjects in part 1 and part 2 given 4- or 12-h infusions of ciprofol, respectively. Propofol was used as the positive control. The primary purpose was to assess the safety of continuous infusion of ciprofol for sedation of healthy subjects. The second purpose was to compare the safety and PK/PD properties of ciprofol with those of propofol.
Methods
Study Design
The phase 1 trial following the part 1 study (4-h continuous infusion) and part 2 study (12-h continuous infusion) results was designed as an open-label, randomized, two-period, two-way cross-over, propofol-positive controlled study. Eight healthy subjects were enrolled to receive a 4-h ciprofol/propofol infusion in the part 1 study. If subjects tolerated the drug well, another eight subjects were assigned to receive a 12-h ciprofol/propofol infusion in part 2. The washout period between the two periods in each part of the study was at least 40 h (Fig. 1).

Flowchart of the trial
The trial was conducted at the Clinical Trials Center, West China Hospital, Sichuan University, from 9 January 2019 to 8 November 2019 and was approved by the Independent Ethics Committee of West China Hospital (approval no. 2018-146). Written informed consent forms were signed by all subjects who participated in the trial, which was prospectively registered at Clinicaltrials.gov (NCT03745625). The study was conducted in compliance with the Declaration of Helsinki and ICH-GCP guidelines for experiments on humans.
Sentinel Subjects
For safety reasons, the first two subjects in both the part 1 and part 2 studies were designated as sentinel subjects. The remaining six subjects in the two parts of the study were not enrolled until the two sentinels had completed two periods of study and a further 24-h safety observation with a safety conclusion. Dosage/mode of administration for the remaining six subjects and the sample collection time points for the PK and PD study were adjusted based on the results of the two sentinels, while the dosage/mode of administration in part 2 was adjusted appropriately according to the results of the part 1 study.
Study Population
Subjects (male and female) were included if they met the following criteria: aged 18–49 years; weight ≥ 45 kg with a body mass index between 19.0 and 26.0 kg/m2; in good health based on a physical examination, laboratory tests, a 12-lead electrocardiography (ECG) and vital signs; no potentially difficult airways (Modified Mallampati Score degree 1–3) and SpO2 > 95%; capable of understanding the study.
The exclusion criteria were: allergic to the active drugs or their excipients; having a history of hypersensitivity or any allergic disease and clinically significant medical disease; drug or alcohol abuse; use of any anesthetic taken from 3 days prior to screening or use of any drug treatment taken 14 days prior to randomization. Pregnant or nursing women were excluded from the study.
Study Drug and Administration
Ciprofol lipid emulsion injection was manufactured by Haisco Pharmaceutical Group Co., Ltd, (Shenyang, Liaoning, China). Propofol was manufactured by AstraZeneca Pharmaceuticals (Wilmington, DE, USA).
In part 1 of the study, first two sentinel subjects received an initial infusion of ciprofol at 0.75 mg/kg/h or propofol at 3 mg/kg/h for 0.5 h, followed by a maintenance dose of ciprofol at 0.5 mg/kg/h or propofol at 2 mg/kg/h for 3.5 h. The result showed that it took between 15 min and > 1 h for sedation of subjects who received ciprofol and propofol, respectively, which did not meet the clinical requirements for rapid sedation. Therefore, the dosage regimen was adjusted for the other six subjects in the part 1 study who received an initial infusion of ciprofol at 1 mg/kg/h or propofol at 5 mg/kg/h for 0.5 h. If the bispectral index (BIS) was between 60–80 and the Richmond Agitation Sedation Scale (RASS) was equal to − 3 twice in a row or RASS ≤ − 4, the maintenance dosage was reduced to 0.4 mg/kg/h (ciprofol) or 2 mg/kg/h (propofol), for a total of 4 h.
In the part 2 study, each subject was given a bolus-loading dose of ciprofol 0.4 mg/kg or propofol 2 mg/kg over 2 min, followed by a maintenance infusion dose of ciprofol at 0.4 mg/kg/h or propofol at 2 mg/kg/h, for a total of 12 h. Within the first 30 min, if RASS ≥ 0, RASS = − 1, BIS ≥ 70 or agitation occurred after the bolus-loading dose, the investigator could decide whether to administer a top-up dose of 0.05–0.1 mg/kg of ciprofol or 0.25–0.5 mg/kg/h of propofol within 30 s. At most, three doses were administered at a minimum interval of 2 min for each increasing dose. If BIS ≤ 45 or RASS ≤ − 4 occurred during the loading dose or additional administration, the investigator stopped the loading dose.
Randomization
Subjects who met the inclusion/exclusion criteria were assigned study IDs in the order of their screen number. The study ID determined the allocation of the study drugs according to the randomization list, which was produced by SAS (version 9.4).
Plasma Sample Collections for PK Analysis
In the part 1 study, 3 ml venous blood samples were drawn before the drug infusion was initiated, at 5 min and 10 min during the initial infusion, at the end of the initial infusion, and 5 min, 15 min, 30 min, 1 h and 2 h during the maintenance dose infusion, as well as at the end of the maintenance infusion, 5 min, 10 min, 20 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, 20 h and 24 h after the end of infusion for both ciprofol and propofol. In the part 2 study for both ciprofol and propofol, 3 ml venous blood samples was drawn before and at the end of the bolus-loading dose and at 3 min, 5 min, 10 min, 20 min, 30 min, 45 min, 1 h, 2 h, 4 h and 8 h during the maintenance dose infusion, as well as at the end of the infusion and 5 min, 10 min, 20 min, 30 min, 1 h, 2 h, 3 h, 12 h, 16 h, 20 h and 24 h after the infusion was stopped.
Measurements
Plasma Concentrations of Ciprofol and Propofol
Ciprofol and propofol concentrations were determined by validated liquid chromatography methods with tandem mass spectrometric detection (LC/MS–MS). A Shimadzu LC-30 AD system coupled to an AB SciexTriple Quad 6500+ mass spectrometer and AB SciexAnalyst™ 1.6.3 software were used. The internal standards of ciprofol and propofol were HSK23287 and propofol-d17, respectively.
PK Parameters
The PK evaluations of ciprofol and propofol were performed by standard non-compartmental analysis using Phoenix WinNonlin version 7.0. The following parameters were calculated from the individual concentration-time data: maximum plasma concentration (Cmax), plasma concentration at the end of infusion (C4h for part 1 study and C12h for part 2 study) and time to maximum plasma concentration (Tmax). The area under the plasma concentration-time curve from 0 to the last quantifiable concentration (AUC0–t) was computed using the linear trapezoidal rule. AUC from time zero extrapolated to infinity (AUC0–∞) was calculated as the sum of AUC0-t and a residual part extrapolated to infinite time. Terminal elimination half-life (t1/2), total clearance (CL), distribution volume (Vd), steady-state distribution volume (Vss) and mean residence time (MRT) were also calculated.
PD Parameters
In the part 1 study, RASS and BIS were measured before and at 5-min intervals after the beginning of initial dosing, at 15-min intervals after the beginning of the maintenance dose and at 2-min intervals from the end of the infusion until RASS returned to 0 three times in a row.
In part 2 of the study, RASS and BIS were measured before and at 2-min intervals for 10 min, at 5-min intervals for 10 to 30 min, at 15-min intervals from 30 min to the end of the infusion and at 2-min intervals from the end of the infusion to RASS returning to 0 three times in a row.
The following parameters were carefully documented: average time to onset of sedation (time from the beginning of study drug administration to the first time RASS was ≤ − 1); average duration of sedation (TRASS, cumulative time of − 3 ≤ RASS ≤ − 1; TBIS, 60 ≤ BIS ≤ 80); average time of recovery from sedation (time from the end of drug administration to three measurements of RASS in a row = 0); average time of starting the maintenance dose (time from the beginning of drug administration to the first RASS ≤ − 3 twice in a row or RASS ≤ − 4); average time of RASS first reaching − 1, − 2 and − 3. The number of dose adjustments and sedation satisfaction scores were also recorded.
Safety
All subjects were under continuous medical supervision throughout the study. All AEs were recorded and blood pressure, heart and respiratory rates measured at 5-min intervals. ECG monitoring was conducted throughout the administration period. Injection pain was evaluated and special attention was paid to hypotension, bradycardia, apnea and hypoxemia. All information including unexpected symptoms or medical conditions was recorded regardless of the relationship to the study drug. Numerous clinical laboratory tests including biochemical tests were performed.
Statistical Analysis
Statistical Analysis System (SAS®) software was used to analyze the study data; descriptive statistics were carried out for demographic, PK and PD data. All data are presented as the mean ± standard deviation (SD) or mean (CV, coefficient of variance) unless indicated otherwise. Comparison of PK and PD parameters between ciprofol and propofol was performed using analysis of variance (ANOVA), with effects for subjects nested within sequence, period and formulation. The subject effect was random, and all other effects were fixed. Cmax, C4h, C12h, AUC0–t and AUC0–∞ were normalized for the dose and log-transformed before ANOVA. The Wilcoxon rank sum test was used when appropriate. All AEs summaries were restricted only to treatment-emergent AEs (TEAEs). Subjects who experienced the same AE (in terms of the MedDRA preferred term) more than once per treatment were only counted once.
Results
Disposition in Phase 1 and the Study Population
A total of 16 healthy subjects (8 in part 1 and 8 in part 2) were enrolled in the study. In part 1, data for the two sentinel subjects were excluded from the PK, PD analyses as the dosage regimen was different from the other six subjects. Therefore, six subjects in the part 1 study and eight subjects in the part 2 study were evaluable for PK and PD data, respectively. The safety analyses were performed with eight subjects in the part 1 and part 2 studies. Demographic data of the subjects are presented in Table S1. There were no significant differences in the demographic characteristics of the subjects in the two parts of the study.
Dosage and Dose Adjustments
The median doses of ciprofol and propofol used, median cumulative doses per unit body weight and average cumulative doses per unit body weight for ciprofol and propofol are listed in Table 1. The median times of top-ups in the ciprofol and propofol groups were similar for the two drugs in the part 1 and part 2 studies, but the top-up times in the part 2 study were higher than those in the part 1 study due to the 12-h infusion (Table 1).
Primary Outcomes: Safety and Tolerability
There were no significant protocol violations among the 16 subjects who were all included in the safety analysis set.
Adverse Events
All subjects reported TEAEs (Table 2), which were grade 1 or grade 2, and subjects recovered completely without clinical intervention. After ciprofol administration, one and two subjects reported grade 1 injection pain in the part 1 and part 2 studies, respectively. After propofol administration, seven and seven subjects reported grade 1–3 pain on injection in the part 1 and part 2 studies, respectively.
Vital Signs
In the part 1 study, the heart rate-time curve was analyzed for all eight enrolled subjects, which showed that the mean heart rate of the eight subjects who received ciprofol or propofol fluctuated between 65.5 and 73.5 beats/min or 61.1 and 73.4 beats/min before and after drug administration, respectively (Fig. 2A). In the part 2 study, the heart rate-time curve revealed that the mean heart rate of the eight subjects who received ciprofol or propofol fluctuated between 52.6 and 68.4 beats/min or 52.5 and 71.9 beats/min before and after drug administration (Fig. 2B).

Changes of the A, B mean heart rate (HR) C, D mean systolic blood pressure (SBP) and E, F mean saturation of blood oxygen (SpO2) with different time points in eight subjects injected with ciprofol and propofol
In the part 1 study, the systolic blood pressure-time curve revealed that the mean range of systolic blood pressure (SBP) after ciprofol ranged from 90.8 to 106.6 mmHg, which was a slightly higher level than SBP measured in subjects who received propofol (Fig. 2C) during the first 10 min after infusion. In part 2, the mean range of SBP before and after administration of ciprofol ranged from 91.10 to 106.40 mmHg in the eight subjects. SBP ranged from 91.10 to 107.50 mmHg in the subjects who received propofol (Fig. 2D).
In the part 1 study, the oxygen saturation-time curve showed that average oxygen saturation ranged from 97.7 to 99.4% for the eight subjects who received ciprofol and ranged from 98.0 to 99.5% in subjects who received propofol (Fig. 2E).
In the part 2 study, the oxygen saturation-time curve showed that the average range of oxygen saturation was 97.5–100.0% for the eight subjects who received ciprofol and 97.4–99.4% for subjects who received propofol (Fig. 2F).
Five subjects experienced hypotension after ciprofol administration in the part 1 study (continuous 4-h infusion), which were all grade 1 special interest TEAEs (TEAEsi), with a minimum duration of 0.75 h and a maximum duration of 3.25 h. After propofol administration, six subjects (75.0%) developed drug-related hypotension (6 times in total), with all being grade 1 TEAEsi. The shortest persistent duration of hypotension was 0.17 h and the longest duration 2.50 h. In both the ciprofol and propofol groups, the impacts of AEs on vital signs were all mild, and all subjects recovered without any intervention treatment.
In the part 2 study, there were ten TEAEsi in the eight subjects who received ciprofol, including hypotension, apnea, bradycardia and hypoxemia. There were eight cases of hypotension in eight subjects, with the shortest duration being 0.17 h and the longest 12.09 h. One subject (B015) experienced bradycardia on one occasion of 0.07-h duration, and one subject (B015) experienced apnea once with a duration of 30 s. In the propofol group, seven subjects experienced eight hypotension episodes, with the shortest duration being 4.91 h and the longest 11.67 h. A total of five apnea events occurred in five subjects, with the shortest duration being 30 s and the longest 85 s. One subject once experienced a TEAEsi of hypoxemia with a duration of 35 s.
During the part 2 study, when comparisons were made between the two groups, the incidence of vital signs TEAEs, drug-related TEAEs and TEAEsi in the ciprofol group had a higher trend than in the propofol group. Respiratory-related TEAEs only occurred in the propofol group, which probably explains why ciprofol is better than propofol in eliciting respiratory depression. However, in the part 2 study it was noteworthy that all AEs were grade 1 or grade 2 and were relieved without any clinical intervention.
Secondary Outcomes
Pharmacokinetic Properties
The plasma concentration at the end of the 4- and 12-h infusions were not statistically significantly different (P > 0.05) for ciprofol or propofol.
The concentration-time curves for all subjects are presented in Fig. 3. The plasma concentration-time profiles of ciprofol were similar to those of propofol but with a lower concentration. The plasma concentration of ciprofol and propofol increased rapidly after the initial dose and initially decreased after the maintenance infusion dose, followed by a slow increase. Neither ciprofol or propofol reached a steady-state concentration in the part 1 and part 2 studies.

Change in plasma concentrations of ciprofol and propofol in subjects in the part 1 and part 2 studies. A, B Part 1 study; C, D part 2 study
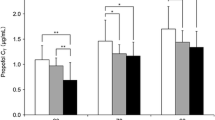
The dose-normalized Cmax (part 1 study only), C4h, C12h and AUCs of ciprofol were greater than for propofol, and the difference between ciprofol and propofol was statistically significant (Table 3). In the part 1 study, most PK parameters for ciprofol and propofol were similar except for CL. In the part 2 study, CL, Vd and Vss of ciprofol and propofol were statistically significantly different but tmax, t1/2, λz and MRT were similar. The inter-subject variability in the PK parameters of ciprofol was higher than for propofol.
Pharmacodynamic Properties
RASS and BIS
Sedation was induced in all 14 subjects in the 2 studies. The onset of and recovery from sedation profiles were similar for ciprofol and propofol. PD parameters showed no statistically significant differences between ciprofol and propofol (Table 4). The individual PD parameters are presented in Figure S1 and Fig. 4. The time of onset of sedation, starting maintenance dose and recovery from sedation showed large inter-subject variability.

BIS-time profiles and BIS-RASS profiles of ciprofol and propofol in subjects in the part 1 and part 2 studies. A, C, E Part 1 study; B, D, F part 2 study
The onset of sedation occurred in the concentration range of 181–520 ng/ml for ciprofol and 734–1630 ng/ml for propofol in the part 1 study. In the part 2 study, subjects quickly reached a deep sedation with RASS equal to − 3 after the bolus loading dose (at the first sampling time point of 2 min). The concentrations of ciprofol and propofol were maintained between 149–540 ng/ml and 913–1690 ng/ml during the maintenance dose infusion, respectively. After the end of the maintenance dose infusion, the plasma concentration rapidly decreased, with a 50% decline within 10 min in the part 1 study accompanied by subjects regaining consciousness. In the part 2 study, subjects returned to consciousness within 6 min, while the time for the plasma concentration to decrease by 50% was between 5 min and 2 h (Fig. 4A–C).
The mean RASS-time and BIS-time profiles of ciprofol were similar to those of propofol (Fig. 4A–D). The change trend of mean RASS time was consistent with the BIS time and TRASS was similar to TBIS (Fig. 4E, F).
Discussion
Ciprofol was developed to induce and maintain anesthesia and sedation in clinical practice. Its sedation effect lasting 30 min, demonstrated by RASS and BIS changes, was proven in previous clinical trials (NCT04029766 on submission), which showed that the onset of sedation was rapid, the depth of sedation was deepened with an increase in the ciprofol dose/plasma exposure and the duration and recovery times of subjects were prolonged [17].
The present study is the first to assess the safety and PK/PD properties of continuous infusions of ciprofol for sedation in healthy subjects for 4 or 12 h. The safety characteristics including the incidence of drug-related TEAEs of ciprofol were slightly lower than for propofol and hypotension; bradycardia, apnea and hypoxemia were common AEs during sedation/anesthesia. Special attention should be paid to these by physicians in future clinical practice. In addition, our study demonstrated that ciprofol had a slightly better effect on respiratory depression, and the effects on blood pressure and heart rate were not inferior to those of propofol. Apnea happened very soon (1–5 min) after the initial bolus dose, which indicated that increasing doses in a short time could quickly achieve sedation but were prone to induce apnea. Therefore, the initial bolus dose should be decreased in the next phase 2 study. We suggest that the loading dose of propofol should be 0.5–1 mg/kg for a 0.5–5-min infusion, the initial maintenance dose reduced to 1.5 mg/kg/h, with an adjustment dose range of 0.25–0.5 mg/kg for 0.5–5 min and 0.3–4 mg/kg/h for the maintenance dose. The loading dose of ciprofol was 0.1–0.2 mg/kg for a 1–5-min infusion, with an adjustment dose range of 0.05–0.1 mg/kg/h, an initial maintenance dose of 0.3 mg/kg/h, with the subsequent maintenance dose 0.06–0.8 mg/kg/h and the adjustment range 0.05–0.1 mg/kg/h. During the study period, vital signs and the electrocardiogram should be monitored in real time.
It was also clear that the incidences of TEAEs, drug-related TEAEs and TEAEsi in subjects reported in the part 2 study were higher than in the part 1 study, which indicated that with the loading dose and the prolonged infusion time, the incidence of TEAE increased accordingly. These findings should alert physician caution, although the severity of AEs was only grade 1–2 and subjects recovered quickly without any intervention treatment being required.
Pain on injection is a common drug-related TEAE during propofol administration [20], and severe injection pain increases the patient's tension and anxiety and directly or indirectly affects the stability of anesthesia induction [21]. The high concentration of propofol in the aqueous phase causes the pain on injection [5]. Previous studies demonstrated that the pain on injection was reduced when the propofol concentration was lower in the aqueous phase of the emulsion [13, 22, 23]. In the present study, the pain on ciprofol injection was mild, probably because ciprofol was formulated as an oil-in-water emulsion because of its aqueous insolubility [4]. In addition, the higher hydrophobicity and lower plasma concentration of ciprofol compared to propofol may have led to the reduction in injection pain.
For the secondary endpoints, we discussed the plasma concentrations and PK/PD profiles, RASS and BIS for both of the study drugs. The plasma concentrations were similar at the end of continuous infusion for 4 and 12 h. There was no drug accumulation during continuous infusions. t1/2 and Vd were reported to be increased during prolonged infusions [24,25,26,27] because of the slow return of the drug from poorly perfused tissue back into the blood stream. This phenomenon was not observed in our study, a finding that might be explained by the fact that the steady-state plasma concentration was not reached. The time for propofol to reach steady-state plasma concentration has been reported to be about 24–30 h [28,29,30].
In the part 1 study, subjects quickly recovered from sedation within 10 min after the end of drug infusion, as previously reported [31]. This finding was in accordance with the rapid decline of drug concentration, dropping below that necessary to maintain sedation (1000 µg/ml) [32, 33], with an overall mean decrease of 50% over the first 10 min [34, 35]. This likely was the result of the rapid redistribution of the drug into peripheral tissues. In the part 2 study, the time at which the plasma concentration decreased to 50% was 5 min to 2 h. Even so, subjects in the part 2 study also quickly recovered from sedation. These observations can be explained by the fact that the plasma concentration at the end of the 12 h infusion was lower than after the 4-h infusion, and we woke the subjects at 2-min intervals from the end of infusion and found that they easily recovered from sedation.
To simulate mild-to-moderate level sedation, the target range value of BIS and RASS should be preset between 60 to 80 and − 1 to − 3. The BIS-time profile was in accordance with the RASS-time profile and BIS comparable to RASS reaching − 3, − 2 and − 1, gradually increasing, with the average scores being close to 60, 70 and 80, respectively. TRASS and TBIS were similar with a good correlation (r = 0.742–0.862) in this study, which confirmed the sedation indexes were selected appropriately. The average cumulative dose per unit weight of propofol administered was 5.28 and 5.78 times that of ciprofol. The average onset and recovery times and the average duration of sedation induced by ciprofol appeared to be slightly briefer than for propofol, but the difference did not reach statistical significance. The concentration of ciprofol and propofol producing sedation ranged between 149–540 and 913–1690 ng/ml. This result indicated that the potency of ciprofol was about five times that of propofol, a finding consistent with the results of preclinical animal efficacy experiments and early clinical efficacy studies.
In the present study, the PK parameters of ciprofol in part 1 showed large inter-subject variability especially for Vd and t1/2, while lower inter-subject variability was found in the part 2 study; perhaps the lesser numbers of subjects in part 1 might have contributed to this result. The inter-individual variability of the average time to onset of sedation, average time of starting the maintenance dose, average time of recovery from sedation and the number of dose adjustments were significant, which could be explained by individual sensitivity to ciprofol and propofol. Other PD parameters showed lower inter-individual variability but larger inter-individual variability in PK parameters. The possible reasons might be that dosing adjustments and continuous stimulation for evaluating the sedation effect were necessary to keep the PD parameters stable, while the variability of the drug distribution and elimination characteristics in different individuals was relatively great.
Conclusions
Both ciprofol and propofol induced a rapid onset of sedation, and subjects quickly woke after continuous infusion ceased. Ciprofol has potential clinical application for administration via continuous intravenous infusion to maintain sedation for at least 12 h and has similar safety and tolerability to propofol.
References
Bryson HM, Fulton BR, Faulds D. Propofol. An update of its use in anaesthesia and conscious sedation. Drugs. 1995;50(3):513–59. https://doi.org/10.2165/00003495-199550030-00008.
Fulton B, Sorkin EM. Propofol. An overview of its pharmacology and a review of its clinical efficacy in intensive care sedation. Drugs. 1995;50(4):636–57. https://doi.org/10.2165/00003495-199550040-00006.
Vanlersberghe C, Camu F. Propofol. Handb Exp Pharmacol. 2008;182:227–52. https://doi.org/10.1007/978-3-540-74806-9_11.
Baker MT, Naguib M. Propofol: the challenges of formulation. Anesthesiology. 2005;103(4):860–76. https://doi.org/10.1097/00000542-200510000-00026.
Jalota L, Kalira V, George E, Shi YY, Hornuss C, Radke O, Pace NL, Apfel CC. Prevention of pain on injection of propofol: systematic review and meta-analysis. BMJ. 2011;342: d1110. https://doi.org/10.1136/bmj.d1110.
McLeskey CH, Walawander CA, Nahrwold ML, Roizen MF, Stanley TH, Thisted RA, White PF, Apfelbaum JL, Grasela TH, Hug CC Jr. Adverse events in a multicenter phase IV study of propofol: evaluation by anesthesiologists and postanesthesia care unit nurses. Anesth Analg. 1993;77(4 Suppl):S3-9.
Tan CH, Onsiong MK. Pain on injection of propofol. Anaesthesia. 1998;53(5):468–76. https://doi.org/10.1046/j.1365-2044.1998.00405.x.
White PF. Propofol formulation and pain on injection. Anesth Analg. 2002;94(4):1042. https://doi.org/10.1097/00000539-200204000-00058.
Meincken M, Norman C, Arevalo O, Saman DM, Bejarano T. Anesthesia onset time and injection pain between buffered and unbuffered lidocaine used as local anesthetic for dental care in children. Pediatr Dent. 2019;41(5):354–7.
Fix WC, Chiesa-Fuxench ZC, Shin T, Etzkorn J, Howe N, Miller CJ, Sobanko JF. Use of a vibrating kinetic anesthesia device reduces the pain of lidocaine injections: a randomized split-body trial. J Am Acad Dermatol. 2019;80(1):58–9. https://doi.org/10.1016/j.jaad.2018.08.011.
Nimmaanrat S, Jongjidpranitarn M, Prathep S, Oofuvong M. Premedication with oral paracetamol for reduction of propofol injection pain: a randomized placebo-controlled trial. BMC Anesthesiol. 2019;19(1):100. https://doi.org/10.1186/s12871-019-0758-y.
Claeys MA, Gepts E, Camu F. Haemodynamic changes during anaesthesia induced and maintained with propofol. Br J Anaesth. 1988;60(1):3–9. https://doi.org/10.1093/bja/60.1.3.
Doenicke AW, Roizen MF, Rau J, Kellermann W, Babl J. Reducing pain during propofol injection: the role of the solvent. Anesth Analg. 1996;82(3):472–4. https://doi.org/10.1097/00000539-199603000-00007.
Langley MS, Heel RC. Propofol. A review of its pharmacodynamic and pharmacokinetic properties and use as an intravenous anaesthetic. Drugs. 1988;35(4):334–72. https://doi.org/10.2165/00003495-198835040-00002.
Robinson BJ, Ebert TJ, O’Brien TJ, Colinco MD, Muzi M. Mechanisms whereby propofol mediates peripheral vasodilation in humans. Sympathoinhibition or direct vascular relaxation? Anesthesiology. 1997;86(1):64–72. https://doi.org/10.1097/00000542-199701000-00010.
Qin L, Ren L, Wan S, Liu G, Luo X, Liu Z, Li F, Yu Y, Liu J, Wei Y. Design, synthesis, and evaluation of novel 2,6-disubstituted phenol derivatives as general anesthetics. J Med Chem. 2017;60(9):3606–17. https://doi.org/10.1021/acs.jmedchem.7b00254.
Teng Y, Ou M, Wang X, Zhang W, Liu X, Liang Y, Zuo Y, Zhu T, Liu B, Liu J. Pharmacokinetic and pharmacodynamic properties of ciprofol emulsion in Chinese subjects: a single center, open-label, single-arm, dose-escalation phase 1 study. Am J Transl Res. 2021 (in press).
Wei Y, Qiu G, Lei B, Qin L, Chu H, Lu Y, Zhu G, Gao Q, Huang Q, Qian G, Liao P, Luo X, Zhang X, Zhang C, Li Y, Zheng S, Yu Y, Tang P, Ni J, Yan P, Zhou Y, Li P, Huang X, Gong A, Liu J. Oral delivery of propofol with methoxymethylphosphonic acid as the delivery vehicle. J Med Chem. 2017;60(20):8580–90. https://doi.org/10.1021/acs.jmedchem.7b01133.
Teng Y, Ou M, Wang X, Zhang W, Liu X, Liang Y, Li K, Wang Y, Ouyang W, Weng H, Li J, Yao S, Meng J, Shangguan W, Zuo Y, Zhu T, Liu B, Liu J. Efficacy and safety of ciprofol for the sedation/anesthesia in patients undergoing colonoscopy: phase IIa and IIb multi-center clinical trials. Eur J Pharm Sci. 2021;164: 105904. https://doi.org/10.1016/j.ejps.2021.105904.
Cheng D, Liu L, Hu Z. Prevention of anesthesia-induced injection pain of propofol in pediatric anesthesia. Pak J Med Sci. 2017;33(3):752–6. https://doi.org/10.12669/pjms.333.12026.
Martinez RA, Hurff A, DeGeorge KC, DeGeorge BR Jr. How to minimize the pain of local anesthetic administration. J Fam Pract. 2020;69(4):172–8.
McIntosh MP, Rajewski RA. Comparative canine pharmacokinetics-pharmacodynamics of fospropofol disodium injection, propofol emulsion, and cyclodextrin-enabled propofol solution following bolus parenteral administration. J Pharm Sci. 2012;101(9):3547–52. https://doi.org/10.1002/jps.23195.
Moore GD, Walker AM, MacLaren R. Fospropofol: a new sedative-hypnotic agent for monitored anesthesia care. Ann Pharmacother. 2009;43(11):1802–8. https://doi.org/10.1345/aph.1M290.
Albanèse J, Auffray JP, Lacarelle B, Martin C, Gouin F. Pharmacokinetics of propofol administrated by prolonged continuous intravenous infusion in man. Anesthesiology. 1987;67(3):A667–A667. https://doi.org/10.1097/00000542-198709001-00667.
Cockshott ID. Propofol ('Diprivan’) pharmacokinetics and metabolism—an overview. Postgrad Med J. 1985;61(Suppl 3):45–50.
Cockshott ID, Briggs LP, Douglas EJ, White M. Pharmacokinetics of propofol in female patients. Studies using single bolus injections. Br J Anaesth. 1987;59(9):1103–10. https://doi.org/10.1093/bja/59.9.1103.
Frenkel C, Schuttler J, Ihmsen H, Heye H, Rommelsheim K. Pharmacokinetics and pharmacodynamics of propofol/alfentanil infusions for sedation in ICU patients. Intensive Care Med. 1995;21(12):981–8. https://doi.org/10.1007/bf01700659.
Albanese J, Martin C, Lacarelle B, Saux P, Durand A, Gouin F. Pharmacokinetics of long-term propofol infusion used for sedation in ICU patients. Anesthesiology. 1990;73(2):214–7. https://doi.org/10.1097/00000542-199008000-00004.
Elfstrom J. Drug pharmacokinetics in the postoperative period. Clin Pharmacokinet. 1979;4(1):16–22. https://doi.org/10.2165/00003088-197904010-00002.
Kanto J, Gepts E. Pharmacokinetic implications for the clinical use of propofol. Clin Pharmacokinet. 1989;17(5):308–26. https://doi.org/10.2165/00003088-198917050-00002.
McKeage K, Perry CM. Propofol: a review of its use in intensive care sedation of adults. CNS Drugs. 2003;17(4):235–72. https://doi.org/10.2165/00023210-200317040-00003.
Beller JP, Pottecher T, Lugnier A, Mangin P, Otteni JC. Prolonged sedation with propofol in ICU patients: recovery and blood concentration changes during periodic interruptions in infusion. Br J Anaesth. 1988;61(5):583–8. https://doi.org/10.1093/bja/61.5.583.
Murdoch JA, Grant SA, Kenny GN. Safety of patient-maintained propofol sedation using a target-controlled system in healthy volunteers. Br J Anaesth. 2000;85(2):299–301. https://doi.org/10.1093/bja/85.2.299.
Bailie GR, Cockshott ID, Douglas EJ, Bowles BJ. Pharmacokinetics of propofol during and after long-term continuous infusion for maintenance of sedation in ICU patients. Br J Anaesth. 1992;68(5):486–91. https://doi.org/10.1093/bja/68.5.486.
Barr J, Egan TD, Sandoval NF, Zomorodi K, Cohane C, Gambus PL, Shafer SL. Propofol dosing regimens for ICU sedation based upon an integrated pharmacokinetic-pharmacodynamic model. Anesthesiology. 2001;95(2):324–33. https://doi.org/10.1097/00000542-200108000-00011.
Acknowledgements
We thank Shanghai Xihua Scientific Co., Ltd., for their efforts in pharmacokinetic analysis. We also appreciate the contribution of the Mosim Co., Ltd., for their valuable support of the statistical analyses.
Funding
The study and the Journal’s Rapid Service and Open Access Fees were funded by Sichuan Haisco Pharmaceutical Group Co., Ltd. This study was supported by the Clinical Research Incubation Project, West China Hospital, Sichuan University (grant number 2019HXFH032), National Major Science and Technology Projects of China (grant number 2017ZX09304023). The funding bodies had no role in data collection, analysis and interpretation of data or writing of the manuscript.
Authorship
All named authors met the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole and have given their approval for this version to be published.
Author Contributions
Conceptualization: YK and JM. Investigation: CH, XO, YT, SS, YW, XZ, YK, JM. Visualization: JM. Writing-original draft: CH. Supervision: JM. Funding acquisition: YK, JM.
Disclosures
Chao Hu, Xiaofeng Ou, Yi Teng, Shiqing Shu, Ying Wang, Xiaohong Zhu, Yan Kang and Jia Miao declare that they have nothing to disclose.
Compliance with Ethics Guidelines
The trial was approved by the Independent Ethics Committee of West China Hospital (approval no. 2018-146). Written informed consent forms were signed by all subjects who participated in the trial. The study was conducted in compliance with the Declaration of Helsinki and ICH-GCP guidelines for experiments on humans.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding authors
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Hu, C., Ou, X., Teng, Y. et al. Sedation Effects Produced by a Ciprofol Initial Infusion or Bolus Dose Followed by Continuous Maintenance Infusion in Healthy Subjects: A Phase 1 Trial. Adv Ther 38, 5484–5500 (2021). https://doi.org/10.1007/s12325-021-01914-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-021-01914-4