
Abstract
Different pathogenic variants in the DNA polymerase-gamma2 (POLG2) gene cause a rare, clinically heterogeneous mitochondrial disease. We detected a novel POLG2 variant (c.1270 T > C, p.Ser424Pro) in a family with adult-onset cerebellar ataxia and progressive ophthalmoplegia. We demonstrated altered mitochondrial integrity in patients’ fibroblast cultures but no changes of the mitochondrial DNA were found when compared to controls. We consider this novel, segregating POLG2 variant as disease-causing in this family. Moreover, we systematically screened the literature for POLG2-linked phenotypes and re-evaluated all mutations published to date for pathogenicity according to current knowledge. Thereby, we identified twelve published, likely disease-causing variants in 19 patients only. The core features included progressive ophthalmoplegia and cerebellar ataxia; parkinsonism, neuropathy, cognitive decline, and seizures were also repeatedly found in adult-onset heterozygous POLG2-related disease. A severe phenotype relates to biallelic pathogenic variants in POLG2, i.e., newborn-onset liver failure, referred to as mitochondrial depletion syndrome. Our work underlines the broad clinical spectrum of POLG2-related disease and highlights the importance of functional characterization of variants of uncertain significance to enable meaningful genetic counseling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mitochondria are important cell organelles fulfilling essential tasks in cells, including energy production. They have their own genetic material (mitochondrial DNA, mtDNA) encoding components of the respiratory chain. Mutations in mtDNA, either inherited or acquired, result in diverse clinical phenotypes [1]. mtDNA is replicated by DNA polymerase gamma, which is composed of a catalytic (encoded by POLG) and an accessory subunit (encoded by POLG2) [2]. Mutations in both nuclear genes lead to variable expression of inherited movement and multisystem disorders associated with mitochondrial dysfunction. While POLG mutations are rather common and have been reviewed in 2019 [3], available information about POLG2 mutations is scarce. To date, the few reported heterozygous POLG2 mutations showed a disease onset in adulthood and were linked to (chronic) progressive external ophthalmoplegia ((c)PEO) and cerebellar ataxia leading to the syndrome of “progressive external ophthalmoplegia with mtDNA deletions, autosomal dominant ataxia type 4” (PEOA4, OMIM #610,131) [4]. Furthermore, biallelic mutations in POLG2 cause a severe infancy-onset phenotype referred to as “mitochondrial DNA depletion syndrome” (OMIM #618,528 or #619,425), leading to liver failure and death in infancy [5]. Here, we describe clinical features and the functional characterization of two siblings with a novel, heterozygous likely pathogenic POLG2 missense variant (c.1270 T > C; p.Ser424Pro). We further compare the phenotype to other mutation carriers by reviewing all POLG2 mutations, including the re-evaluation of the pathogenicity of variants from the published literature.
Methods
Patients
The study was approved by the local ethics committee of the University of Lübeck and all participants gave written informed consent. Blood samples from three affected family members (including the index patient [L-10343], her brother [L-11048], and their mother [L-10914]) were genetically tested by exome sequencing. The index patient and her brother underwent a detailed clinical examination and skin biopsies were taken to establish primary skin fibroblast lines. No biopsy could be taken from the meanwhile deceased mother.
Genetic Testing
After exclusion of several spinocerebellar ataxias due to repeat expansions, exome sequencing was performed at CENTOGENE GmbH (Rostock, Germany) in a diagnostic setting. The candidate variant in POLG2 was confirmed by Sanger sequencing in all three affected family members.
Functional Analysis
Cell Culture
Two healthy individuals (L-2152, L-2153) without pathogenic POLG2 variants were included as controls in the functional characterization. Dermal skin fibroblast cultures were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Life technologies) supplemented with 10% fetal bovine serum (Life technologies) and 1% penicillin/streptomycin (Life technologies) at 37 °C with 5% CO2.
Mitochondrial DNA and Replication Analyses
For deep mitochondrial sequencing, we used blood samples from three patients and DNA from fibroblast cultures of both available patients. We also included four age- and sex-matched controls for each patient and DNA source. mtDNA was amplified by two overlapping long-range PCRs (FampA: 5′-AAATCTTACCCCGCCTGTTT-3′, FampB: 5′-GCCATACTAGTCTTTGCCGC-3′, RampA: 5′-AATTAGGCTGTGGGTGGTT-3′, RampB: 5′-GGCAGGTCAATTTCACTGGT-3′ [6]). Sequence analysis was performed at > 1000 × coverage on a GridION machine (Oxford Nanopore, Oxford, UK) as described [7], using Mutserve2 (v2.0.0) for mtDNA variant calling [8]. Furthermore, a real‐time PCR assay for detection of three targets within the mitochondrial genome (ND1 [NADH dehydrogenase 1], ND4, and D‐loop) was performed using blood samples from the three POLG2 mutation carriers and three healthy controls as described [9, 10]. For mtDNA copy number quantification, the ND1 concentration per unit area was calculated. To detect mtDNA major arc deletions, the ND1:ND4 ratio was determined. During transcription and replication, a linear DNA molecule, that is, 7S DNA, is incorporated in this particular region of the mitochondrial genome, resulting in a triple‐stranded structure denominated as “D‐loop.” The D‐loop:ND1 ratio is therefore representative of the mtDNA replication status in a given cell, with a higher ratio representing more actively transcribed or replicating mtDNA molecules. All measurements were performed three times in duplicates. We also used long-range PCR of the mtDNA to test for the presence of shorter fragments due to partial mtDNA deletions.
Immunofluorescence Staining and Image Analysis
For immunocytochemical analysis, cells on coverslips were fixed in 4% formaldehyde for 15 min, permeabilized, and blocked with 0.1% Triton X-100 in 4% normal goat serum in PBS for 1 h. Mitochondrial network interconnectivity was analyzed in cells stained for mitochondrial GRP75. Immunofluorescence staining was performed with primary antibody against GRP75 (1:1000; Abcam) and respective secondary fluorescence antibody (1:400; Life technologies). The images were taken as z-stacks using a confocal microscope. The form factor was calculated using the formula [Pm2]/[4πAm], with Pm being the length of the mitochondrial outline and Am being the area of the mitochondrion. At least ten cells from two coverslips per individual were analyzed and a mean form factor was calculated as a measure for mitochondrial interconnectivity using ImageJ (NIH software) as previously described [11].
Western Blot Analysis
Cell pellets were extracted using SDS extraction buffer (50 mM Tris–HCl pH 7.6, 150 mM NaCl, 1% DOC, 1% NP-40, and 0.1% SDS) or 1% Triton X-100 lysis buffer (containing 10% glycerol, 150 mM NaCl, 25 mM Hepes pH7.4, 1 mM EDTA, 1.5 mM MgCl2, proteinase inhibitor cocktail) and gels were blotted onto nitrocellulose membranes. Antibodies used for immunoblotting were anti-TOMM20 (translocase of the outer mitochondrial membrane 20; 1:1,000; Santa Cruz) and anti-β-actin (1:1,000,000; Sigma).
Mitochondrial Membrane Potential (MMP) Analysis
Fluorescence-activated cell sorting (FACS) was used to assess MMP. Primary skin fibroblasts were incubated with tetramethylrhodamine methyl ester perchlorate (TMRM, 10 nM). Subsequently, samples were washed to remove unbound dye. Carbonyl cyanide-p-trifluorometoxyphenylhydrazone (FCCP, 10 µM) was added to a part of the cells to suppress MMP. The TMRM-staining was measured using a BD LSR II® flow cytometer. First, living cells (4′,6-diamidino-2-phenylindole (DAPI)negative) were identified and used for the gating strategy. Then, the mean fluorescence intensity (MFI) of TMRM-stained cells was assessed in FCCP-treated fibroblasts compared to fibroblasts just diluted in medium. Results were calculated using the following formula: MFI TMRMmedium – MFI TMRMFCCP = MMP. Data were analyzed with FlowJo® Software (Treestar).
Statistical Analysis
Differences were analyzed using unpaired t-tests or analyses of variance (ANOVA) with a Bonferroni-Dunn post hoc test. The error bars indicate SEM of n ≥ 3 independent experiments. Age at onset, age at examination, and disease duration in the review part are shown as mean ± standard deviation. Statistical analyses were performed with GraphPad Prism 9.
Review of the Literature
To identify the clinical signs and symptoms associated with POLG2 mutations, we conducted a literature review in PubMed with the search term “POLG2” on October 23, 2022, identifying 82 publications (Fig. 1). We extracted clinical as well as genetic information systematically from all publications. For all identified POLG2 variants, the frequency was analyzed using the GnomAD database (https://gnomad.broadinstitute.org/), the CADD (Combined Annotation Dependent Depletion) score [12] (https://cadd.gs.washington.edu/) was calculated, and a pathogenicity scoring was performed according to the MDSGene workflow as described (https://www.mdsgene.org/methods). Moreover, all variants were rated according to the ACMG (American College of Medical Genetics and Genomics) guidelines [13] using Franklin by Genoox (https://franklin.genoox.com/clinical-db/home).

Flow chart of the search strategy of the literature review. The flow diagram according to the PRISMA guidelines presents the steps of literature research, the respective numbers of papers as well as reasons of exclusion
Individuals harboring variants with a MAF > 0.01 at least in a certain population and/or variants rated as benign according to the ACMG criteria or by MDSGene scoring were disregarded as well as individuals with more than one POLG2 variant on the same allele or additional variants in genes linked to similar phenotypes (Table S1).
Results
Case Reports
We report two siblings with a heterozygous POLG2 variant exhibiting cerebellar ataxia and progressive ophthalmoplegia in one sibling and dysmetria of saccades in the other sibling. The index patient L-10343 (Fig. 2A, Video 1), a 56-year-old female patient of German origin, initially experienced gait imbalance at the age of 30 years. The disease has been slowly progressive: 10 years after disease onset, the patient developed diplopia. At the age of 50 years, diplopia was so pronounced that the patient tended to keep one eye closed to avoid disturbing visual impressions. Moreover, speech and gait also deteriorated. Cognitive problems, as well as autonomic symptoms, were denied. The neurological examination revealed bilateral ptosis and external ophthalmoplegia, manifesting by reduced eye bulb adduction more strongly pronounced on the right side. The patient had slowed and hypometric horizontal saccades whereas vertical saccades were normal. She exhibited dysarthria and severe cerebellar ataxia, including dysmetria during finger-chase and finger-nose testing, and an abnormal heel-shin slide, the latter more pronounced on the left side. She was unable to walk and to stand unaided requiring a wheelchair. Moreover, there was dystonic posturing of the hands and mild cervical dystonia with torticollis and laterocollis to the right side. She scored 27/40 points on the Scale for the assessment and rating of ataxia (SARA). Cognitive function was not impaired as revealed by 28/30 points on the Montreal cognitive assessment (MoCA). The patient had neither spasticity nor signs of neuropathy or parkinsonism. A cranial MRI was unremarkable.

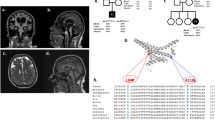
A novel heterozygous variant in POLG2. A Pedigree of the presented family harboring a heterozygous POLG2 variant. Squares represent male individuals; circles represent female family members. The index patient is marked with an arrow. Dark gray squares represent clinically affected family members with genetic confirmation of the c.1270 T > C (p.Ser424Pro) variant. The striped circle indicates that the index patient’s grandmother was reported to be affected. However, no clinical examination and no genetic testing were performed. Age at onset (AAO) and age at examination (AAE) are stated for affected family members if available (NA: not available). B Genomic DNA sequences of the c.1270 T > C variant are shown from blood. The traces are representative of 2 sequencing runs. C Mitochondrial DNA (mtDNA) sequence variants in POLG2 mutation carriers. MtDNA was extracted from blood or cultured fibroblasts. The number of low-frequency heteroplasmic variants (1–15% heteroplasmy frequency [HF]) (upper panels) and higher frequency variants (HF > 5%) (lower panels). Each dot represents one sample, and median and 1st and 3rd quartile are indicated
Her youngest brother L-11048 (Fig. 2A, Video 2), a construction worker, developed balance problems at the age of 40 years, first manifesting while working on scaffolds. In the following years, he developed gait impairment, slightly slurred speech, and problems with fine motor tasks. The neurological examination at the age of 50 years revealed hypermetric saccades and macro square wave jerks but no ophthalmoplegia. Moreover, he had dysarthria, dysdiadochokinesia, hypermetric finger chase, and a slightly abnormal finger-nose test (SARA 10/40). His gait was broad-based and ataxic. Signs of neuropathy were present clinically (reduced sense of vibration) and in nerve conduction studies (mild sensory axonal neuropathy).
In both siblings, standard laboratory analysis, including creatinine kinase, liver enzymes (alanine aminotransferase and aspartate aminotransferase), and bilirubin, were unremarkable, as well as electroencephalography.
Another older sister and one younger brother were reported to be unaffected whereas the patients’ mother presented features of cerebellar ataxia commencing at the age of 50 years. Of note, also her maternal mother was reported to have suffered from ataxia collectively indicating an autosomal dominant or maternal mode of inheritance (Fig. 2A).
Spinocerebellar ataxias due to repeat expansions and pathogenic variants in 188 ataxia genes, including POLG but not POLG2, were excluded prior to exome sequencing in the index patient and her affected brother. By filtering the exome data for rare (minor allele frequency (MAF) < 0.01), heterozygous variants shared by the three affected individuals in genes previously linked to a mitochondrial phenotype, we detected a POLG2 missense variant (NM_007215.3: c.1270 T > C; p.Ser424Pro) as the only candidate. This variant was confirmed by Sanger sequencing (Fig. 2B) and has not yet been reported in any patient but is adjacent to the previously reported p.Ser423Tyr variant [14]. It has also not been reported in gnomAD (~ 140,000 samples; https://gnomad.broadinstitute.org/gene/ENSG00000256525?dataset=gnomad_r2_1). The CADD score was 24.8 pointing to deleteriousness. Due to the rarity of the variant and the segregation in the family but in the absence of any published functional data, the variant was classified initially as variant of uncertain significance (VUS, ACMG criteria: PM2 PP1, PP3, PP4 [13]).
Functional Analysis
To evaluate pathogenicity in vitro, we performed functional analyses to test for mitochondrial integrity in the POLG2 variant carriers. We first looked for changes of the mtDNA. Notably, deep mtDNA sequencing did not reveal any differences when compared to controls (Fig. 2C), neither for low-frequency (somatic) heteroplasmic nor for high-frequency variants. Furthermore, we did also not identify large mtDNA deletions neither by quantitative PCR nor by long-range PCR (data not shown). Likewise, we did not observe changes in mtDNA copy number and replication status in the POLG2 mutation carriers compared to healthy controls (data not shown).
Next, we analyzed the effect of mutant POLG2 on the integrity of the mitochondrial network by calculating the form factor in cultured POLG2-mutant fibroblasts and controls. The analysis revealed decreased mitochondrial branching and interconnectivity in POLG2-mutant fibroblasts from the index patient and her brother (Fig. 3A, B) when compared to controls (p < 0.05). In addition, we analyzed protein levels of the mitochondrial translocase TOMM20, located in the outer membrane, by Western blotting. TOMM20 protein levels were decreased in POLG2-mutant fibroblasts from both available patients compared to healthy controls (Fig. 3C). Furthermore, POLG2-mutant fibroblasts exhibited a decreased mitochondrial membrane potential (p < 0.05), analyzed by flow cytometry, compared to healthy controls (Fig. 3D, E). This suggests the variant c.1270 T > C (p.Ser424Pro) in POLG2 as the cause of the complex neurological phenotype due to mitochondrial dysfunction in this family despite the absence of any detectable changes on mtDNA derived from blood and fibroblasts. As the diagnosis was made by genetic testing, a muscular biopsy was not performed.

Altered mitochondrial network, decreased TOMM20 levels, and decreased mitochondrial membrane potential (MMP) in POLG2-mutant fibroblasts. POLG2-mutant fibroblasts from the index patient (L-10343), and her brother (L-11048), and two healthy control fibroblasts (L-2152, L-2153) were examined. A The mitochondrial network is shown using confocal microscopy in cells immunostained with anti-GRP75. The scales correspond 20 µm. B Form factor as a measure for mitochondrial network interconnectivity (GRP75 immunostaining) was calculated for the controls and the POLG2-mutants. Each dot represents the measurement in a single cell (n = 14). The mean values and the standard deviations of the investigated individuals are shown (p < 0.05, comparing POLG2-mutant fibroblasts with healthy controls). C Western blot analysis shows decreased mitochondrial TOMM20 protein levels in POLG2-mutant fibroblasts in comparison to controls with β-actin as loading control. D The number of tetramethylrhodamin-methylester (TMRM; 10 nM)-stained cells was assessed by flow cytometry under basal condition (left panel) or upon treatment with a mitochondrial uncoupler (depolarizer) carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; 10 µM) (right panel). Representative dot plots from two POLG2-mutant fibroblasts (L-11048, L-10343) and two healthy controls (L-2152, L-2153) are depicted. Each dot represents the fluorescence measurement in a single cell (n = 10,000). E Quantification of the TMRM-positive cells normalized to FCCP-treated cells by using FlowJo. The analysis revealed reduced number of cells stained with the TMRM dye, indicating decreased MMP in POLG2-mutant fibroblasts. The mean values and the standard deviations of the investigated individuals are shown (n = 3 experiments, p < 0.05, comparing MMP in POLG2-mutant fibroblasts with MPP in control fibroblasts)
Due to the detected functional changes in the mitochondrial integrity, a strong criterion from the ACMG recommendations was now fulfilled, we classified the variant as likely pathogenic (ACMG criteria: PS3, PM2 PP1, PP3, PP4 [13]).
Review of the Literature
The literature search revealed only 17 reports containing clinical information for individual monoallelic (heterozygous) or biallelic (homozygous) POLG2 variant carriers. After excluding patients with multiple variants on the same allele in POLG2 or other genes linked to a similar phenotype [15,16,17,18,19], 24 patients with suggested POLG2-related disease carrying 14 different variants in the heterozygous state [4, 14, 20,21,22,23,24,25] remained. Five of these 14 variants (39%) did not fulfill the criteria of pathogenicity by MDSGene and/or scored “benign” or “likely benign” according to the ACMG guidelines and were excluded (Table S1). Out of the five excluded variants, three had a MAF > 0.01, at least in certain populations. The rating of variants according to state-of-the-art criteria of pathogenicity led to the exclusion of eight patients (Table S2). The remaining nine POLG2 variants scored at least as possibly pathogenic and comprised three missense changes, three nonsense, one frameshift, one splice site variant, and one in-frame insertion (Table S2). Furthermore, all three homozygous variants reported in independent patients with recessive POLG2-related disease were scored at least possibly pathogenic according to the MDSGene and VUS/likely pathogenic according to ACMG criteria, respectively (Table S3).
Five publications focused on twelve adult-onset patients, while one paper reported on a patient examined at the age of 26 years that had exercise intolerance and myoclonus as core symptoms but was excluded from further analysis due to missing information on the age of onset [5, 17, 20,21,22] (Table S2). Among the remaining adult-onset cases (n = 12, 10 females), the age at onset was 52.2 ± 9.5 years, age at examination 66.2 ± 9.5 years, and disease duration 13.9 ± 5.9 years, respectively. Cerebellar ataxia was the most common feature reported in nine individuals (100% of cases with information on this item (n = 9)), followed by neuropathy in six of seven (86%), and PEO in eight of twelve (67%) patients. Ptosis was reported in three of eight (38%) and seizures were observed in three patients, while neither gastric nor liver problems were reported in any of the patients with onset in adulthood. Moreover, POLG2 mutations manifested with parkinsonism in three individuals from the same Belgian pedigree [22] and one additional independent female patient [25], and “extrapyramidal signs” were reported in two, and “pyramidal signs” in four individuals from a recently published family [23] (Table 1, Table S2). In one case, camptocormia was the core symptom [24] (Table S2). Regarding muscle biopsy results, all six individuals with data on cytochrome C oxidase (COX) staining had COX-negative fibers and mtDNA deletions in muscle tissue were observed in all six patients in whom this feature was investigated (Table 1).
One single screening study investigating 112 POLG-negative unrelated individuals with possible early-onset mitochondrial disease identified eleven childhood-onset cases with heterozygous POLG2 mutations [14], of which three individuals (age at examination 5.7 ± 4.0 years, 1 female) had variants scored at least possibly pathogenic according to the criteria applied in our study. Regarding the phenotype, muscle weakness/hypotonia and liver disease were present in two cases, accompanied by gastric complaints and developmental delay in one individual.
The literature review also revealed three patients with homozygous POLG2 missense variants. The first patient was a 3-month-old boy carrying the homozygous missense variant c.544C > T (p.Arg182Trp). He suffered from a mitochondrial DNA depletion syndrome manifesting as a fulminant liver failure with consecutive death [6, 26]. Other biallelic POLG2 mutations in two independent individuals have been linked to epilepsy [27] and a combination of childhood-onset optic atrophy, cerebellar ataxia, peripheral neuropathy, psychiatric comorbidities, and premature ovarian failure [28], respectively (Table S3).
Discussion
Here, we report three affected family members from an autosomal-dominant ataxia pedigree all harboring a novel likely pathogenic heterozygous POLG2 variant. Two of these patients underwent extensive clinical examination. Their clinical presentation was in line with the previously observed phenotype of POLG2-related disease, including PEO in the index patient and cerebellar ataxia in both siblings.
Since the missense variant was of uncertain pathogenicity, we tested the mitochondrial integrity in blood and patient-derived cultured fibroblasts. We demonstrated that the mitochondrial network was altered, and TOMM20 protein levels as well as MMP were decreased in POLG2-mutant fibroblasts. The use of fibroblast cultures to model POLG2-related disease is supported by previous reports showing a reduction of MMP in the mutant [20, 23]. Notably, we did not observe changes in mtDNA, neither an increased number of low- or high-frequency single nucleotide variants when compared to age- and sex-matched controls, nor copy number changes or mtDNA deletions in our patients. The latter might be restricted to muscle cells. Interestingly, it was previously shown that even after ethidium bromide-induced mtDNA depletion, the restoration of mtDNA levels was not delayed in fibroblasts from POLG2 patients [29]. This suggests that cells compensate for the mtDNA replication defect by slowing down mtDNA degradation, possibly explaining the lack of detectable changes on the mtDNA in blood and fibroblasts. Based on the segregation of the variant within the family, the characteristic phenotype, and the altered mitochondrial integrity, our data suggest that this variant is likely pathogenic and causes the mitochondrial phenotype in this family. Of note, functional analyses, as performed in the context of the novel variant presented here, are needed to classify newly identified variants for which pathogenicity is uncertain.
A review of the literature revealed that in contrast to POLG-linked disorders, POLG2-related disease is very rare — only 16 cases with heterozygous and three individuals with biallelic likely disease-causing POLG2 variants have been published to date. Of note, more than a third of the variants described as causative for dominantly inherited POLG2-related disease in the literature did not meet the state-of-the-art criteria for pathogenicity scoring, stressing the urgent need to re-evaluate the impact of reported variants as soon as further data and new scoring recommendations become available. Importantly, the classification of variants according to the MDSGene criteria was consistent with the ACMG guidelines.
Similar to POLG-related disease, the clinical spectrum of heterozygous POLG2 mutations comprises cerebellar ataxia and PEO in adulthood-onset and metabolic abnormalities and seizures in childhood-onset cases. Moreover, seizures may also be present in adult-onset patients, as well as parkinsonism and camptocormia. As expected, biallelic POLG2 variants can lead to a more severe phenotype with early-onset liver failure and death, referred to as mtDNA depletion syndrome. Notably, cases with a later onset and a less severe phenotype with biallelic POLG2 mutations have been published as well. MtDNA deletions and COX-negative fibers in muscle tissue have been reported in a few tested patients with POLG2-related disease due to heterozygous mutations. However, COX-negative fibers, as well as the clinical symptoms, have a significant overlap with the clinical picture of POLG-related disease and other mitochondrial conditions, which appears plausible due to the joint function of the proteins.
The detection of a POLG2 variant might have implications for patient care: Individuals with mitochondriopathies are at an increased risk of complications related to volatile anesthetics, muscle relaxants, and some drugs such as valproic acid, statins, and metformin. Furthermore, certain antibiotics should be avoided or only be used with caution, as those substances might deteriorate mitochondrial disease [30]. However, whether these precautions also apply to POLG2-related disease has not yet been reported. Concerning the treatment, proven disease-modifying drugs are not available, and no data exist on whether mitochondrial enhancers, e.g., coenzyme Q10 or carnitine, help alleviate the symptoms.
Taken together, we provide evidence that the novel POLG2 variant leads to impaired mitochondrial integrity suggesting its pathogenicity for the mitochondrial phenotype (ataxia and progressive external ophthalmoplegia) in the present family. Overall, the phenotypic spectrum in POLG2-related disease is broad and distinct in childhood- and adult-onset cases. Our work emphasizes the relevance of investigating POLG2 as a nuclear gene in patients with cerebellar ataxia — as POLG2 might currently not be included in all commercially available diagnostic ataxia panels — and further highlights the importance of the functional characterization of variants of uncertain significance to allow for meaningful genetic counseling.
Data Availability
Additional raw data supporting the conclusions of this article will be made available by the authors upon reasonable request.
References
Chapman J, Ng YS, Nicholls TJ. The maintenance of mitochondrial DNA integrity and dynamics by mitochondrial membranes. Life (Basel). 2020;10(9):164.
Kaguni LS. DNA polymerase gamma, the mitochondrial replicase. Annu Rev Biochem. 2004;73:293–320.
Rahman S, Copeland WC. POLG-related disorders and their neurological manifestations. Nat Rev Neurol. 2019;15(1):40–52.
Longley MJ, Clark S, Yu Wai Man C, Hudson G, Durham SE, Taylor RW, et al. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet. 2006;78(6):1026–34.
Varma H, Faust PL, Iglesias AD, Lagana SM, Wou K, Hirano M, et al. Whole exome sequencing identifies a homozygous POLG2 missense variant in an infant with fulminant hepatic failure and mitochondrial DNA depletion. Eur J Med Genet. 2016;59(10):540–5.
Fendt L, Zimmermann B, Daniaux M, Parson W. Sequencing strategy for the whole mitochondrial genome resulting in high quality sequences. BMC Genomics. 2009;10:139.
Lüth T, Schaake S, Grünewald A, May P, Trinh J, Weissensteiner H. Benchmarking low-frequency variant calling with long-read data on mitochondrial DNA. Front Genet. 2022;13: 887644.
Weissensteiner H, Forer L, Fuchsberger C, Schöpf B, Kloss-Brandstätter A, et al. mtDNA-Server: next-generation sequencing data analysis of human mitochondrial DNA in the cloud. Nucleic Acids Res. 2016; 8;44(W1):W64–9.
Rygiel KA, Grady JP, Taylor RW, Tuppen HAL, Turnbull DM. Triplex real-time PCR - an improved method to detect a wide spectrum of mitochondrial DNA deletions in single cells. Sci Rep. 2015;5(1):9906.
Grünewald A, Rygiel KA, Hepplewhite PD, Morris CM, Picard M, Turnbull DM. Mitochondrial DNA depletion in respiratory chain-deficient Parkinson disease neurons. Ann Neurol. 2016;79(3):366–78.
Grünewald A, Voges L, Rakovic A, Kasten M, Vandebona H, Hemmelmann C, et al. Mutant Parkin impairs mitochondrial function and morphology in human fibroblasts. PLoS ONE. 2010;5(9): e12962.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Young MJ, Longley MJ, Li F-Y, Kasiviswanathan R, Wong L-J, Copeland WC. Biochemical analysis of human POLG2 variants associated with mitochondrial disease. Hum Mol Genet. 2011;20(15):3052–66.
Ferraris S, Clark S, Garelli E, Davidzon G, Moore SA, Kardon RH, et al. Progressive external ophthalmoplegia and vision and hearing loss in a patient with mutations in POLG2 and OPA1. Arch Neurol. 2008;65:125–31.
Nagappa M, Bindu PS, Taly AB, Sonam K, Shwetha C, Kumar R, et al. Palatal tremor in POLG-associated ataxia. Mov Disord Clin Pract. 2015;2:318–20.
Valencia CA, Wang X, Wang J, Peters A, Simmons JR, Moran MC, et al. Deep sequencing reveals novel genetic variants in children with acute liver failure and tissue evidence of impaired energy metabolism. PLoS ONE. 2016;11(8): e0156738.
Walter MC, Czermin B, Muller-Ziermann S, Bulst S, Steward JD, Hudsonn G, et al. Late-onset ptosis and myopathy in a patient with a heterozygous insertion in POLG2. J Neurol. 2010;257(9):1517–23.
Siegert S, Mindler GT, Brücke C, Kranzl A, Patsch J, Ritter M, et al. Expanding the phenotype of the FAM149B1-related ciliopathy and identification of three neurogenetic disorders in a single family. Genes (Basel). 2021;12(11):1648.
Craig K, Young MJ, Blakely EL, Longley MJ, Turnbull DM, Copeland WC et al. A p.R369G POLG2 mutation associated with adPEO and multiple mtDNA deletions causes decreased affinity between polymerase γ subunits. Mitochondrion. 2012;12(2):313–19.
Lieber DS, Calvo SE, Shanahan K, Slate NG, Shangtao L, Hershman SG, et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology. 2013;80(19):1762–70.
Van Maldergem L, Besse A, De Paepe B, Blakely EL, Appadurai V, Humble MM, et al. POLG2 deficiency causes adult-onset syndromic sensory neuropathy, ataxia and parkinsonism. Ann Clin Transl Neurol. 2017;4(1):4–14.
Kim M, Kim AR, Kim JS, Park J, Youn J, Ahn JH, et al. Clarification of undiagnosed ataxia using whole-exome sequencing with clinical implications. Parkinsonism Relat Disord. 2021;80:50–64.
Lehmann Urban D, Motlagh Scholle L, Alt K, Ludolph AC, Rosenbohm A. Camptocormia as a novel phenotype in a heterozygous POLG2 mutation. Diagnostics (Basel). 2020;10(2).
Hou Y, Zhao X, Xie Z, Yu M, Lv h, Zhang W, et al. Novel and recurrent nuclear gene variations in a cohort of Chinese progressive external ophthalmoplegia patients with multiple mtDNA deletions. Mol Genet Genomic Med. 2022; 10:e1921.
Hoff KE, DeBalsi KL, Sanchez-Quintero MJ, Longley MJ, Hirano M, Naini AB, et al. Characterization of the human homozygous R182W POLG2 mutation in mitochondrial DNA depletion syndrome. PLoS ONE. 2018;13(8): e0203198.
Lee SJ, Kanwal S, Yoo DH, Park HR, Choi B-O, Chung KW. A POLG2 homozygous mutation in an autosomal recessive epilepsy family without ophthalmoplegia. J Clin Neurol. 2019;15(3):418–20.
Dosekova P, Dubiel A, Karlowicz A, Zietkiewicz S, Rydzanicz M, Habalova V, et al. Whole exome sequencing identifies a homozygous POLG2 missense variant in an adult patient presenting with optic atrophy, movement disorders, premature ovarian failure and mitochondrial DNA depletion. Eur J Med Genet. 2019;103821.
Stewart JD, Schoeler S, Sitarz KS, Horvath R, Hallmann K, Pyle A, et al. POLG mutations cause decreased mitochondrial DNA repopulation rates following induced depletion in human fibroblasts. Biochim Biophys Acta. 2011;1812(3):321–5.
Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2015;17(9):689–701.
Acknowledgements
The authors thank the patients for participation in examinations for research purposes.
Funding
Open Access funding enabled and organized by Projekt DEAL. The study was funded by the Foundation of the University Hospital of Schleswig–Holstein “Gutes Tun,” the Damp Foundation (to Alexander Münchau and Katja Lohmann), and by a Habilitation Fellowship for Women Researchers from the University of Lübeck (to Marija Dulovic-Mahlow).
Author information
Authors and Affiliations
Contributions
Max Borsche, Marija Dulovic-Mahlow, Katja Lohmann, and Norbert Brüggemann contributed to the study conception and design. Max Borsche, Sinem Tunc, Alexander Münchau, and Norbert Brüggemann contributed to the collection and the analysis of the clinical data. Marija Dulovic-Mahlow, Hauke Baumann, Theresa Lüth, Susen Schaake, Selin Özcakir, Ana Westenberger, Joanne Trinh, Evelyn Knappe, and Katja Lohmann conceptualized and performed the laboratory experiments. Max Borsche performed the literature search. The first draft of the manuscript was written by Marija Dulovic-Mahlow and Max Borsche. Norbert Brüggemann and Katja Lohmann contributed significantly to the revision of the manuscript. All authors commented on previous versions of the manuscript and read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
This study was conducted with the approval by the ethics committee of the University of Lübeck.
Patient Consent
Obtained.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file2 Video 1: Representative excerpts of the neurological examination of the index patient, a 56-years old female, presenting with external ophthalmoplegia and severe cerebellar ataxia. (MOV 4921 KB)
Supplementary file3 Video 2: The patient shows milder cerebellar ataxia compared to his sister, no external ophthalmoplegia, but dysmetria of saccades. (MOV 4839 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Borsche, M., Dulovic-Mahlow, M., Baumann, H. et al. POLG2-Linked Mitochondrial Disease: Functional Insights from New Mutation Carriers and Review of the Literature. Cerebellum 23, 479–488 (2024). https://doi.org/10.1007/s12311-023-01557-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-023-01557-x