
Abstract
Introduction
The objective of this study was to estimate the relative efficacy and safety of fixed-dose combination aclidinium/formoterol 400/12 μg twice daily compared to tiotropium 18 μg once daily in adult patients with moderate-to-severe chronic obstructive pulmonary disease (COPD).
Methods
A systematic literature review performed in March 2014, using a predefined search strategy in MEDLINE, EMBASE and Cochrane Library, identified 17 randomized placebo-controlled trials, (tiotropium n = 15; aclidinium/formoterol n = 2). Outcomes of interest were: bronchodilation (peak and trough forced expiratory volume in 1 s (FEV1)), COPD symptoms [Transition Dyspnea Index (TDI) focal score and % of responders (>1 unit improvement)] and Health Related Quality of Life (HRQoL) [St. George’s Respiratory Questionnaire (SGRQ) total score and % responders (>4 unit improvement)], % of patients with ≥1 exacerbations, adverse events (AE), serious adverse events (SAE), hospitalization and mortality, all at 24 weeks. In the absence of head-to-head trials between aclidinium/formoterol and tiotropium, a Bayesian indirect treatment comparison (ITC) was used with placebo as common control.
Results
Regarding bronchodilation, aclidinium/formoterol was found to be more efficacious than tiotropium at peak FEV1, with mean difference in change from baseline (DCFB) 143 mL [95% credible interval (CrI): 112, 174] and at trough FEV1 [DCFB 26 mL (95% CrI −2, 55)]. Aclidinium/formoterol is expected to be more efficacious than tiotropium in improving dyspnea symptoms measured by TDI [DCFB 0.54 points (95% CrI 0.09, 0.99); odds ratio (OR) of responders 1.51 (95% CrI 1.11, 2.06)]. SGRQ results are comparable for aclidinium/formoterol versus tiotropium [DCFB −0.52 (95% CrI −2.21, 1.17); OR of responders 1.16 (95% CrI 0.47, 2.87)]. The ITC results suggest similar safety profiles regarding AEs, SAEs and hospitalization.
Conclusion
Based on the ITC, aclidinium/formoterol is expected to be more efficacious than tiotropium in terms of lung function and symptom control while providing comparable HRQoL results and safety profile.
Funding
AstraZeneca.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic obstructive pulmonary disease (COPD) is a disorder characterized by the progressive development of airway obstruction, which manifests as an accelerated decline in lung function, with symptoms such as breathlessness on physical exertion, deteriorating health status and exacerbations [1].
Currently COPD is the fourth leading cause of death globally [2], a major cause of morbidity and mortality, projected to become the world’s third leading cause of mortality by 2020 [3]. Characterized by progressive airflow limitation, COPD also has a major economic impact [4].
According to the COPD Guidelines from 2011, which were updated in 2015, it is recommended to combine two long-acting bronchodilators in moderate-to-severe COPD patient groups [5]. The combination of two bronchodilators with different mechanisms of action, such as long-acting muscarinic antagonists (LAMAs) and long-acting β2-agonists (LABAs), are a successful treatment option for patients with COPD. Compared to single bronchodilators, the combination of LAMAs and LABAs demonstrates significant improvements in lung function without increasing the risk for adverse events [5–8]. The use of fixed-dose combinations (FDCs) of LABAs and LAMAs provide the opportunity to improve the accessibility and conformity compared to separate inhalers. Also, the dose of each substance used in the combination can be enhanced. An objection related to the development of an FDC is the arrangement of improved bronchodilation over monotherapy segments, while adjusting the associated adverse effects with efficacy [9]. The safety and efficacy profiles of both LAMAs and LABAs are well accepted. However, it is important to recognize both the similarities and differences in both efficacy and safety, when combining two substances.
A new LABA/LAMA FDC, aclidinium/formoterol 400/12 μg twice-daily (BD), has recently been introduced in the management of COPD. The FDC, aclidinium/formoterol, is compared to placebo and aclidinium and formoterol as monotherapies in two pivotal, randomized, placebo-controlled studies [ACLIFORM (ClinicalTrials.gov identifier NCT01462942) and AUGMENT (Clinicaltrials.gov identifier NCT01437397)] [10, 11]. Results from both studies show a significant improvement in 24 h symptom control compared with placebo and aclidinium and formoterol monotherapies. Furthermore, in the aclidinium/formoterol group, the frequency of exacerbations is also reduced compared to placebo [6].
Tiotropium 18 µg is a once-daily treatment and has been the first and most widely prescribed LAMA for COPD, considered as the standard of care in many countries [12]. Based on the outcomes of the AUGMENT and ACLIFORM studies, it is expected that an FDC of aclidinium/formoterol will be more efficient on key COPD outcomes, compared to LAMA monotherapies. As there are no published direct head-to-head comparisons on the clinical efficacy and safety between FDC aclidinium/formoterol and tiotropium, alternative methodologies need to be employed to inform health-care practitioners.
For this reason, a systematic literature review and Bayesian indirect treatment comparison (ITC) were undertaken to assess the relative efficacy and safety of aclidinium/formoterol 400/12 μg BD versus tiotropium 18 μg once daily (OD) for the treatment of adult patients with moderate-to-severe COPD.
Methods
Data Sources
A systematic literature review was performed to identify randomized placebo-controlled trials (RCTs) reporting the safety and efficacy of aclidinium/formoterol 400/12 μg and tiotropium 18 μg compared to each other or placebo. Using a predefined strategy, MEDLINE®, MEDLINE in-process and EMBASE® databases were searched simultaneously through the OVID platform, while the Cochrane Central Register of Controlled Trials was searched separately. The American Thoracic Society International Conference (2013) and European Respiratory Society International Congress (2013) were hand-searched for relevant abstracts. In addition, the search was also performed in ClinicalTrials.gov website. The searches were performed on March 24, 2014, for studies in English language with a time restriction from the year 1989 to March 2014. The predefined search strategies used were tailored for each database and are presented in Supplementary Table 1. This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Study Selection Process
The relevance of each citation identified was assessed according to predefined abstract selection criteria (Supplementary Table S2). First, titles and abstracts were screened for eligibility, and then full texts of the selected articles were assessed by one researcher and checked against the original study by another. Those that met the inclusion criteria were included for data extraction.
The studies of interest were RCTs with duration of 22–26 weeks, including adults with moderate-to-severe COPD, reporting on aclidinium/formoterol 400/12 μg BD (using the Genuair® device [AstraZeneca AB, Södertälje, Sweden]) or tiotropium 18 μg OD (using the Handihaler® device [Boehringer Ingelheim, Ridgefield, USA]) compared with each other or placebo. The efficacy outcomes of interest were: trough forced expiratory volume in 1 s (FEV1) (pre-bronchodilatory), peak FEV1 (post-bronchodilatory), St. George’s Respiratory Questionnaire (SGRQ) score, Transition Dyspnea Index (TDI) focal score and the % of patients with ≥1 exacerbations. The safety outcomes of interest were: adverse events, serious adverse events, hospitalization and mortality. In all cases, outcomes reported in the range of 22–26 weeks were grouped as 24 weeks.
Data Abstraction and Quality Assessment
For the studies identified that met the inclusion criteria, details were extracted on population characteristics, interventions, outcomes and the study design of interest at 24 weeks (22–26 weeks) (Tables 1, 2, Supplementary Table S5, Supplementary Table S6). Data abstraction was performed by one researcher and verified against the original study publication by another. Data of interest presented in graphs were extracted using DigitizeIT version 4.1 software (DigitizeIT, Braunschweig, Germany).
For continuous outcomes, the change from baseline (CFB) and the associated sampling variance were extracted or calculated based on the available data. For dichotomous outcomes, the number of patients experiencing an event was extracted or estimated based on the reported percentages and intention to treat population, and the total patient-years of follow-up were calculated.
The validity of each trial used in the ITC was assessed using the National Institute of Health and Clinical Excellence (NICE) checklist. The results of this assessment were not explicitly used in the ITC, but serve as additional information to determine the quality of the evidence base when interpreting the results (Supplementary Table S3).
Data Synthesis: Indirect Treatment Comparison
The existence of a connection between the treatments of interest via a common control (placebo), as well as the study design and patient characteristics of the identified studies, was used to assess the feasibility of a valid ITC [13]. Subsequently, the identified evidence was used to perform an ITC within a Bayesian framework to simultaneously synthesize the results of the included studies and obtain relative treatment effects [14, 15]. A linear model with normal likelihood distribution was used for continuous outcomes, and a Poisson likelihood with a log link for the dichotomous outcomes [16]. Flat (non-informative) prior distributions, normal with zero mean and variance of 10,000, were assumed for the relative treatment effects of all outcomes. A uniform distribution with range 0–5 was used as the prior of the between-study standard deviation.
For each outcome, a fixed and a random effects model was evaluated. The goodness of fit of each model to the data was assessed using the deviance information criterion [17]. The posterior densities were estimated using the Markov chain Monte Carlo (MCMC) simulations based on 80,000 iterations on three chains, with a burn-in of 20,000 iterations. Convergence assessment was based on visual inspection of trace plots and accuracy of the posterior estimates using the Monte Carlo error for each parameter. WinBUGS 1.4.3 (MRC Biostatistics Unit, Cambridge, UK) statistical software was used for the analyses and the models were based on those defined by Dias et al. [18]. The posterior distributions were summarized with the median to reflect the most likely value of the estimate, and the 2.5th and 97.5th percentile to capture the 95% credible interval (CrI). For each end point, the probability that each treatment was better than a certain comparator was established.
Results
Search and Selection Results
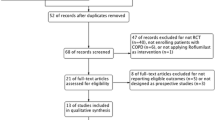
After searching, a total of 2401 abstracts from the databases and 88 clinical trials from ClinicalTrials.gov were identified (Fig. 1). Following the abstracts and full-text publication screening stages, 17 full-text publications [8, 12, 19–33] were identified and 3 clinical study reports [34–36] were provided by AstraZeneca. In total, the evidence base comprised 15 different studies; 13 studies [8, 12, 19–33, 36] compared tiotropium 18 μg to placebo (14,697 patients) and two studies [34, 35] compared aclidinium/formoterol 400/12 μg to placebo (1246 patients).

Flowchart of the study selection process. ATS American Thoracic Society, CSR clinical study report, ERS European Respiratory Society
Study Characteristics
An overview of the study characteristics is presented in Table 1. All studies were multicenter, placebo-controlled RCTs. Twelve studies [8, 12, 19–22, 24–28, 30, 31, 33, 36] were double-blind and three [23, 29, 32] included tiotropium as an open-label arm. The included studies varied in terms of the number of patients randomized to each treatment, ranging from 117 [31] to 3006 [12, 22]. The trial duration varied from 96 weeks [33] to 24 weeks [23–27, 34, 35]. The use of ICS (inhaled corticosteroids) as a background treatment was allowed in all studies and patients were permitted a short-acting beta-agonist as rescue medication (salbutamol or albuterol). The studies were of comparable quality, according to the results of the assessment using NICE questionnaire (Supplementary Table S3). In general, the method of randomization and concealment of treatment allocation was well reported.
Patient Characteristics
An overview of the main patient characteristics is provided in Table 2. The enrolled patients were adults with a COPD diagnosis. The studies included a predominantly male population, ranging from 50% [35] to 99% [24], while in three studies [21, 24, 31] more than 80% of the included patients were male in both arms. The patients’ average age across all the studies was similar (range 63–68 years). Overall, spirometry measures were fairly consistent at baseline. According to the inclusion criteria, most studies required an FEV1/forced vital capacity (FVC) of less than or equal to 0.70 and an FEV1% predicted range between 30% and 80%. The mean FEV1% predicted at baseline ranged between 35.6% and 56.4%. FEV1 at baseline ranged from 0.96 liter (L) to 1.55 L. The FEV1/FVC at baseline was reported to be between 41.3% and 55.3%. Across all the included studies, the percentage of patients per arm that used ICS at baseline ranged between 7% and 71%. The percentage was lower in both aclidinium/formoterol trials (7% to 9% for LAC-MD-31 and 19–22% for M/40464/30R) than in the other studies (35–71%). Furthermore, all studies included patients who were current or ex-smokers. In studies where the percentage of current smokers was reported, it ranged from 40% to 53%. Six studies (reported in 7 publications: [23, 25–29, 31]) did not report the percentage of current smokers. The mean number of pack-years ranged from 35.0 to 69.4 years.
Indirect Treatment Comparison
Despite some differences identified across the studies in terms of study design and patient characteristics, the 15 RCTs (reported in 20 publications) are considered to be broadly comparable and the ITC was feasible [13]. The diagram of the trials included in the ITC is shown in Fig. 2.

Network of studies included in the indirect treatment comparison
Efficacy Outcomes
Individual study results for efficacy outcomes are presented in Supplementary Table S5, where data not reported but estimated are denoted by an asterisk. The results of the ITC analysis are presented in Table 3. Regarding lung function, for both outcomes considered in this study, i.e., peak and trough FEV1, aclidinium/formoterol 400/12 µg appeared to be more efficacious compared to tiotropium 18 µg at 24 weeks.
Regarding health-related quality of life, as measured by SGRQ total score, the individual study results for aclidinium/formoterol demonstrate high variation between the M/40464/30R and LAC-MD-31 studies. The cause of this variation is unknown and cannot be explained by differences in study design or patient characteristics. The heterogeneity is reflected in the results of the ITC by means of wide credible intervals with a difference in CFB of −0.52 (95% CrI −2.21, 1.17). Similarly, for the % of responders (patients with >4 units reduction), the relative effect is heterogeneous with variation between M/40464/30R and LAC-MD-31 studies with an odds ratio (OR) 1.16 [95% CrI (0.47, 2.87)]. Due to this high variation, the results should be interpreted with caution; aclidinium/formoterol appeared to be comparable to tiotropium for both SGRQ total score and % responders.
Aclidinium/formoterol was more efficacious than tiotropium in improving breathlessness measured by TDI and % responders (i.e., patients with >1 point increase from baseline).
With regard to the percentage of patients with at least one exacerbation, aclidinium/formoterol was likely to be better compared to placebo with OR of 0.78 [95% CrI (0.56, 1.08)] and comparable to tiotropium with OR 1.03 (95% CrI [0.73, 1.47]). For this outcome, the time period of 24 weeks is relatively short, as the results are heavily dependent on the recent history of the patients recruited (e.g., if they had an exacerbation within the last months before recruitment, see inclusion/exclusion criteria in Table 1). Furthermore, the percentage of patients with at least one exacerbation in the placebo arm is almost 3.5 times higher in Donohue et al. 2002 and 2003 [26, 27] (45.8%) than in M/40464/30R (13.4%) [34], suggesting differences in COPD severity, in exacerbation-related study inclusion criteria or in the way the exacerbations were defined/reported. For these reasons, the results of the ITC shall be interpreted with caution.
Safety Outcomes
For the safety outcomes, the individual study results are presented as: number of patients with an event (n); number of patients included in the analysis (N); and proportion of patients with an event per treatment arm (Supplementary Table S6).
Compared to placebo, aclidinium/formoterol [OR 1.19; 95% CrI (0.95, 1.49)], and tiotropium [OR 1.03; 95% CrI (0.85, 1.24)] resulted in a mean OR above 1, suggesting an advantage for placebo, although not a significant one as the CrI included 1 in all cases. In both cases, the results of this analysis should be interpreted with extreme caution, first due to the limited number of studies, the time of assessment (24 weeks is a rather short period for safety outcomes) and potential differences in the way this outcome is reported in each study.
In the results of the ITC for serious adverse events for active treatments compared to placebo, the median OR for all active treatments was above 1 suggesting an advantage for placebo, but in all cases the credible intervals included 1; thus, the difference cannot be considered as significant. In line with this in pairwise comparisons between the active treatments, the CrI include 1 in all cases. Regarding AEs, the results of this analysis should be interpreted with extreme caution, due to the limited number of studies and the time of assessment (24 weeks is a rather short period for safety outcomes).
The results of the ITC regarding the proportion of patients with hospitalization within 24 weeks are uncertain for aclidinium/formoterol versus placebo with an OR 0.65 [95% CrI (0.22, 1.64)], mainly due to the lack of data, while for tiotropium the OR was 0.59 [95% CrI (0.46, 0.74)] versus placebo. Similarly, aclidinium/formoterol was comparable to tiotropium with OR 1.03 [95% Crl (0.37, 2.90)], but with high uncertainty.
The ITC for mortality was not (computationally) feasible, as the majority of the studies reported zero events (deaths) which lead the algorithm (MCMC) to numerical overflow, even when applying a continuity correction of 0.5. With such a large proportion of trials with zero events, estimation of a treatment effect and its variance becomes practically impossible. The individual study results for mortality are presented in Supplementary Table S6.
Discussion
Based on the results of the ITC, aclidinium/formoterol is expected to be more efficacious than tiotropium in terms of peak FEV1, TDI focal score and TDI responders. Regarding trough FEV1, aclidinium/formoterol is expected to be favorable compared to tiotropium. In all other efficacy and safety end points, aclidinium/formoterol and tiotropium are expected to result in similar (comparable) outcomes. The analysis for mortality was not feasible because the majority of the studies reported zero events.
A few other studies compared LABA/LAMA combinations versus tiotropium in a head-to-head trial. Both the SHINE study (ClinicalTrials.gov identifier, NCT01202188) [8] and SPARK study (ClinicalTrials.gov identifier, NCT01120691) [37] compared QVA149 (indacaterol 110 mg/glycopyrronium 50 mg) to tiotropium. The SHINE study reports comparable results to our study with superior improvements in lung function for the QVA149 group compared to tiotropium. The safety results are comparable to placebo and with no additional safety signal compared to tiotropium [8]. The SPARK study also shows similar results, with a significant reduction in the rate of all exacerbations, and a significant improvement in trough FEV1 and health status favoring the dual LABA/LAMA bronchodilator QVA149 versus tiotropium. Furthermore, no safety differences between the dual LABA/LAMA bronchodilator and tiotropium are found [37]. Decramer et al. [38] and Maleki-Yazdi et al. [39] both compared umeclidinium plus vilanterol versus tiotropium. Both studies report a significant improvement in lung function compared to tiotropium, and no safety differences are found between the groups.
It is challenging to demonstrate the relevance of the end points on COPD studies comparing combination therapy to monotherapy. To determine the clinical effectiveness, the minimum clinically important difference (MCID) is often used to acknowledge a clinically significant effect. This measure is however focused on the comparison of a monotherapy versus placebo. When comparing a combination therapy to a monotherapy, uncertainty has occurred if the MCID is a valid measure, because the differences in effects tend to be smaller since both arms receive active therapy. Jones et al. [40] discuss this issue and have introduced the ‘minimum worthwhile incremental advantage’ which can be used to describe the percentage of patients experiencing improvement at or above MCID when adding active treatment on top of another active treatment or when comparing two active treatments to each other [6, 40].
Furthermore, there are a number of other potential limitations to this analysis. First, as for any meta-analysis, inherent limitations are related to the potential for within-study bias and publication bias. Furthermore, there are differences in the definitions of exacerbations and in study methodology, populations that could introduce bias. For example, across all the included studies, the percentage of patients per arm that used ICS at baseline ranged between 7% and 71%. The percentage was lower in both aclidinium/formoterol trials (7–9% for LAC-MD-31 and 19–22% for M/40464/30R) than in the other studies (35–71%). Also, for the SGRQ total score outcome, the CFB versus placebo reported for aclidinium/formoterol 400/12 demonstrated high variation between the M/40464/30R and LAC-MD-31 studies. The cause of this variation is unknown and cannot be explained by the study designs or patient characteristics.
In addition, bias could be introduced due to the imbalances in potential treatment effect modifiers (e.g., FEV1 predicted at baseline) and differences in the background medications. Due to the lack of access to individual patient data and the low number of studies (especially for aclidinium/formoterol), it was not feasible to further explore these differences.
Furthermore, it is considered complicated to include safety in indirect comparisons, since this is not a straightforward approach. However, we decided to include safety next to efficacy outcomes, since a benefit–risk assessment will add important data about the intervention.
Conclusion
The results of this analysis suggest that aclidinium/formoterol is more efficacious with a similar safety profile compared to tiotropium.
References
Bhowmik A, Seemungal TA, Sapsford RJ, Wedzicha JA. Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax. 2000;55(2):114–20.
World Health Organization: World Health Statistics. Geneva. 2008. http://www.who.int/gho/publications/world_health_statistics/EN_WHS08_Full.pdf. 2008.
Chapman KR, Mannino DM, Soriano JB, Vermeire PA, Buist AS, Thun MJ, et al. Epidemiology and costs of chronic obstructive pulmonary disease. Eur Respir J. 2006;27(1):188–207.
National Institutes of Health. National Heart, Lung and Blood Institute. Morbidity and Mortality: 2012 chart book on cardiovascular, lung, and blood diseases. Bethesda, MD. https://www.nhlbi.nih.gov/files/docs/research/2012_ChartBook_508.pdf. 2012.
Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the diagnosis, management, and prevention of COPD. http://www.goldcopd.org/uploads/users/files/GOLD_Report_2015.pdf. 2015.
Bateman E, Chapman KR, Singh D, D’Urzo AD, Molins E, Leselbaum A, Garcia Gil E Aclidinium bromide and formoterol fumarate as a fixed-dose combination in COPD: pooled analysis of symptoms and exacerbations from two six-month, multicentre, randomised studies (ACLIFORM and AUGMENT). Respiratory Research. 2015;16(92):1–13.
Donohue JF, Maleki-Yazdi MR, Kilbride S, Mehta R, Kalberg C, Church A. Efficacy and safety of once-daily umeclidinium/vilanterol 62.5/25 mcg in COPD. Respir Med. 2013;107(10):1538–46.
Bateman ED, Ferguson GT, Barnes N, Gallagher N, Green Y, Henley M, et al. Dual bronchodilation with QVA149 versus single bronchodilator therapy: the SHINE study. Eur Respir J. 2013;42(6):1484–94.
European Medicines Agency. Guideline on the non-clinical development of fixed combinations of medicinal products. 2009. London. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/10/WC500003976.pdf. 2008.
Singh D, Jones PW, Bateman ED, Korn S, Serra C, Molins E, Caracta C, Garcia Gil E, Leselbaum A Efficacy and safety of aclidinium bromide/formoterol fumarate fixed-dose combinations compared with individual components and placebo in patients with COPD (ACLIFORM-COPD): a mutlicentre, randomised study. BMC Pulm Med. 2014;14(178):1–11.
D’Urzo AD, Rennard SI, Kerwin EM, Mergel V, Leselbaum AR, Caracta CF Efficacy and safety of fixed-dose combinations of aclidnium bromide/formoterol fumarate: the 24 week, randomized, placebo-controlled AUGMENT COPD study. Respir Res. 2014;15(123):1–18.
Tashkin DP, Celli B, Senn S, Burkhart D, Kesten S, Menjoge S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med. 2008;359(15):1543–54.
Cope S, Zhang J, Saletan S, Smiechowski B, Jansen JP, Schmid P. A process for assessing the feasibility of a network meta-analysis: a case study of everolimus in combination with hormonal therapy versus chemotherapy for advanced breast cancer. BMC Med. 2014;12:93.
Salanti G. Indirect and mixed-treatment comparison, network, or multiple-treatments meta-analysis: many names, many benefits, many concerns for the next generation evidence synthesis tool. Res Synth Methods. 2012;3(2):80–97.
Hoaglin DC, Hawkins N, Jansen JP, Scott DA, Itzler R, Cappelleri JC, et al. Conducting indirect-treatment-comparison and network-meta-analysis studies: report of the ISPOR Task Force on Indirect Treatment Comparisons Good Research Practices: part 2. Value Health J Int Soc Pharmacoeconomics Outcomes Res. 2011;14(4):429–37.
Dias S, Sutton AJ, Ades AE, Welton NJ. Evidence synthesis for decision making 2: a generalized linear modeling framework for pairwise and network meta-analysis of randomized controlled trials. Med Decis Mak. 2013;33(5):607–17.
Spiegelhalter DJBN, Carlin BR, van der Linde A. Bayesian measures of model complexity and fit. J R Stat Soc Ser B Stat Methodol. 2002;64:583–616.
Dias S, Welton NJ, Sutton AJ, Ades AE NICE DSU technical support document 2: a generalised linear modelling framework for pairwise and network meta-analysis of randomised controlled trials. Decision Support Unit. http://www.nicedsu.org.uk/TSD2%20General%20meta%20analysis%20corrected% 2015April2014.pdf. 2014.
Chan CK, Maltais F, Sigouin C, Haddon JM, Ford GT, SS Group. A randomized controlled trial to assess the efficacy of tiotropium in Canadian patients with chronic obstructive pulmonary disease. Can Respir J. 2007;14(8):465–72.
Boehringer I Spiriva assessment of FEV1 (SAFE). The effect of inhaled tiotropium bromide (18 mcg once daily) on the change in FEV1 during long-term treatment in patients with COPD. A 1-year parallel group, double-blind, randomised, placebo-controlled study. Boehringer Ingelheim Clinical TrialRegister. 2005.
Tonnel AB, Perez T, Grosbois JM, Verkindre C, Bravo ML, Brun M, et al. Effect of tiotropium on health-related quality of life as a primary efficacy endpoint in COPD. Int J COPD. 2008;3(2):301–10.
Celli B, Decramer M, Kesten S, Liu D, Mehra S, Tashkin DP, et al. Mortality in the 4-year trial of tiotropium (UPLIFT) in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;180(10):948–55.
Vogelmeier C, Kardos P, Harari S, Gans SJ, Stenglein S, Thirlwell J, et al. Formoterol mono- and combination therapy with tiotropium in patients with COPD: a 6-month study. Respir Med. 2008;102(11):1511–20.
Niewoehner DE, Rice K, Cote C, Paulson D, Cooper JA Jr, Korducki L, et al. Prevention of exacerbations of chronic obstructive pulmonary disease with tiotropium, a once-daily inhaled anticholinergic bronchodilator: a randomized trial.[Summary for patients in Ann Intern Med. 2005 Sep 6;143(5):I20; PMID: 16144886]. Ann Intern Med. 2005;143(5):317–26.
Brusasco V, Hodder R, Miravitlles M, Korducki L, Towse L, Kesten S, et al. Health outcomes following treatment for 6 months with once-daily tiotropium compared with twice daily salmeterol in patients with COPD.[Erratum appears in Thorax. 2005 Feb; 60(2):105]. Thorax. 2003;58(5):399–404.
Donohue JF, van Noord JA, Bateman ED, Langley SJ, Lee A, Witek TJ Jr, et al. A 6-month, placebo-controlled study comparing lung function and health status changes in COPD patients treated with tiotropium or salmeterol. Chest. 2002;122(1):47–55.
Donohue JF, Menjoge S, Kesten S, Donohue JF, Menjoge S, Kesten S. Tolerance to bronchodilating effects of salmeterol in COPD. Respir Med. 2003;97(9):1014–20.
Casaburi R, Mahler DA, Jones PW, Wanner A, San PG, ZuWallack RL, et al. A long-term evaluation of once-daily inhaled tiotropium in chronic obstructive pulmonary disease. Eur Respir J. 2002;19(2):217–24.
Donohue JF, Fogarty C, Lotvall J, Mahler DA, Worth H, Yorgancioglu A, et al. Once-daily bronchodilators for chronic obstructive pulmonary disease: indacaterol versus tiotropium. Am J Respir Crit Care Med. 2010;182(2):155–62.
Frith PT Benefits of dual bronchodilation with QVA149 once daily versus placebo, indacaterol, NVA237 and tiotropium in patients with COPD: The shine study. Respirology. 2013;Conference(var.pagings):April.
Ambrosino N, Foglio K, Balzano G, Paggiaro PL, Lessi P, Kesten S, et al. Tiotropium and exercise training in COPD patients: effects on dyspnea and exercise tolerance. Int J COPD. 2008;3(4):771–80.
Kerwin E, Hebert J, Gallagher N, Martin C, Overend T, Alagappan VK, et al. Efficacy and safety of NVA237 versus placebo and tiotropium in patients with COPD: the GLOW2 study. Eur Respir J. 2012;40(5):1106–14.
Cooper CB, Celli BR, Wise R, Brito J Jr, Guo J, Legg D. Relationship between quality of life and exercise endurance in a long-term study of tiotropium in COPD patients [abstract]. Am J Respir Crit Care Med. 2012;185:A1534.
Clinical Study Report: M/40464/30R. 2013.
Clinical Study Report: LAC-MD-31. 2013.
Clinical Study Report: SHINE study (Novartis) 2013.
Wedzicha JA, Decramer M, Ficker JH, Niewoehner DE, Sandstrom T, Taylor AF, et al. Analysis of chronic obstructive pulmonary disease exacerbations with the dual bronchodilator QVA149 compared with glycopyrronium and tiotropium (SPARK): a randomised, double-blind, parallel-group study. Lancet Respir Med. 2013;1(3):199–209.
Decramer M, Anzueto A, Kerwin E, Kaelin T, Richard N, Crater G, et al. Efficacy and safety of umeclidinium plus vilanterol versus tiotropium, vilanterol, or umeclidinium monotherapies over 24 weeks in patients with chronic obstructive pulmonary disease: results from two multicentre, blinded, randomised controlled trials. Lancet Respir Med. 2014;2(6):472–86.
Maleki-Yazdi MR, Kaelin T, Richard N, Zvarich M, Church A. Efficacy and safety of umeclidinium/vilanterol 62.5/25 mcg and tiotropium 18 mcg in chronic obstructive pulmonary disease: results of a 24-week, randomized, controlled trial. Respir Med. 2014;108(12):1752–60.
Jones PW, Beeh KM, Chapman KR, Decramer M, Mahler DA, Wedzicha JA. Minimal clinically important differences in pharmacological trials. Am J Respir Crit Care Med. 2014;189(3):250–5.
Acknowledgments
This study was conducted by Mapi on behalf of AstraZeneca, who funded the study and the writing of this manuscript. Article processing charges for this study were also funded by AstraZeneca. All authors participated in the design and conduct of the study, as well as drafting and revising the manuscript. Leandro Lindner is an employee of AstraZeneca. Miriam van der Weijden, Andreas Karabis and Goran Medic are employees of Mapi and served as paid consultants to AstraZeneca during the conduct of this study and the preparation of this manuscript.
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole and have given final approval for the version to be published.
Disclosures
Miriam van der Weijden, Goran Medic and Andreas Karabis are all employees of Mapi and served as paid consultants to AstraZeneca during the conduct of this study. Leandro Lindner is an employee of AstraZeneca.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Medic, G., Lindner, L., van der Weijden, M. et al. Efficacy and Safety of Aclidinium/Formoterol versus Tiotropium in COPD: Results of an Indirect Treatment Comparison. Adv Ther 33, 379–399 (2016). https://doi.org/10.1007/s12325-016-0299-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-016-0299-4