
Abstract
Introduction
The aims of this study were to characterize the single-dose pharmacokinetics (PK) of the major active metabolites of ozanimod, CC112273 and CC1084037, and to evaluate the effect of gemfibrozil (a strong inhibitor of cytochrome P450 [CYP] 2C8), itraconazole (a strong inhibitor of CYP3A and P-glycoprotein [P-gp]), and rifampin (a strong inducer of CYP3A/P-gp and moderate inducer of CYP2C8) on the single-dose PK of ozanimod and its major active metabolites in healthy subjects.
Methods
This was a phase 1, randomized, parallel-group, open-label study with two parts. In part 1, 40 subjects were randomized to receive a single oral dose of ozanimod 0.46 mg (group A, n = 20) or oral doses of gemfibrozil 600 mg twice daily for 17 days with a single oral dose of ozanimod 0.46 mg on day 4 (group B, n = 20). In part 2, 60 subjects were randomized to receive a single oral dose of ozanimod 0.92 mg (group C, n = 20), oral doses of itraconazole 200 mg once daily for 17 days with a single oral dose of ozanimod 0.92 mg on day 4 (group D, n = 20), or oral doses of rifampin 600 mg once daily for 21 days with a single oral dose of ozanimod 0.92 mg on day 8 (group E, n = 20). Plasma PK parameters for ozanimod, CC112273, and CC1084037 were estimated using noncompartmental methods.
Results
Dose-proportional increases in maximum observed concentration (Cmax) and area under the concentration–time curve (AUC) were observed for ozanimod, CC112273, and CC1084037. The mean terminal elimination half-life (t1/2) for ozanimod was approximately 20–22 h while the mean t1/2 for CC112273 and CC1084037 were approximately 10 days. CC112273 and CC1084037 exposures were highly correlated with or without interacting drugs. Itraconazole increased ozanimod AUC by approximately 13% while rifampin reduced ozanimod AUC by approximately 24%, suggesting a minor role of CYP3A and P-gp in the overall disposition of ozanimod. Gemfibrozil increased the AUC for CC112273 and CC1084037 by approximately 47% and 69%, respectively. Rifampin reduced the AUC for CC112273 and CC1084037, primarily via CYP2C8 induction, by approximately 60% and 55%, respectively.
Conclusions
Ozanimod’s major active metabolites, CC112273 and CC1084037, exhibited similar single-dose PK properties and their exposures were highly correlated. CYP2C8 is one of the important enzymes in the overall disposition of CC112273 and subsequently its direct metabolite CC1084037.
Trial Registration
Clinical trial: NCT03624959
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Ozanimod is extensively metabolized to form two major active metabolites CC112273 and CC1084037. Multiple biotransformation pathways are involved in the disposition of ozanimod and its major active metabolites, including cytochrome P450 (CYP) 2C8 and 3A |
We conducted a phase 1 study to characterize the pharmacokinetics of the major active metabolites of ozanimod and to evaluate the effect of the index inhibitors or inducers of CYP2C8 and CYP3A |
What was learned from this study? |
Itraconazole, a strong CYP3A inhibitor, modestly increased ozanimod exposure by approximately 13%. Gemfibrozil, a strong CYP2C8 inhibitor, increased the AUC for CC112273 and CC1084037 by approximately 47% and 69%, respectively. Rifampin, reduced the AUC for CC112273 and CC1084037, primarily via CYP2C8 induction, by approximately 60% and 55%, respectively |
CYP2C8 has an important role in the disposition of CC112273 and consequently its direct, interconverting metabolite CC1084037 |
Introduction
Ozanimod is an orally administered sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity selectively to S1P receptors 1 (S1P1) and 5 (S1P5). Ozanimod blocks the capacity of lymphocytes to egress from lymph nodes, reducing the number of lymphocytes in peripheral blood [1]. Ozanimod is approved in the USA for the treatment of adults with relapsing forms of multiple sclerosis (MS) and in Europe for the treatment of adults with relapsing–remitting MS. The mechanism by which ozanimod exerts therapeutic effects in MS is unknown but may involve the reduction of lymphocyte migration into the central nervous system. Ozanimod also is in clinical development for the treatment of moderate to severe ulcerative colitis and Crohn’s disease [2, 3].
Ozanimod is extensively metabolized to form a number of circulating active metabolites, including two major and interconverting active metabolites, CC112273 and CC1084037, which both display similar activity and selectivity for S1P1 and S1P5 [4]. Ozanimod is metabolized via multiple biotransformation pathways, including aldehyde dehydrogenase (ADH) and alcohol dehydrogenase (ALDH), cytochrome P450 (CYP) isoforms 1A1 and 3A, and gut microflora. Ozanimod is also a weak substrate for P-glycoprotein (P-gp). CC112273 is metabolized by CYP2C8 to form RP101509 or reduced to form CC1084037 via carbonyl reductase (CBR). CC1084037 is metabolized by aldo–keto reductases (AKR) 1C1/1C2 and/or 3β- and 11β-hydroxysteroid dehydrogenase (HSD) to form CC112273. The interconversion between CC112273 and CC1084037 favors CC112273. Both CC112273 and CC1084037 are not substrates of drug transporters.
Ozanimod hydrochloride 0.5 mg and 1 mg (equivalent to ozanimod 0.46 mg and 0.92 mg, respectively) once daily (QD) were evaluated in phase 2 and 3 clinical studies in patients with MS [5, 6]. The pharmacokinetics (PK) of ozanimod was previously characterized in healthy subjects [1]; however, the PK characteristics of the major active metabolites CC112273 and CC1084037 have not been described.
It is important to understand the impact of concomitant medications on the elimination pathways of ozanimod and its major active metabolites to guide dosing recommendations. Clinical modulators of CBR, AKR, or HSD enzyme have not been identified. However, concomitant medications can inhibit or induce CYP3A and/or CYP2C8 activities. Regulatory guidance recommends using itraconazole and gemfibrozil as strong inhibitors of CYP3A and CYP2C8, respectively, and rifampin as a strong inducer of CYP3A and moderate inducer of CYP2C8 [7, 8]. No strong index inducer of CYP2C8 has been identified.
This study was conducted to characterize the single-dose PK properties of the major active metabolites of ozanimod and to evaluate the effect of steady-state gemfibrozil, itraconazole, and rifampin on the single-dose PK of ozanimod and its major active metabolites in healthy adult subjects.
Methods
Ethics
The study protocol and informed consent were reviewed and approved by an institutional review board (IntegReview, Austin, TX, USA). All subjects provided written informed consent before study entry. The study was conducted in accordance with the ethical principles of Good Clinical Practice and the Declaration of Helsinki.
Study Population
Healthy female or male subjects aged 18–55 years, with a body weight of at least 50 kg and body mass index of 18.0–30.0 kg/m2 were included in the study. Subjects were excluded if they had seated systolic blood pressure outside 90 to 140 mmHg; seated diastolic blood pressure 50–90 mmHg; pulse rate outside 55–90 bpm; a positive drug/alcohol test; a history of alcoholism, drug abuse, or addiction within 24 months; or a history of any abnormality or illness that, in the opinion of the investigator, may affect absorption, distribution, metabolism, or elimination of the study drugs or would limit the subject’s ability to complete the study. Other exclusion criteria included use of any systemic over-the-counter medication (excluding acetaminophen up to 1 g/day), dietary or herbal supplements within 7 days (28 days for St. John’s wort) prior to the first dose of study drug, and any systemic prescription medication (excluding hormonal contraceptives) within 28 days or five times the elimination half-life, whichever is longer, prior to the first dose of study drug and during the study.
Study Design
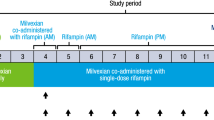
This was a phase 1, randomized, parallel-group, open-label study with two parts (Fig. 1). Sex was used as a stratifying factor within each study part. In part 1, 40 subjects were randomized into one of the two treatment groups A or B (20 subjects per group). Subjects in group A (reference) received a single oral dose of ozanimod 0.46 mg on day 1. Subjects in group B (test) received oral doses of gemfibrozil 600 mg twice daily on days 1 through 17, with a single oral dose of ozanimod 0.46 mg co-administered with gemfibrozil on day 4. In part 2, 60 subjects were randomized into one of the three treatment groups C, D, or E (20 subjects per group). Subjects in group C (reference) received a single oral dose of ozanimod 0.92 mg on day 1. Subjects in group D (test) received oral doses of itraconazole 200 mg QD on days 1 through 17, with a single oral dose of ozanimod 0.92 mg co-administered with oral doses of itraconazole on day 4. Subjects in group E (test) received oral doses of rifampin 600 mg QD on days 1 through 21, with a single oral dose of ozanimod 0.92 mg co-administered with oral doses of rifampin on day 8. While food has no effect on ozanimod exposure [9], to standardize conditions between treatment groups, ozanimod (with and without the CYP modulators) was administered following an overnight fast of at least 10 h before dosing. Subjects remained fasted for 4 h after ozanimod dosing. Gemfibrozil was administered 30 min before morning and evening meals, except when co-administered with ozanimod (day 4), when both medications were administered following an overnight fast. Itraconazole was administered with food, except for when co-administered with ozanimod (day 4), when both medications were administered following an overnight fast. Rifampin was administered on an empty stomach (either 1 h before or 2 h after a meal), except for when co-administered with ozanimod (day 8), when both medications were administered following an overnight fast.

Study design
Subjects were screened 28 days prior to the first dose. Eligible subjects were domiciled in the clinical research unit (CRU) from day −1 until after the last PK sample collection. Following discharge from the CRU, follow-up safety phone calls were conducted at approximately 28 and 75 days after the last dose of ozanimod.
PK Blood Sampling Schedule and Bioanalysis
PK blood samples were collected predose (0 h) and at 1, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, 48, 72, 96, 120, 144, 168, 192, 216, 240, 264, 288, 312, and 336 h after ozanimod dosing. At each time point, approximately 8 mL of blood was collected.
Plasma concentrations of ozanimod and its major active metabolites (CC112273 and CC1084037) were determined by using the validated liquid chromatography–tandem mass spectrometry methods by ICON (Whitesboro, NY). To measure plasma concentrations, ozanimod and its major active metabolites were extracted from 400-μL aliquots of human K2 EDTA plasma by support-liquid extraction and then analyzed using reversed-phase high-performance liquid chromatography with electrospray MS/MS detection. The calibrations were accomplished by weighted linear regressions (1/x2) of the ratio of the peak area of the analyte to that of the added deuterated internal standards and plotting against the corresponding nominal concentrations of ozanimod and its metabolites, respectively. The methods were validated over the concentration ranges of 4–2000 pg/mL for both ozanimod and CC1084037, and 25 to 10,000 pg/mL for CC112273. The inter-assay precision values (%CV) for ozanimod, CC112273, and CC1084037 were 9.8% or less. The accuracy (%bias) values for ozanimod and its metabolites were within − 0.8% to 4.2%.
PK and Safety Assessments
Pharmacokinetic parameters were calculated using noncompartmental analysis (Phoenix™ WinNonlin®, version 8.0.0.3176) and actual sample collection time. The following plasma PK parameters were estimated for ozanimod, CC112273, and CC1084037: maximum observed plasma concentration (Cmax), time to reach Cmax (Tmax), area under the concentration–time curve (AUC) from time 0 to last quantifiable concentration (AUClast), AUC from time 0 to infinity (AUC∞), and terminal elimination half-life (t1/2). In addition, apparent oral clearance and apparent volume of distribution associated with the terminal phase were estimated for ozanimod. Plasma concentration values below the limits of quantitation (BLQ) were replaced with zero for the mean plasma concentration–time profiles. For PK parameter calculation, BLQ values prior to Cmax were replaced with zero and BLQ values after Cmax were treated as missing.
Physical examinations, 12-lead electrocardiograms (ECGs), vital sign measurements, and clinical laboratory tests were performed, and adverse events (AEs) and concomitant medications were monitored throughout the study to assess safety.
Statistical Evaluation
The sample size estimation was conducted using a precision approach as no formal hypothesis testing was planned. The precision estimates of AUClast and Cmax were calculated using the historical variability estimate of 0.33 based on log transformed data for the analyte CC112273, the largest of the estimated variabilities of parent and metabolites. The sample size of 16 subjects for each of the treatment groups was expected to produce a two-sided 90% confidence interval (CI) with a distance from the difference in means to the limits that is equal to 0.198 when the estimated standard deviation is 0.33 in the natural log scale. Assuming a ratio of 1, this would back-transform to an approximate CI of 0.81 to 1.22. Assuming up to a 20% dropout, 20 subjects were enrolled in each group to ensure 16 evaluable subjects in each group.
For each of the comparisons, an analysis of variance (ANOVA) model with fixed effects for treatment group and sex were used to analyze the natural log transformed AUClast and Cmax. A point estimate and 90% CIs were provided for the geometric least squares mean ratio upon back-transformation. Model assumptions were evaluated through residual as a function of predicted plots, residual quantile plots, and residual normality plots.
The incidence, severity, and relationship of AEs and clinically significant changes from baseline in clinical laboratory tests, vital signs, and ECGs were summarized by treatment group using descriptive statistics. All statistical tabulations and analyses were performed using SAS, version 9.3 (SAS Institute, Cary, NC).
Results
Subject Disposition and Baseline Characteristics
One hundred subjects were enrolled and 96 subjects (96.0%) completed the study. Four subjects (4.0%) did not complete the study as a result of AEs (one subject in group B and one subject in group E), subject’s decision (one subject in group C because of a family emergency), and investigator’s decision (one subject in group D with a behavioral problem). Fifty-seven percent of subjects were male and 43% were female. The mean (standard deviation, SD) age and body mass index were 34.5 (9.55) years and 26.19 (2.598) kg/m2, respectively. The demographic and baseline characteristics are presented in Table 1. Overall, demographics and baseline characteristics were generally similar across the treatment groups.
Single-Dose PK of Ozanimod and Major Active Metabolites
The mean (SD) plasma concentration–time profiles for ozanimod and its major active metabolites following a single oral dose of ozanimod 0.46 mg or 0.92 mg are shown in Fig. 2. Plasma concentrations of ozanimod, CC112273, and CC1084037 appeared to decline monoexponentially after the absorption or formation phase. Plasma PK parameters for ozanimod and its major active metabolites are summarized in Table 2. Following single dose oral administration, ozanimod absorption was slow, with a median Tmax of 8 h. Median Tmax for CC112273 and CC1084037 were approximately 10 and 16 h, respectively. Dose-proportional increases in Cmax and AUC were observed for ozanimod, CC112273, and CC1084037. The metabolite/parent (M/P) AUClast ratio for CC112273 and CC1084037 were approximately 11 and 2, respectively. The mean t1/2 for ozanimod was approximately 20–22 h while the mean t1/2 for CC112273 and CC1084037 were approximately 10 days.

Mean (SD) plasma concentration–time profiles for ozanimod, CC112273, and CC1084037 following oral administration of a single dose of ozanimod 0.46 mg (a) or 0.92 mg (b)
Scatter plots with linear regression lines to illustrate the relationship of exposure (Cmax and AUClast) between the two major interconverting metabolites CC112273 and CC1084037 are presented in Fig. 3. CC112273 and CC1084037 exposures were highly correlated with or without extrinsic factors (i.e., interacting drugs). The coefficients of determination for Cmax and AUClast were approximately 0.959–0.969 and 0.968–0.982, respectively.

Relationship of CC112273 and CC1084037 exposures when ozanimod was administered alone or in combination with CYP modulators. AUClast AUC from time 0 to last quantifiable concentration, Cmax maximum observed plasma concentration, R2 coefficient of determination, x CC112273 Cmax or AUClast, y CC1084037 Cmax or AUClast. P value is for test of slope = 0
Effect of Gemfibrozil, Itraconazole, and Rifampin on Single-Dose PK of Ozanimod and Major Active Metabolites
The mean (SD) plasma concentration–time profiles for ozanimod, CC112273, and CC1084037 following a single oral dose of ozanimod alone or in combination with CYP modulators are shown in Figs. 4, 5, and 6. The statistical analysis to assess the effect of gemfibrozil on the PK of ozanimod and its metabolites is presented in Table 3. Gemfibrozil, a strong inhibitor of CYP2C8, had no effect on ozanimod as shown by the 90% CI for the ratio of geometric least squares means for ozanimod Cmax and AUC∞ within the default equivalence limits of 0.80–1.25. Gemfibrozil, however, increased Cmax and AUClast of the major active metabolites by approximately 27–35% and 47–68%, respectively. Itraconazole, a strong inhibitor of CYP3A, had no effect on ozanimod Cmax (90% CI within the limits of 0.80–1.25) and increased ozanimod AUC∞ by approximately 13%. Itraconazole reduced Cmax of the major active metabolites by approximately 22% while it had no apparent effect on the AUClast of the major active metabolites. Rifampin, a strong inducer of CYP3A and a moderate inducer of CYP2C8, reduced ozanimod Cmax and AUC∞ by approximately 21–24%. While rifampin modestly reduced Cmax of the major active metabolites by approximately 13–15%, it reduced AUClast of the major active metabolites by approximately 55–60%.

Mean (SD) plasma concentration–time profiles for ozanimod following oral administration of a single dose of ozanimod alone (a) and in combination with steady-state gemfibrozil, itraconazole, or rifampin (b)

Mean (SD) plasma concentration–time profiles for CC112273 following oral administration of a single dose of ozanimod alone (a) and in combination with steady-state gemfibrozil, itraconazole, or rifampin (b)

Mean (SD) plasma concentration–time profiles for CC1084037 following oral administration of a single dose of ozanimod alone (a) and in combination with steady-state gemfibrozil, itraconazole, or rifampin (b)
Safety
No serious AEs were reported. Two subjects were discontinued from the study as a result of AEs suspected by the investigator not to be related to ozanimod treatment: one subject in group B (gemfibrozil + ozanimod) for synovitis and one subject in group E (rifampin + ozanimod) for urticaria and pruritus. Overall, 32 subjects (32.0%) had at least one treatment-emergent AE. When ozanimod was administered alone, three subjects (15%) and six subjects (30%) who received ozanimod 0.46 mg and 0.92 mg, respectively, experienced treatment-emergent AEs. When ozanimod was co-administered with gemfibrozil, itraconazole, or rifampin, five subjects (25%), two subjects (10%), and eight subjects (40%), respectively, experienced treatment-emergent AEs. All AEs were mild or moderate in severity. The commonly reported treatment-emergent AEs are shown in Table 4 which included constipation (ozanimod alone, ozanimod + gemfibrozil, and ozanimod + itraconazole), headache (gemfibrozil alone, gemfibrozil + ozanimod, and rifampin alone), and chromaturia (rifampin alone, rifampin + ozanimod). There were no clinically significant changes from baseline for vital signs and clinical laboratory results and no abnormal physical examination findings. One subject in group C (ozanimod 0.92 mg) had an asymptomatic second-degree atrioventricular (AV) block (Mobitz type 1) approximately 6 h after dosing. A repeat ECG conducted approximately 90 min later did not show any clinically significant findings. The event resolved without additional intervention or therapy.
Discussion
This study was conducted to characterize the single-dose PK of the major active metabolites of ozanimod and to evaluate the effect of index inhibitors or inducers of CYP2C8 and/or CYP3A on the PK of ozanimod and its major active metabolites. Two single oral doses of ozanimod 0.46 mg and 0.92 mg were studied since the phase 2 and 3 studies evaluated two ozanimod maintenance dosing regimens of 0.46 mg and 0.92 mg QD. A two-way crossover design for drug interaction evaluation is preferred over parallel-study designs. However, as a result of the long t1/2 of the major active metabolites (approx. 10 days), a crossover study design is not feasible and therefore a parallel-group design was used. In this study, the index CYP modulators were administered multiple doses prior to ozanimod co-administration (i.e., lead-in dosing phase) to ensure the maximum inhibition or induction effect on CYP enzymes [10,11,12,13] and continued during the 14-day PK sample collections to maintain CYP enzyme inhibition or induction effect. Dosing of rifampin, itraconazole, and gemfibrozil for up to 3 weeks in duration was chosen to minimize the potential safety risks associated with longer exposure in healthy volunteers. PK sample collections up to 14 days postdose allowed reliable determination of the terminal elimination phase of ozanimod and its metabolites. If PK samples were to be collected for 3–5 times the t1/2 of the major active metabolites (i.e., approx. 30–50 days of PK sampling), dosing of rifampin, itraconazole, and gemfibrozil would be 33–58 days (including the lead-in dosing phase), which is not practical and may not be safe for healthy volunteers.
The single-dose PK properties of ozanimod in this study are consistent with a recent study [1]. Ozanimod exhibited dose-proportional increase in exposure, high volume of distribution, moderate apparent oral clearance, and a mean t1/2 of 20–22 h. Similar to the parent ozanimod, exposure for the major active metabolites, CC112273 and CC1084037, increased dose proportionally following ozanimod single oral doses of 0.46–0.92 mg. Since CC1084037 is a direct and interconverting metabolite of CC112273, and CC1084037 is not subject to other elimination pathways, CC112273 and CC1084037 exposures (Cmax and AUC) were highly correlated with or without extrinsic factors (i.e., interacting drugs). The mean t1/2 for CC112273 and CC1084037 were also similar at approximately 10 days. Following a single oral dose, the M/P ratios for CC112273 and CC1084037 were approximately 11 and 2, respectively. However, with multiple dosing the M/P ratios for CC112273 and CC108437 are expected to be substantially higher because of the anticipated accumulation based on their long t1/2.
In vitro data showed that ozanimod is metabolized by multiple biotransformation pathways including ALDH and ADH, CYPs 1A1 and 3A, and gut microflora. Ozanimod is also a weak substrate for P-gp. In this study, itraconazole, a strong inhibitor of CYP3A and P-gp, increased ozanimod AUC∞ by approximately 13% and rifampin, a strong inducer of CYP3A and P-gp, reduced ozanimod AUC∞ by approximately 24%. These modest changes are not clinically meaningful and indicated a minor role of CYP3A and P-gp in the overall disposition of ozanimod.
In vitro data showed that CYP2C8 and oxido-reductases are involved in the metabolism of the major active metabolite CC112273. The active metabolite CC1084037 is formed directly from CC112273, and the interconversion between these two active metabolites is mediated by CBR, AKR 1C1/1C2, and/or 3β- and 11β-HSD. In this study, gemfibrozil, a strong inhibitor of CYP2C8, increased the AUClast for CC112273 and CC1084037 by approximately 47% and 69%, respectively. Rifampin, a strong inducer of CYP3A and a moderate inducer of CYP2C8, reduced the AUClast for CC112273 and CC1084037 by approximately 60% and 55%, respectively. The effect of rifampin on CC112273 and CC1084037 is primarily caused by induction of CYP2C8 since CYP3A is not directly involved in the formation or elimination of these two active metabolites and itraconazole, a strong CYP3A4 inhibitor, had no clinically meaningful effect on the PK of CC112273 and CC1084037. It should be noted that there is no strong inducer of CYP2C8 for drug interaction studies. Results from this study support that CYP2C8 is one of the important enzymes in the overall disposition of CC112273 and subsequently its direct metabolite CC1084037.
A single oral dose of ozanimod (0.46 mg or 0.92 mg) alone or in combination with multiple doses of gemfibrozil, itraconazole, or rifampin in healthy subjects was generally well tolerated. All AEs were mild to moderate in severity. Chromaturia, reported in three subjects in group E (rifampin + ozanimod), is a well-documented and benign side effect of rifampin [14, 15]. S1P receptor modulators may cause a transient decrease in heart rate (HR) and AV conduction delays upon initiation of dosing [16]. However, these effects may attenuate over time, secondary to S1P receptor desensitization, internalization, and degradation on atrial myocytes [17]. An up-titration schedule of ozanimod 0.23 mg QD for 4 days followed by doses of 0.46 mg QD for 3 days prior to the maintenance dose of 0.92 mg attenuates the first-dose HR and AV conduction effects in patients wtih MS [18]. One subject in this study who received a single dose of ozanimod 0.92 mg (without dose titration) had an asymptomatic episode of second-degree AV block (Mobitz type 1) approximately 6 h after dosing.
Conclusions
Results from this study showed that ozanimod’s major active metabolites CC112273 and CC1084037 exhibited similar t1/2 and their exposures were highly correlated. Results also indicated that CYP3A and P-gp have a minor contribution to the overall disposition of ozanimod, while CYP2C8 has an important role in the disposition of CC112273 and consequently its direct, interconverting metabolite CC1084037.
References
Tran JQ, Hartung JP, Peach RJ, et al. Results from the first-in-human study with ozanimod, a novel, selective sphingosine-1-phosphate receptor modulator. J Clin Pharmacol. 2017;57:988–96.
Sandborn WJ, Feagan BG, Wolf DC, et al. Ozanimod induction and maintenance treatment for ulcerative colitis. N Engl J Med. 2016;374:1754–62.
Feagan BG, Sandborn WJ, Danese S, et al. Ozanimod induction therapy for patients with moderate to severe Crohn's disease: a single-arm, phase 2, prospective observer-blinded endpoint study. Lancet Gastroenterol Hepatol. 2020. https://doi.org/10.1016/S2468-1253(20)30188-6.
Tran JQ, Zhang P, Surapaneni S, Selkirk J, Yan G, Palmisano M. Absorption, metabolism, and excretion, in vitro pharmacology, and clinical pharmacokinetics of ozanimod, a novel sphingosine 1-phosphate receptor agonist [abstract P993]. In: Triennial Joint Meeting of the European Committee for Treatment and Research in Multiple Sclerosis and Rehabilitation in Multiple Sclerosis; 2019 September 11–13, 2019; Stockholm, Sweden; 2019.
Comi G, Kappos L, Selmaj KW, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): a multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. 2019;18:1009–200.
Cohen JA, Arnold DL, Comi G, et al. Safety and efficacy of the selective sphingosine 1-phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15:373–81.
Food and Drug Administration. Guidance for Industry Clinical Drug Interaction Studies—Cytochrome P450 enzyme- and transporter-mediated drug interactions. 2020 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions. Accessed 8 Apr 2020.
European Medicines Agency. Guideline on the investigation of drug interactions. 2012 https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf. Accessed April 8, 2020.
Tran JQ, Hartung JP, Tompkins CA, Frohna PA. Effects of high- and low-fat meals on the pharmacokinetics of ozanimod, a novel sphingosine-1-phosphate receptor modulator. Clin Pharmacol Drug Dev. 2018;7:634–40.
Liu L, Bello A, Dresser MJ, et al. Best practices for the use of itraconazole as a replacement for ketoconazole in drug–drug interaction studies. J Clin Pharmacol. 2016;56:143–51.
Inui N, Akamatsu T, Uchida S, et al. Chronological effects of rifampicin discontinuation on cytochrome P450 activity in healthy Japanese volunteers, using the cocktail method. Clin Pharmacol Ther. 2013;94:702–8.
Tran JQ, Kovacs SJ, McIntosh TS, Davis HM, Martin DE. Morning spot and 24-hour urinary 6 beta-hydroxycortisol to cortisol ratios: intraindividual variability and correlation under basal conditions and conditions of CYP 3A4 induction. J Clin Pharmacol. 1999;39:487–94.
Bruderer S, Petersen-Sylla M, Boehler M, Remenova T, Halabi A, Dingemanse J. Effect of gemfibrozil and rifampicin on the pharmacokinetics of selexipag and its active metabolite in healthy subjects. Br J Clin Pharmacol. 2017;83:2778–88.
Douglas JG, McLeod MJ. Pharmacokinetic factors in the modern drug treatment of tuberculosis. Clin Pharmacokinet. 1999;37:127–46.
Ford SL, Sutton K, Lou Y, et al. Effect of rifampin on the single-dose pharmacokinetics of oral cabotegravir in healthy subjects. Antimicrob Agents Chemother. 2017;61:e00487-17.
Juif PE, Kraehenbuehl S, Dingemanse J. Clinical pharmacology, efficacy, and safety aspects of sphingosine-1-phosphate receptor modulators. Expert Opin Drug Metab Toxicol. 2016;12:879–95.
Brossard P, Scherz M, Halabi A, Maatouk H, Krause A, Dingemanse J. Multiple-dose tolerability, pharmacokinetics, and pharmacodynamics of ponesimod, an S1P1 receptor modulator: favorable impact of dose up-titration. J Clin Pharmacol. 2014;54:179–88.
Zeposia [package insert]. Summit, NJ: Celgene Corporation; 2020.
Acknowledgements
We thank the participants of this study.
Funding
This study was funded by Celgene Corporation. Bristol-Myers Squibb Company funded the journal’s Rapid Service and Open Access Fees.
Medical Writing and/or Editorial Assistance
Editorial assistance was provided by Lorraine Sweeney of Peloton Advantage, LLC, an OPEN Health company, with funding from Bristol-Myers Squibb Company.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Writing (original draft preparation): JQT; Writing (review and editing): JQT, PZ, AG, LL, MS, XW, and MP; Research design: JQT, PZ, LL, XW, MS, and MP; Performing research: JQT, PZ, XW, and MS; Data analysis: JQT, PZ, and AG.
Disclosures
Jonathan Q. Tran is a former employee of Bristol-Myers Squibb Company and current employee of Mirati Therapeutics. Peijin Zhang, Atalanta Ghosh, Liangang Liu, Mary Syto, Xiaomin Wang, and Maria Palmisano are employees of Bristol-Myers Squibb Company and hold stock in Bristol-Myers Squibb Company.
Compliance with Ethics Guidelines
The study protocol and informed consent were reviewed and approved by an institutional review board (IntegReview, Austin, TX, USA). The study was conducted in accordance with the ethical principles of Good Clinical Practice and the Declaration of Helsinki. All subjects provided written informed consent before study entry.
Data Availability
Celgene, a Bristol-Myers Squibb Company, is committed to responsible and transparent sharing of clinical trial data with patients, healthcare practitioners, and independent researchers for the purpose of improving scientific and medical knowledge as well as fostering innovative treatment approaches. Data requests may be submitted to Celgene, a Bristol-Myers Squibb Company, at https://vivli.org/ourmember/celgene/ and must include a description of the research proposal.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital Features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12789758.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Tran, J.Q., Zhang, P., Ghosh, A. et al. Single-Dose Pharmacokinetics of Ozanimod and its Major Active Metabolites Alone and in Combination with Gemfibrozil, Itraconazole, or Rifampin in Healthy Subjects: A Randomized, Parallel-Group, Open-Label Study. Adv Ther 37, 4381–4395 (2020). https://doi.org/10.1007/s12325-020-01473-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01473-0