
Abstract
Background
Psoriasis vulgaris is commonly treated with topical corticosteroids and vitamin D analogues. Although potent and super-potent topical corticosteroids are very effective at clearing psoriasis, with short-term reactive treatment durations, symptoms usually recur after treatment discontinuation, necessitating long-term disease management strategies. A foam formulation of calcipotriol and betamethasone dipropionate (Cal/BD foam), consisting of calcipotriol 50 μg/g and betamethasone dipropionate 0.5 mg/g, is approved for the daily treatment of psoriasis for up to 4 weeks. Here, we describe a clinical trial protocol for evaluating the long-term safety and efficacy of twice-weekly Cal/BD foam as a proactive topical maintenance therapy for plaque psoriasis for up to 52 weeks.
Objective
The aim of this trial was to evaluate the safety and efficacy of Cal/BD foam when applied twice weekly for up to 52 weeks as proactive maintenance therapy, with the goal of preventing or delaying disease relapse as long as possible while minimizing adverse effects.
Methods
Once-daily Cal/BD foam treatment responders from an initial 4-week open-label period were randomized to receive Cal/BD foam or foam vehicle applied to previously cleared plaques twice weekly for up to 52 weeks. In case of relapse, affected subjects in either group received rescue therapy with once-daily Cal/BD foam for 4 weeks on active areas. Thus, the trial (NCT02899962) compared the long-term use of Cal/BD foam in a proactive approach with a conventional, reactive approach.
Planned Outcomes
Efficacy endpoints included the time to first relapse, the number of relapse-free days, and the number of relapses during the maintenance phase. Safety assessments included adverse events, incidence of rebound, local safety and tolerability scores, and effects on calcium metabolism and hypothalamic–pituitary–adrenal axis function.
Trial registration
ClinicalTrials.gov identifier, NCT02899962.
Similar content being viewed by others


Why carry out this study? |
Optimal management of psoriasis should include strategies for both the initial, rapid relief of symptoms and the long-term prevention of disease relapse; however, long-term use of potent and super-potent steroids as monotherapy is associated with safety concerns |
We sought to determine if the use of twice-weekly Cal/BD aerosol foam in a novel, proactive treatment regimen for up to 52 weeks could prevent or delay disease relapse in patients with psoriasis vulgaris while minimizing local and systemic adverse events |
The PSO-LONG trial is the first randomized, double-blind, long-term study of its kind to use a topical treatment to maintain psoriasis status as “clear” or “almost clear” for up to 1 year using a patient-friendly approach with just twice-weekly application of a single, fixed-combination product |
What was learned from the study? |
We hypothesized that the time to first relapse in patients with previously cleared skin would be prolonged in those who received proactive therapy with twice-weekly Cal/BD foam as compared with the outcome for patients treated with a conventional, as-needed, reactive approach in which active drug was only administered after relapse occurred |
Superior efficacy of the proactive management with Cal/BD foam was anticipated to be evidenced by a greater number of relapse-free days and fewer relapses compared with controls; a favorable long-term safety profile was also projected |
The outcomes of this novel study will provide valuable insights into the ability of Cal/BD foam to provide a solution for the long-term management of psoriasis in patients with all disease severities and thereby advance the treatment options that can maintain clear skin for patients with this chronic, recurring disease |
Digital Features
This article is published with digital features to facilitate understanding of the article. You can access the digital features on the article’s associated Figshare page. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12834647.
Introduction
Background on Psoriasis
Psoriasis is a chronic, immune-mediated, multisystem disease that primarily affects the skin and joints [1, 2]. The most common form of this condition, plaque psoriasis, manifests as sharply demarcated, scaling, and erythematous lesions that may vary in shape and size [1, 3]. Plaques are predominantly located on the extensor sides of the knees and elbows, but they may also be anywhere on the body [1, 3].
Epidemiology
Estimates suggest that psoriasis affects as many as 7.5 million people in the USA and more than 125 million people worldwide [4,5,6]. Within this population, approximately 80% of patients have mild-to-moderate and/or localized disease that can be successfully treated with topical medications; approximately 20% of patients have moderate-to-severe disease that often requires treatment with systemic medications [7,8,9]. Despite the numerous treatment options that are currently available for psoriasis, more than half of patients report being dissatisfied with their treatment, and a significant number of patients continue to have residual plaques that require long-term maintenance therapy [7].
Current Treatment Strategies
Because there is no cure for psoriasis, treatment strategies aim to clear active disease sites and prolong symptom-free periods [7]. Currently available topical agents for mild-to-moderate psoriasis include corticosteroids, vitamin D analogues, tazarotene, anthralin, calcineurin inhibitors, and tar [8, 9]. Potent biologic therapies have been developed for use in patients with moderate-to-severe psoriasis [10]; nevertheless, topical therapies continue to be an effective means to complement systemic therapies in this hard-to-treat population [8, 9]. In fact, almost all patients utilizing systemic therapy continue the use of topical therapies, which highlights the importance of topicals for effective management of chronic psoriasis. Topical corticosteroids are the mainstay of therapy for the management of mild-to-moderate psoriasis [8, 9]. These agents, available in many strengths, formulations, and combinations, exert their therapeutic effects via anti-inflammatory, antiproliferative, and immunosuppressive pathways that contribute to their use for psoriasis and other immune-mediated disorders [8, 9, 11]. Their therapeutic advantages notwithstanding, topical corticosteroids carry the risk of local and systemic adverse events (AEs), such as suppression of the hypothalamic–pituitary–adrenal (HPA) axis, which may complicate long-term use [8, 9, 11]. Furthermore, some patients have a phobia about using topical corticosteroids, and their anxiety concerning possible overapplication of a topical steroid may lead to reduced compliance and thus poor treatment outcomes [12].
Various studies have demonstrated the therapeutic benefits of alternating between different topical agents on different days, such as adding a weekday nonsteroidal agent to a weekend pulse therapy regimen [13,14,15,16,17]. A common sequential regimen with topical therapy consists of applying halobetasol propionate (HP), a potent topical corticosteroid, once daily in the morning, and using calcipotriol ointment, a vitamin D analogue, once daily in the evening for approximately 1 month [18]. Subsequently, calcipotriol is administered twice daily on weekdays, and HP is administered twice daily on weekends, until symptoms fully resolve, at which point treatment will be tapered [18].
Although evidence supports the use of sequential therapy in maintaining clinical improvements [19,20,21], patient adherence can be problematic. Adherence to topical therapy may improve with the use of a single, fixed-combination treatment, rather than expecting the patient to apply two or more distinct topical agents in the right order [9, 22,23,24]. In addition, the ability to effectively maintain relapse-free periods for psoriasis may not be possible using an as-needed, reactive treatment approach, whereas implementing a proactive strategy may reduce relapse rates [25, 26]. The concept of proactive therapy, defined as the administration of a long-term, low-dose, intermittent treatment, has been successfully used for atopic dermatitis [27] and scalp psoriasis [26, 28], but it has not yet been rigorously examined for psoriasis affecting the body. We conducted this trial on the use of a fixed-combination treatment for long-term proactive maintenance therapy for psoriasis, with the objective of obtaining rigorous scientific data to support this therapeutic approach and its potential use in clinical practice as a part of chronic psoriasis management.
Long-Term Clinical Studies of Topical Agents for Treatment of Psoriasis
Although topical steroids play a key role in the treatment of psoriasis, it is generally recommended not to exceed 4 weeks of daily topical steroid monotherapy because of the potential risk of AEs. This limitation on treatment duration of topical steroids can be problematic for the long-term management of psoriasis, and it has led to the common use of a reactive, as-needed treatment approach that fails to address relapse prevention.
The need for long-term treatment strategies is acknowledged, and such strategies might combine treatment with nonsteroidal agents, such as vitamin D analogues, to minimize steroid exposure. However, there is no standard as to which topical combinations are best, resulting in highly variable treatment plans. Proactive strategies are an additional long-term treatment option [25, 26, 29], but this novel approach has only recently begun to be formally tested, and there is some uncertainty as to the optimal treatment parameters. A consequence of this piecemeal approach to long-term psoriasis treatment is that it is unclear which of these methods is optimal, as well as how long the efficacy and safety of such treatments can be maintained. There is thus a considerable need for clinical trials that are designed to address relapse prevention.
Long-term studies of topical agents for the treatment of psoriasis that are currently available have certain limitations. These include, but are not limited to, the nature of open-label studies, relatively small patient populations, the absence of a placebo arm, and the reliance on self-reported patient measures as the final study outcomes [30,31,32]. Moreover, the studies may be characterized by inadequate safety assessments, nonstandardized treatment dosing and administration, and lack of consensus on definitions for treatment success and endpoints. A recent review and position statement on the treatment of psoriasis with vitamin D3 analogues, corticosteroids, and compound formulations revealed that few, large, long-term, randomized, blinded clinical trials have been conducted [30].
Among the few long-term studies that have been done, one found that an ointment containing the vitamin D analogue calcipotriol 50 μg/g plus the corticosteroid betamethasone dipropionate 0.5 mg/g (Cal/BD) was safe and well tolerated when administered over a 52-week period [33]. Specifically, this study found that Cal/BD ointment demonstrated a favorable safety profile when used on its own or alternating every 4 weeks with calcipotriol [33]. The incidence rate of AEs known to be associated with long-term corticosteroid use was low, and adrenal function was assessed in a subset of subjects [33]. An additional study demonstrated that as-needed use of the fixed-combination ointment for up to 52 weeks was not associated with signs of adrenal suppression [34]. However, rather than comparing a proactive vs a reactive treatment strategy, the objectives of these studies were to compare the safety [33] and efficacy [35] of three different treatment regimens including (1) use of the two-compound formulation by itself for 52 consecutive weeks, (2) use of the two-compound product and calcipotriol alone in alternating 4-week periods for up to 52 weeks, and (3) use of the two-compound product for 4 weeks followed by 48 weeks of calcipotriol ointment [35]. Assessments of efficacy were limited to investigator global assessments of disease severity and patient-reported treatment satisfaction [35].
In a long-term study examining scalp psoriasis, the safety and efficacy of a topical suspension of Cal/BD that was formulated for use on the scalp were compared with calcipotriol alone during a reactive, 52-week paradigm in which patients stopped and restarted treatment as needed [36]. When administered reactively over a 52-week period, the two-compound scalp formulation did not increase the rate of adverse drug reactions or AEs associated with long-term use of topical corticosteroids, and it further demonstrated increased efficacy relative to reactive treatment with calcipotriol alone [36]. However, proactive therapy was not assessed, as this study used the conventional, as-needed reactive approach to treating scalp psoriasis, and the efficacy assessments were limited to the Physician’s Global Assessment (PGA) of disease severity and patient ratings of treatment efficacy [36].
In the PSO-TOP study, patients applied a Cal/BD topical suspension once daily for 8 weeks followed by as-needed application for an additional 56 weeks [37]. The objective of the study was to compare the clinical response related to treatment optimization vs standard of care, not to assess the safety and efficacy of topical therapy per se.
Finally, in a long-term open-label study, subjects with moderate-to-severe plaque psoriasis were treated for 8 weeks with HP plus tazarotene (HP/TAZ) lotion, with additional treatment being administered in 4-week cycles as needed for up to 1 year [38]. This study reported on the long-term safety profile of HP/TAZ, including assessments of AEs and tolerability. However, neither HPA axis function nor efficacy was assessed using this reactive treatment regimen, and therefore no conclusions can be drawn regarding these aspects of long-term treatment with HP/TAZ, the long-term effects of HP/TAZ on adrenal suppression, or its long-term efficacy.
As these long-term studies of topical treatments for psoriasis vulgaris indicate, there remains a significant gap in the available data for comparing the conventional reactive approach vs proactive treatment strategies. This will require robust randomized clinical trials that incorporate longer treatment durations and appropriate sample sizes to achieve adequate power and improve clinical relevance. Such trials will need to include well-defined and extensive assessments of efficacy and safety, because the available data from existing long-term studies are limited.
Calcipotriene Plus Betamethasone Dipropionate (Cal/BD) Foam as a Treatment Option for Psoriasis Vulgaris
An alcohol-free aerosol foam formulation of the fixed-combination Cal/BD that incorporates a non-skin-drying emollient vehicle was recently developed to improve treatment for patients with psoriasis [23]. Compared with the Cal/BD ointment [9, 23], the Cal/BD foam has enhanced penetration and is considered to be more cosmetically acceptable [9]. These features could increase patient adherence and lead to improved efficacy in a real-world setting [23]. Currently, Cal/BD foam is indicated for the daily topical treatment of plaque psoriasis in patients 12 years of age and older for up to 4 weeks [39].
The efficacy and safety of Cal/BD foam have been evaluated in three 4-week clinical trials involving approximately 1100 randomized subjects [23, 40, 41]. In these short-term studies, once-daily Cal/BD foam demonstrated greater efficacy in treating psoriasis than did Cal/BD ointment [40] and also greater efficacy than foams containing the individual active ingredients [41]. Importantly, Cal/BD aerosolized foam was well tolerated, with a favorable safety and tolerability profile that was similar to that of Cal/BD ointment and of the individual active ingredients [23, 40, 41]. A 12-week study in approximately 460 subjects revealed greater disease improvements with Cal/BD foam than with Cal/BD gel, and without detection of any new safety signals [42]. Together, these studies demonstrate the benefits of Cal/BD foam for the topical treatment of mild, moderate, and severe plaque psoriasis without compromise in safety and tolerability.
Trial Rationale
In the PSO-LONG trial, the use of Cal/BD foam for maintenance therapy was investigated in a novel, randomized, double-blind, long-term (12-month) study. The long-term safety and efficacy profile were rigorously assessed, and the trial compared a proactive approach of a fixed, twice-weekly maintenance regimen with use of a conventional, reactive approach (i.e., the vehicle control arm). Specifically, during the long-term maintenance phase of the trial, subjects were randomly assigned to apply either Cal/BD foam or vehicle twice weekly to a disease site that was previously cleared (or almost cleared) during the preceding open-label phase. In the event of relapse, which was defined as an increase in the PGA to a score of at least “mild” (PGA = 2), subjects were directed to apply Cal/BD foam to the active area once daily for 4 weeks, regardless of group assignment. This phase 3 clinical trial was thus the first long-term comparison of the standard, as-needed approach that is utilized in clinical practice with a novel regimen that was designed to increase patient adherence (and thus treatment outcomes) by including only twice-weekly proactive application of a single, dual-combination product.
The objective of the PSO-LONG trial was to assess the ability of Cal/BD foam to maintain initial clinical efficacy for a longer period of time than twice-weekly application of a vehicle. In addition, this trial tested the hypothesis that proactive treatment would prolong the time to relapse and reduce relapse frequency while maintaining safety, and that patients receiving long-term maintenance therapy would demonstrate a superior response compared with vehicle patients in terms of the number of relapse-free days.
Although topical corticosteroids and vitamin D analogues are commonly used psoriasis treatments, there are a number of safety considerations associated with their application, particularly on large surface areas or over extended periods of time [9]. Local cutaneous side effects of topical corticosteroids include skin atrophy, telangiectasia, striae distensae, acne, folliculitis, and purpura [8]. The systemic side effects associated with topical corticosteroids are relatively infrequent but can include Cushing’s syndrome, osteonecrosis of the femoral head, cataracts, glaucoma, and HPA axis suppression [8]. Implementing more sophisticated treatment regimens involving corticosteroids, such as with proactive, fixed, twice-weekly application, may allow for longer treatment durations with reduced concern for adrenal suppression [43]. Similarly, although the side effect profile of vitamin D is generally mild, systemic side effects such as hypercalcemia and parathyroid hormone suppression may arise in extreme cases in which patients are applying more than the recommended dosage or have an underlying condition such as impaired renal function or calcium metabolism [8, 9]. Another possible concern with topical corticosteroids is rebound, wherein the disease recurs worse than the pretreatment baseline after the treatment is discontinued [8]. Although rebound has been reported to occur when topical corticosteroids are abruptly discontinued, this phenomenon is poorly characterized and requires further study [8]. Given these important considerations, another goal of the PSO-LONG trial was to evaluate the long-term safety of Cal/BD foam over extended management of psoriasis in a comprehensive manner. Accordingly, both local and systemic safety assessments were included, such as calcium homeostasis and HPA axis changes related to vitamin D analogue and corticosteroid treatment, respectively, as well as the incidence of rebound.
Methods
Study Design
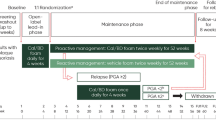
PSO-LONG was a multicenter, prospective trial that consisted of an initial open-label 4-week treatment phase followed by a randomized, double-blind, vehicle-controlled, maintenance phase of up to 52 weeks in subjects with psoriasis vulgaris (Fig. 1). Regular study visits were scheduled every 4 weeks from baseline during the open-label phase through the end of the maintenance phase, as detailed in Table 1.

Trial design. The boxed region and outset highlight the details of the 52-week maintenance phase. ACTH adrenocorticotropic hormone, AE adverse event, Cal/BD calcipotriol 50 μg/g and betamethasone dipropionate 0.5 mg/g, FU follow-up, UNS unscheduled visit, Relapse FU follow-up on relapse
Screening/Washout Phase
Assessment of subject eligibility occurred at a screening visit prior to a washout period. Eligible subjects included adults with psoriasis vulgaris on the trunk and/or limbs rated as at least “mild” according to the PGA, with body surface area (BSA) involvement of 2–30%, and with a modified Psoriasis Area and Severity Index (m-PASI) score of at least 2 (see “Sample Selection” and Table 2). Prior to any trial-related procedure (including washout), signed informed consent was obtained from the subject. A washout period of up to 4 weeks was required for subjects who were currently receiving, or who had recently been treated with, anti-psoriatic treatments or other relevant medication not permitted as defined by the exclusion criteria (see Tables 3 and 4). On completion of the washout period, the subject’s ongoing eligibility for the trial was confirmed at visit 1. If no washout was needed, the subject could enter visit 1 directly.
Open-Label Treatment Phase
After the screening/washout phase, subjects deemed eligible for the trial were enrolled in the initial open-label phase, during which they applied Cal/BD foam once daily on psoriatic lesions on the trunk and/or limbs for 4 weeks. All medication needed for 4 weeks was dispensed at visit 1, with the first application made during the initial visit under the supervision and instruction of the trial staff. At this time, the subjects were given a treatment instruction sheet describing the scenarios to be considered depending on the different treatment phases, and they also received verbal instruction about how to apply the trial medication. Note that there were different types of instructions for open-label, maintenance, and relapse treatment methods, and subjects were always reminded to follow one of the particular instructions to avoid confusion among the different treatment phases. Subjects who achieved treatment success (PGA score of “clear” or “almost clear” with at least a 2-grade improvement from baseline) at week 4 (visit 2) were then randomized in a 1:1 ratio to a maintenance phase of up to 52 weeks with Cal/BD foam or vehicle (maintenance investigational product, IP) twice weekly. Subjects who did not achieve treatment success were discontinued from the trial.
Maintenance Phase
During the randomized maintenance phase, subjects received three cans of maintenance IP every 4 weeks, which was sufficient for the visit interval when used twice per week, as described below. At each visit, the IP, including empty containers dispensed at the previous visit, were returned by the subject. The subjects applied maintenance IP twice weekly 3 or 4 days apart (according to subject preference, and on fixed days throughout the trial, e.g., Thursday and Sunday) on all areas on the trunk and limbs where lesions had cleared or almost cleared during the initial open-label phase or after treatment of a relapse. To improve medication adherence, electronic reminders were sent to the subject every week on the selected application days. Subjects were assessed regularly at clinical visits every 4 weeks. In addition, if, in the opinion of the subject, a relapse (exacerbation of psoriasis) occurred between two regular monthly visits, the subject was assessed by the investigator at an unscheduled visit.
Following confirmation of a relapse, the subject was provided with rescue IP and asked to apply it to any active area that was judged to be at least “mild” according to the PGA, irrespective of whether those areas were active at baseline or were new lesions. The rescue IP was applied once daily on the active areas for 4 weeks, with all seven cans of rescue IP being dispensed to the subjects upon initial confirmation of relapse to ensure adequate supply throughout the 4-week rescue period. If additional areas became active during rescue treatment, those were also treated once daily with rescue IP. During a relapse, areas that were not active continued to receive twice-weekly maintenance treatment, as detailed above. If a score of “clear” or “almost clear” was regained after 4 weeks, the twice-weekly maintenance regimen was restarted on the now “clear” or “almost clear” area(s) according to the original randomization scheme. If a score of “clear” or “almost clear” was not regained after the 4 weeks of once-daily rescue treatment, then the subject exited the trial.
Follow-up Phase
After the maintenance phase, there were three types of follow-up visits (Table 1). Follow-up visit 1 (FU1) was only relevant for subjects with either an ongoing AE or abnormal laboratory findings at the end of treatment. Follow-up visit 2 (FU2) was only conducted for subjects who underwent the adrenocorticotropic hormone (ACTH) challenge test and exhibited signs of altered HPA axis function (serum cortisol concentrations ≤ 18 μg/dL at 30 min after ACTH challenge). If signs of adrenal suppression were present at FU2, additional ACTH challenge tests were performed until adrenal suppression resolved. All subjects were followed up for events of rebound for up to 8 weeks after the last application of maintenance IP. If a rebound was confirmed at follow-up visit 3 (FU3), the subject was followed up for 14 days or until resolution (whichever happened first), and this follow-up period may have continued beyond the end of the clinical trial if the rebound was considered to be a serious AE (SAE).
Sample Selection
It was determined that between 178 and 190 subjects per group (380 total) were needed in order to obtain a power of 90% for a 5% significance level. To achieve a total of 380 randomized subjects, an estimated 832 subjects were screened. This screening and randomization process allowed for a sufficient number of subjects after accounting for an anticipated exponential decline and dropout rate of 30% over 52 weeks. For the HPA axis test, up to 60 subjects (with BSA > 10%) needed to be enrolled to achieve approximately 30 randomized subjects at visit 2. Randomization of these subjects was stratified by trial site, HPA axis testing, and baseline disease severity (mild, moderate, severe). The maximum number of subjects enrolled with “mild” disease according to the PGA was capped at 20% to ensure similar distribution of disease severity at baseline as in previous Cal/BD foam short-term trials and, hence, a similar trial population.
Reasons for withdrawal from the trial included screening failure, lack of efficacy, AEs, withdrawal by the subject, loss to follow-up, death, or other relevant reasons. In addition, as previously noted, subjects were withdrawn if they did not achieve treatment success after the initial 4-week open-label phase and/or if they were unable to score “clear” or “almost clear” after a 4-week treatment for a relapse.
Measurements
Investigator and patient assessments were recorded during each of the scheduled study visits (Table 1), as well as during all unscheduled visits.
Clinical Assessments
All dermatologic assessments of the treatment area were performed by a dermatologist, certified physician’s assistant, advanced registered nurse practitioner, or general practitioner experienced in treating psoriasis vulgaris. The investigator made a global assessment of the disease severity of psoriasis of the trunk and limbs using the 5-point PGA scale, in which 0 = clear, 1 = almost clear, 2 = mild, 3 = moderate, and 4 = severe. This assessment represented the average lesion severity and was based on the condition of the disease at the time of evaluation.
Investigators also assessed the extent and severity of clinical signs of the subject’s psoriasis. These assessments excluded the head (face and scalp), thus yielding an m-PASI score. The extent of psoriatic involvement was recorded for the arms, trunk, and legs using a scale that ranges from 0 to 6, with 0 indicating no involvement and 6 indicating 90–100% involvement. The severity of the psoriatic lesions in each of the three areas was recorded for clinical signs of redness, thickness, and scaliness, with each clinical sign being scored on a 5-point scale, in which 0 = none, 1 = mild, 2 = moderate, 3 = severe, and 4 = very severe. These individual assessments were used to calculate a single measure of disease extent and severity (i.e., the m-PASI score).
The involvement of psoriasis vulgaris on the trunk and limbs was also measured by the investigator’s assessment of BSA involvement. This measurement was recorded as a percentage of the total BSA, estimating that the surface of the subject’s full, flat palm (with all five fingers) equated to approximately 1% of the total BSA.
Finally, during the baseline visit, the investigator selected a target lesion of at least 3 cm along its longest axis and recorded its location in detail so that it could be easily identified and assessed during all subsequent visits. The severity of the target lesion continued to be assessed throughout the study for clinical signs of redness, thickness, and scaliness, per the scales noted above.
Subject Assessments
Subjects made self-assessments of quality of life (QOL) and health status/symptoms as specified in the schedule of trial procedures (Table 1). These self-assessments included the Dermatology Life Quality Index (DLQI), the psoriasis-specific version of the EuroQoL 5-dimensional questionnaire (EQ-5D-5L-PSO), and the Work Productivity and Activity Impairment: Psoriasis (WPAI:PSO) scale, which were completed at the trial site on an electronic tablet. Subjects completed the Psoriasis Symptom Severity Inventory (PSI), which is an eight-item measure that assesses symptoms of itch, redness, scaling, burning, cracking, stinging, flaking, and pain, as scheduled (Table 1) and at home, on an eDiary device that was provided to them for use throughout the study. The subject’s global assessment of disease severity was made on site at all scheduled and unscheduled visits, and was based on the condition of the disease at the time of the evaluation and not in relation to the condition at an earlier visit or time. To ensure unbiased answers for questionnaires that were completed on site, the patient-reported outcomes (PROs) were completed prior to any other assessments.
Safety and Laboratory Assessments
Safety assessments included AEs, which were collected by medically qualified personnel at all visits throughout the study duration. Subjects were particularly monitored for any cutaneous side effects that can occur with topical corticosteroids and vitamin D analogues. The assessment of local safety and tolerability consisted of signs assessed by the investigator and symptoms reported by the subjects. The investigator assessed application-site reactions for perilesional signs of erythema, edema, dryness, and erosion. The subject assessed the symptom of “application-site burning or pain.”
Blood and urine samples were collected as scheduled (Table 1) for planned laboratory assessments. Blood samples were analyzed for calcium, albumin, and 25-OH vitamin D, as well as for biomarkers related to systemic inflammation, cardiovascular disease, and metabolic syndrome in consenting participants. Additionally, blood samples were used for adrenal function testing in a subset of subjects (approx. 30) whose psoriasis affected more than 10% BSA. HPA axis function was considered as suppressed if serum cortisol was no greater than 18 μg/dL at 30 min after cosyntropin injections administered during testing at weeks 4, 28, and 56 (trial end) or at early withdrawal. Laboratory assessment of the urine samples reported calcium and creatinine levels and the calcium-to-creatinine ratio.
Planned Outcomes
Efficacy Endpoints
The primary efficacy endpoint was the time to first relapse during the maintenance phase, in which relapse was an exacerbation of psoriasis defined by a PGA rating of at least mild. This was calculated as the number of days from randomization to the day at which the subject had the first relapse confirmed. For subjects who either did not experience a relapse or were withdrawn from the trial, the number of days was treated as a censored observation at the day of the end-of-trial visit.
Secondary efficacy endpoints included the number of relapses during the maintenance phase and the number of days each subject was relapse-free, as indicated by a PGA score of “clear” or “almost clear.” Additional exploratory efficacy endpoints included a summary of m-PASI scores by visit, the number of relapse-free subjects at each visit, the time to when PASI-75 was no longer fulfilled, the time to first relapse according to m-PASI, and a summary of the proportion of subjects who obtained “clear” or “almost clear” after treatment relapse separated by the number of relapses. The target lesion scores, the affected BSA over time, the number of active treatment days during the maintenance phase, and the PROs regarding QOL and health status also served as exploratory endpoints.
Safety Endpoints
Planned outcomes from safety assessments included AEs associated with long-term corticosteroid use, the incidence of rebound, and local safety and tolerability assessment scores. Additional safety outcomes included analyses of the effects of treatment on calcium metabolism and HPA axis function.
Data Analysis
Trial Analysis Sets
All subjects enrolled in the trial were accounted for in the clinical trial report. An open-label safety analysis set was defined by including all subjects exposed to treatment with the IP, whether randomized or not. A maintenance phase safety analysis set was defined by excluding subjects from the full analysis set who either received no treatment with IPs following randomization or provided no post-randomization safety evaluations, or both. All randomized subjects were included in the full analysis set and were analyzed for efficacy. A per-protocol analysis set was defined by excluding subjects from the full analysis set who received no treatment with IPs after randomization, provided no efficacy data following the start of maintenance treatment, were known to have taken the wrong IPs throughout the maintenance phase, or did not fulfil the disease-defining criteria.
Subject Disposition
The reasons for leaving the trial during the open-label treatment phase were collected for all included subjects by last visit attended. The reasons for leaving the trial during the maintenance phase were collected for all randomized subjects by treatment group and last visit attended.
Baseline Demographics and Disease Characteristics
Descriptive statistics of baseline demographics and other characteristics were calculated for all subjects included in the trial and for all randomized subjects by treatment group. A summary of age, sex, ethnicity, race, and baseline m-PASI and PGA scores was provided according to study center.
Efficacy Analyses
The primary endpoint was compared between treatments and analyses performed both for the full analysis set (primary) and for the per-protocol analysis set (supportive). The comparison was done using a proportional hazards model with treatment group, trial site, and disease severity at baseline as factors. The estimate of the hazard ratio of Cal/BD relative to vehicle, together with the 95% confidence interval and P value, was calculated. Additionally, the estimated survival curves with confidence intervals were presented graphically for each treatment group, with percentiles of the survival distribution tabulated by treatment group.
The secondary endpoints were analyzed for the full analysis set, with the analysis for the per-protocol analysis set being supportive, as above. Adjustment for multiplicity was done using the Holm–Bonferroni method. Specifically, for the number of relapses during the maintenance phase, the data were analyzed in a Poisson regression model with treatment group, trial site, and disease severity at baseline as factors, with subject as a random effect, and with risk time as an offset. The estimated incidence rate ratio for active treatment group relative to vehicle group with 95% confidence interval and P value was calculated. A sensitivity analysis was also performed in which subjects who at some point did not achieve “clear” or “almost clear” after treatment of a relapse were excluded. For the number of days that each subject was relapse-free, the data were analyzed via analysis of variance (ANOVA) with treatment group, trial site, and disease severity at baseline as factors. The estimated difference between Cal/BD and vehicle groups was calculated with 95% confidence intervals and P value. Two sensitivity analyses were performed with different handling of missing data. In the first sensitivity analysis, the subject’s observed part of the maintenance phase was assumed to be representative for the unobserved part of the maintenance phase when the subject was withdrawn because of a non-drug-related reason. The second sensitivity analysis was based on a subject’s last observation carried forward (LOCF) when the subject was withdrawn because of a non-drug-related reason. If a subject withdrew from the trial for a drug-related reason, then in both sensitivity analyses, the subject was not considered to be in a relapse-free period from the time when the subject left the trial.
All other exploratory analyses of efficacy and PROs were analyzed for the full analysis set. Both m-PASI scores and the number of relapse-free subjects at each visit were summarized by visit and treatment group. PASI-75 was defined as at least a 75% reduction in the m-PASI score from baseline, and the time to when PASI-75 was no longer fulfilled during the maintenance phase was compared between treatments using a proportional hazards model with treatment group, trial site, and disease severity at baseline as factors. Relapse according to m-PASI level was defined as the baseline m-PASI value minus 50% of the reduction in m-PASI score obtained prior to randomization for the maintenance phase. The comparison between treatments of time to first relapse according to m-PASI was made via a proportional hazards model, as above, with the estimated survival curves and confidence intervals calculated for each treatment group. For each treatment group, the number of active treatment days during the maintenance phase was calculated, and the proportion of subjects who obtained “clear” or “almost clear” PGA scores after treatment relapse was summarized by the number of relapses. Target lesion scores and the affected BSA were summarized over time per treatment group.
Analysis of Safety
An analysis of AEs was performed for each phase of the trial using that phase’s corresponding safety population, as described above. AEs were coded during the course of the trial according to the Medical Dictionary for Regulatory Activities and were categorized by preferred terms and system organ class, as well as by treatment group for those recorded during the maintenance phase. An overall summary of the number (percentage) of subjects with any treatment-emergent AE (TEAE), SAE, premature discontinuation from the trial due to an AE, treatment-related AE, and/or severe AE was planned. The severity of each type of AE and the causal relationship to the trial medication for each AE was tabulated by treatment. For the maintenance phase, both the percentage of subjects with AEs and the percentage of subjects with related AEs were compared between treatment groups via chi-square test or Fisher’s exact test (if expected cell count was < 5). AEs associated with long-term topical corticosteroid use were determined along with information about demography and baseline characteristics. Additionally, the number of rebounds occurring both within and after the first 2 months of entering the maintenance phase were summarized by treatment group, and the number of subjects with cases of rebound was calculated.
The local safety and tolerability signs detailed above were summarized for both treatment phases, with data being split by group during the maintenance phase. For the subset of subjects who underwent the ACTH challenge, which was used for the HPA axis test, the number of subjects with serum cortisol concentration ≤ 18 μg/dL at 30 min after the ACTH challenge was summarized over time by treatment group. The change in vital signs from baseline to the end of the open-label phase and from randomization to the end-of-trial visit was also summarized. The change in each of the laboratory parameters from baseline to the end of the open-label phase and from randomization to the end of the trial was summarized as mean, standard deviation, median, minimum, and maximum values, with values classified as “low,” “normal,” or “high” depending on whether they were below, within, or above the reference range.
Strengths and Limitations
The PSO-LONG trial adds data where there is a dearth of evidence from robust, randomized clinical trials that incorporate an extended treatment period. The purpose of the trial was to investigate whether Cal/BD foam can be used by patients to provide safe and efficacious proactive, long-term management of psoriasis. Specifically, the trial investigated whether twice-weekly (i.e., once per day on two nonconsecutive days) application of Cal/BD foam was able to maintain the initial clinical efficacy at PGA 0/1 for an extended period of time, as compared with twice-weekly application of vehicle. Because both treatment groups received Cal/BD foam once daily for 4 weeks for rescue in case of relapse during the maintenance phase, the vehicle arm utilized a treat-as-needed approach and thus reflected the normal clinical use of the product. This allowed for the first-ever comparison of a proactive vs a reactive treatment strategy in a clinical trial evaluating long-term treatment options for disease management in psoriasis.
Comparisons of Cal/BD foam and the two other marketed Cal/BD formulations as an ointment and gel suspension have shown that the systemic safety profile and the short-term AE profile of Cal/BD foam are similar to the profiles of those formulations individually, with no new safety concerns for Cal/BD foam beyond those already described for the ointment and gel suspension. On the basis of this, no specific safety or tolerability concerns were anticipated with the proposed long-term treatment.
In summary, the PSO-LONG study provided evidence for the long-term safety and efficacy of twice-weekly topical Cal/BD foam, the benefits and limitations of proactive therapy, and potential benefits for long-term patient care in clinical practice. It was predicted that proactive management would prolong the time to relapse and reduce the frequency of relapses in patients with psoriasis compared with vehicle, while maintaining a favorable long-term safety profile.
Study Strengths
This was the first randomized, double-blind, 52-week clinical trial that evaluated long-term safety and efficacy outcomes of a proactive treatment regimen. This novel approach to the long-term management of psoriasis was intended to keep patients in a relapse-free status while maintaining a favorable safety profile.
Long-term use of products that contain potent or super-potent topical corticosteroids can lead to adverse cutaneous side effects, such as skin atrophy and striae, as well as adrenocortical suppression and infection [8, 9]. Using intermittent corticosteroid dosing in the maintenance treatment of psoriasis is a commonly recommended approach, as it may control the signs of psoriasis while reducing drug exposure and thereby decreasing the risk of AEs [18]. Thus, the PSO-LONG study significantly augmented knowledge of the therapeutic benefit of Cal/BD foam during the long-term treatment of psoriasis as well as the safety profile of Cal/BD foam, by assessing AEs often associated with long-term corticosteroid treatment for psoriasis.
The present study also evaluated long-term efficacy based on the hypothesis that subjects who received Cal/BD foam twice weekly would achieve superior responses over those receiving vehicle, as indicated by a higher number of relapse-free days during the maintenance phase. The 12-month study duration allowed proper analysis of relapse prevention over a period of time during which subjects would typically be expected to experience relapses. This particular study also extended prior findings on the clinical efficacy of Cal/BD foam [23, 40,41,42].
Finally, this study provided a much-needed comparison of a proactive, twice-weekly topical psoriasis treatment against the reactive approach that is commonly implemented in treatment strategies. This comparison provides valuable evidence on the efficacy and safety of a proactive treatment strategy with a dual-agent topical therapy.
Study Limitations
Currently, there is no clear-cut, evidence-based consensus on the use of a proactive maintenance regimen to keep patients with psoriasis relapse-free. Clinicians may be hesitant to prescribe a proactive, long-term treatment regimen, and patients may be hesitant to comply with such a strategy, because of safety concerns associated with long-term topical corticosteroid use. Further research may be needed to better understand the barriers to use of the proactive treatment method.
Ethics and Dissemination
Written approval for the clinical trial was obtained from the relevant institutional review boards (IRBs), independent ethics committees, and regulatory authorities, as required. The Signatory Investigator (Mark Lebwohl, MD) was overseen by Advarra. The list of IRBs for each individual participating country can be found in the electronic supplementary material. The clinical trial was conducted in accordance with the principles of the World Medical Association, Declaration of Helsinki, and Ethical Principles for Medical Research Involving Human Subjects.
All subjects received written and verbal information concerning the clinical trial. This information emphasized that participation in clinical trials was voluntary and that the subject was permitted to withdraw from the clinical trial at any time and for any reason. All subjects were given an opportunity to ask questions and were given sufficient time to consider before consenting. The subject’s signed and dated informed consent to participate in the clinical trial was obtained prior to any clinical trial-related procedure’s being carried out, in accordance with Good Clinical Practice and all applicable laws and regulations.
The PSO-LONG trial was registered under the National Clinical Trial (NCT) number 02899962 and the EudraCT number 2016-000556-95. Basic information on this clinical trial was posted on the website (https://www.clinicaltrials.gov) before the first subject was enrolled. Results will be made available on the website for LEO Pharma, according to the company’s position on access to clinical trial information. As this was a multicenter clinical trial, no publication by an investigator of his/her trial results shall be made before the first multicenter report is published. LEO subscribes to the joint position of the innovative pharmaceutical industry for public disclosure of clinical trial results in a free, publicly accessible database, regardless of outcome.
References
Langley RG, Krueger GG, Griffiths CE. Psoriasis: epidemiology, clinical features, and quality of life. Ann Rheum Dis. 2005;64(Suppl 2):ii18–23 (discussion ii4–5).
Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol. 2014;70(3):512–6.
Griffiths CE, Christophers E, Barker JN, et al. A classification of psoriasis vulgaris according to phenotype. Br J Dermatol. 2007;156(2):258–62.
Griffiths CEM, van der Walt JM, Ashcroft DM, et al. The global state of psoriasis disease epidemiology: a workshop report. Br J Dermatol. 2017;177(1):e4–e7.
Strober BE, van der Walt JM, Armstrong AW, et al. Clinical goals and barriers to effective psoriasis care. Dermatol Ther (Heidelb). 2019;9(1):5–18.
Takeshita J, Gelfand JM, Li P, et al. Psoriasis in the US Medicare population: prevalence, treatment, and factors associated with biologic use. J Investig Dermatol. 2015;135(12):2955–63.
Bagel J, Gold LS. Combining topical psoriasis treatment to enhance systemic and phototherapy: a review of the literature. J Drugs Dermatol. 2017;16(12):1209–22.
Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis. Section 3. Guidelines of care for the management and treatment of psoriasis with topical therapies. J Am Acad Dermatol. 2009;60(4):643–59.
Stein Gold LF. Topical therapies for psoriasis: improving management strategies and patient adherence. Semin Cutan Med Surg. 2016;35(2 Suppl 2):S36–44 (quiz S5).
Menter A, Strober BE, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol. 2019;80(4):1029–72.
Kim WB, Jerome D, Yeung J. Diagnosis and management of psoriasis. Can Fam Physician. 2017;63(4):278–85.
El Hachem M, Gesualdo F, Ricci G, et al. Topical corticosteroid phobia in parents of pediatric patients with atopic dermatitis: a multicentre survey. Ital J Pediatr. 2017;43(1):22.
Abidi A, Ahmad F, Singh SK. A comparative clinical evaluation of once daily versus alternate day application of topical clobetasol propionate cream in psoriasis. J Clin Diagn Res. 2013;7(1):100–5.
Emer JJ, Frankel A, Sohn A, Lebwohl M. A randomized, double-blind, placebo-controlled study to evaluate the safety and efficacy of ammonium lactate lotion 12% and halobetasol propionate ointment 0.05% in the treatment and maintenance of psoriasis. J Clin Aesthetic Dermatol. 2011;4(2):28–39.
Koo J, Blum RR, Lebwohl M. A randomized, multicenter study of calcipotriene ointment and clobetasol propionate foam in the sequential treatment of localized plaque-type psoriasis: short- and long-term outcomes. J Am Acad Dermatol. 2006;55(4):637–41.
Lebwohl M, Yoles A, Lombardi K, Lou W. Calcipotriene ointment and halobetasol ointment in the long-term treatment of psoriasis: effects on the duration of improvement. J Am Acad Dermatol. 1998;39(3):447–50.
Zampetti A, Barone A, Antuzzi D, et al. Topical preparations for the treatment of psoriasis: results of a retrospective study over 15 years. J Dermatol Treat. 2008;19(3):134–40.
Gupta R, Koo J. Topical sequential therapy. In: Koo J, Levin E, Leon A, Wu J, Lebwohl M, editors. Mild to moderate psoriasis. 3rd ed. Boca Raton: CRC; 2014.
Brodell RT, Bruce S, Hudson CP, et al. A multi-center, open-label study to evaluate the safety and efficacy of a sequential treatment regimen of clobetasol propionate 0.05% spray followed by calcitriol 3 mg/g ointment in the management of plaque psoriasis. J Drugs Dermatol. 2011;10(2):158–64.
Gupta R. Prolonged remission of psoriasis with azathioprine pulse therapy. Indian J Dermatol. 2015;60(4):360–3.
Kubota Y, Yoneda K, Nakai K, et al. Effect of sequential applications of topical tacrolimus and topical corticosteroids in the treatment of pediatric atopic dermatitis: an open-label pilot study. J Am Acad Dermatol. 2009;60(2):212–7.
Girolomoni G, Calzavara Pinton P, Cristaudo A, Cicchetti A. Back to the future: a new topical approach for mild-to-moderate psoriasis. G Ital Dermatol Venereol. 2018;153(3):375–82.
Leonardi C, Bagel J, Yamauchi P, et al. Efficacy and safety of calcipotriene plus betamethasone dipropionate aerosol foam in patients with psoriasis vulgaris–a randomized phase III study (PSO-FAST). J Drugs Dermatol. 2015;14(12):1468–77.
Zaghloul SS, Goodfield MJ. Objective assessment of compliance with psoriasis treatment. Arch Dermatol. 2004;140(4):408–14.
Lavaud J, Mahe E. Proactive treatment in childhood psoriasis. Ann Dermatol Venereol. 2020;147(1):29–35.
Poulin Y, Papp K, Bissonnette R, et al. Clobetasol propionate shampoo 0.05% is efficacious and safe for long-term control of moderate scalp psoriasis. J Dermatol Treat. 2010;21(3):185–92.
Wollenberg A, Ehmann LM. Long term treatment concepts and proactive therapy for atopic eczema. Ann Dermatol. 2012;24(3):253–60.
Saraceno R, Camplone G, D'Agostino M, et al. Efficacy and maintenance strategies of two-compound formulation calcipotriol and betamethasone dipropionate gel (Xamiol® gel) in the treatment of scalp psoriasis: results from a study in 885 patients. J Dermatol Treat. 2014;25(1):30–3.
Ito K, Koga M, Shibayama Y, Tatematsu S, Nakayama J, Imafuku S. Proactive treatment with calcipotriol reduces recurrence of plaque psoriasis. J Dermatol. 2016;43(4):402–5.
Augustin M, Mrowietz U, Bonnekoh B, et al. Topical long-term therapy of psoriasis with vitamin D(3) analogues, corticosteroids and their two compound formulations: position paper on evidence and use in daily practice. J Dtsch Dermatol Ges. 2014;12(8):667–82.
Bagel J, Zapata J, Nelson E. A prospective, open-label study evaluating adjunctive calcipotriene 0.005%/betamethasone dipropionate 0.064% foam in psoriasis patients with inadequate response to biologic therapy. J Drugs Dermatol. 2018;17(8):845–50.
Lambert J, Hol CW, Vink J. Real-life effectiveness of once-daily calcipotriol and betamethasone dipropionate gel vs. ointment formulations in psoriasis vulgaris: final analysis of the 52-week PRO-LONG study. J Eur Acad Dermatol Venereol. 2015;29(12):2349–55.
Kragballe K, Austad J, Barnes L, et al. A 52-week randomized safety study of a calcipotriol/betamethasone dipropionate two-compound product (Dovobet/Daivobet/Taclonex) in the treatment of psoriasis vulgaris. Br J Dermatol. 2006;154(6):1155–60.
Fleming C, Ganslandt C, Leese GP. Short- and long-term safety assessment of a two-compound ointment containing calcipotriene/betamethasone dipropionate (Taclonex/Daivobet/Dovobet ointment): hypothalamic–pituitary–adrenal axis function in patients with psoriasis vulgaris. J Drugs Dermatol. 2010;9(8):969–74.
Kragballe K, Austad J, Barnes L, et al. Efficacy results of a 52-week, randomised, double-blind, safety study of a calcipotriol/betamethasone dipropionate two-compound product (Daivobet/Dovobet/Taclonex) in the treatment of psoriasis vulgaris. Dermatology. 2006;213(4):319–26.
Luger TA, Cambazard F, Larsen FG, et al. A study of the safety and efficacy of calcipotriol and betamethasone dipropionate scalp formulation in the long-term management of scalp psoriasis. Dermatology. 2008;217(4):321–8.
Reich K, Zschocke I, Bachelez H, et al. A Topical Treatment Optimization Programme (TTOP) improves clinical outcome for calcipotriol/betamethasone gel in psoriasis: results of a 64-week multinational randomized phase IV study in 1790 patients (PSO-TOP). Br J Dermatol. 2017;177(1):197–205.
Lebwohl MG, Sugarman JL, Gold LS, et al. Long-term safety results from a phase 3 open-label study of a fixed combination halobetasol propionate 0.01% and tazarotene 0.045% lotion in moderate-to-severe plaque psoriasis. J Am Acad Dermatol. 2019;80(1):282–5.
LEO pharma. Enstilar® [package insert]. Madison: LEO Pharma; 2017.
Koo J, Tyring S, Werschler WP, et al. Superior efficacy of calcipotriene and betamethasone dipropionate aerosol foam versus ointment in patients with psoriasis vulgaris—a randomized phase II study. J Dermatol Treat. 2016;27(2):120–7.
Lebwohl M, Tyring S, Bukhalo M, et al. Fixed combination aerosol foam calcipotriene 0.005% (Cal) plus betamethasone dipropionate 0.064% (BD) is more efficacious than Cal or BD aerosol foam alone for psoriasis vulgaris: a randomized, double-blind, multicenter, three-arm, phase 2 study. J Clin Aesthetic Dermatol. 2016;9(2):34–41.
Paul C, Stein Gold L, Cambazard F, et al. Calcipotriol plus betamethasone dipropionate aerosol foam provides superior efficacy vs. gel in patients with psoriasis vulgaris: randomized, controlled PSO-ABLE study. J Eur Acad Dermatol Venereol. 2017;31(1):119–26.
Levin E, Gupta R, Butler D, Chiang C, Koo JY. Topical steroid risk analysis: differentiating between physiologic and pathologic adrenal suppression. J Dermatol Treat. 2014;25(6):501–6.
Acknowledgements
The authors thank the participants of the study. Dr. Warren is supported by the NIHR Manchester Biomedical Research Centre.
Funding
This study was funded by LEO Pharma. LEO Pharma are also funding the journal’s Rapid Service and Open Access Fees.
Medical Writing, Editorial, and Other Assistance
Writing, editorial support, and formatting assistance were provided by p-value communications. Support for this assistance was funded by LEO Pharma.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contribution
LSG, JA-L, J-PL, RW, SKT, LK, PY, and ML all provided substantial contributions to the conception and design of the work, as well as the acquisition, analysis, and interpretation of data. All authors have contributed toward the preparation of the manuscript, have approved the final submitted version, and have agreed to be listed as authors.
Disclosures
Dr. Stein Gold is a consultant, investigator and/or speaker for LEO Pharma, Arcutis, Dermavant, Pfizer, AbbVie, Lilly, and Ortho Derm. Dr. Alonso-Llamazares is an advisor and investigator for LEO Pharma, Celgene, and speaker for Celgene, Lilly, Dermira, and Ortho Derm. Dr. Lacour is an investigator for Amgen, Boehringer Ingelheim, Celgene, Dermira, AbbVie, Janssen, Lilly, LEO Pharma, Novartis, Pfizer, Regeneron, and Sanofi; a speaker for Celgene, AbbVie, Lilly, LEO Pharma, Novartis, and Sanofi; a consultant for Novartis; and serves as an advisor for Lilly, LEO Pharma, and Sanofi. Dr. Warren reports consulting fees from AbbVie, Almirall, Amgen, Arena, Avillion, Bristol Myers Squibb, Boehringer Ingelhein, Celgene, Janssen, LEO Pharma, Lilly, Novartis, Pfizer, Sanofi, and UCB; and reports research grants from AbbVie, Almirall, Amgen, Celgene, Janssen, Lilly, LEO Pharma, Novartis, Pfizer, and UCB. Dr. Tyring has conducted studies sponsored by LEO Pharma. Dr. Kircik has served as an investigator, speaker, consultant, or advisor for AbbVie, Almirall, Amgen, Arcutis, Bristol Myers Squibb, Dermira, Eli Lilly, Novartis, Dr. Reddys, and Valeant. Dr. Yamauchi is a consultant, investigator, and/or speaker for AbbVie, Arcutis, Amgen, Bristol Myers Squibb, Dermavant, Janssen, LEO Pharma, Lilly, Novartis, Ortho-Dermatologics, Sun Pharma, and UCB.
Compliance with Ethics Guidelines
Written approval for the clinical trial was obtained from the relevant institutional review boards, independent ethics committees, and regulatory authorities, as required. The Signatory Investigator (Mark Lebwohl, MD) was overseen by Advarra. The list of IRBs for each individual participating country can be found in the electronic supplementary material. The clinical trial was conducted in accordance with the principles of the World Medical Association, Declaration of Helsinki, and Ethical Principles for Medical Research Involving Human Subjects. Written informed consent was obtained from subjects prior to enrollment.
Data Availability
Data sharing is not applicable to this article, as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Digital Features
To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.12834647.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Stein Gold, L., Alonso-Llamazares, J., Lacour, JP. et al. PSO-LONG: Design of a Novel, 12-Month Clinical Trial of Topical, Proactive Maintenance with Twice-Weekly Cal/BD Foam in Psoriasis. Adv Ther 37, 4730–4753 (2020). https://doi.org/10.1007/s12325-020-01497-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01497-6