
Abstract
This is a summary of the virtual presentation given at the 2021 meeting of the Society for Research on the Cerebellum and Ataxias, https://www.meetings.be/SRCA2021/, where the therapeutic potential of the CCK-CCK1R pathway for treating diseases involving Purkinje cell degeneration was presented. Spinocerebellar ataxia type 1 (SCA1) is one of a group of almost 50 genetic diseases characterized by the degeneration of cerebellar Purkinje cells. The SCA1 Pcp2-ATXN1[30Q]D776 mouse model displays ataxia, i.e. Purkinje cell dysfunction, but lacks progressive Purkinje cell degeneration. RNA-seq revealed increased expression of cholecystokinin (CCK) in cerebella of Pcp2-ATXN1[30Q]D776 mice. Importantly, the absence of Cck1 receptor (CCK1R) in Pcp2-ATXN1[30Q]D776 mice conferred a progressive degenerative disease with Purkinje cell loss. Administration of a CCK1R agonist to Pcp2-AXTN1[82Q] mice reduced Purkinje cell pathology and associated deficits in motor performance. In addition, administration of the CCK1R agonist improved motor performance of Pcp2-ATXN2[127Q] SCA2 mice. Furthermore, CCK1R activation corrected mTORC1 signaling and improved the expression of calbindin in the cerebella of AXTN1[82Q] and ATXN2[127Q] mice. These results support the Cck-Cck1R pathway is a potential therapeutic target for the treatment of diseases involving Purkinje neuron degeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Spinocerebellar ataxias (SCAs) are a genetically heterogeneous group of over 45 autosomal dominant neurodegenerative diseases. In many SCAs, dysfunction along with degeneration of cerebellar Purkinje cells precedes pathology in other neuronal cells/regions [1, 2]. This feature of the SCAs supports the concept that Purkinje cells are more vulnerable to cellular perturbations than other neurons [3,4,5].
Numerous unique features of Purkinje cells very likely underlie their enhanced vulnerability to perturbations. Purkinje cells are large neurons with an extensive and elaborate dendritic tree that has a large excitatory synaptic input. For example, climbing fiber synapses onto Purkinje cells, which generate complex spike excitatory postsynaptic potentials, are among the most powerful excitatory synapses in the brain. In addition, Purkinje cells generate autonomous pacemaking spikes. Consistent with these electrophysiological and structural properties, Purkinje cells have remarkably high metabolic activity. Furthermore, as noted by Hekman and Gomez (2015), several reports demonstrate that disruptions in protein homeostasis impact Purkinje cells before other neurons in mice and humans [6,7,8,9,10]. This aspect of Purkinje cell vulnerability is particularly relevant to SCAs as deficits in protein homeostasis are a recurring pathogenic theme for these diseases [11].
The utilization of SCA mouse models has been critical for gaining insight into the molecular basis of these disorders [5]. In the case of Purkinje cell vulnerability in SCA1, cerebellar transcriptomic analyses were used for the identification of both disease progression and protection pathways [12]. Notably, it was found that the level of cholecystokinin (CCKs) expression and its subsequent interaction with the CCK1 receptor is a potential Purkinje cell protective pathway in SCA1 mice. Subsequently, it was demonstrated that administration of a Cck1R agonist improves motor performance, corrects mTORC1 signaling, and improves the expression of calbindin in the cerebella of SCA1 and SCA2 transgenic mice. These results indicate that manipulation of the CCK-CCK1R pathway is a potential therapeutic target for the treatment of diseases involving Purkinje neuron degeneration. This minireview discusses the evidence supporting the CCK-CCK1R as a protective pathway in SCA mice and speculates on the broader implications of these findings.
Purkinje Cell Protection by CCK is Dependent on Presence of CCK1 Receptor
The CCK-CCK1R line of investigation began with a study to examine the role of ATXN1-S776 phosphorylation in Purkinje cell SCA1 pathogenesis. It was found that replacing the serine amino acid with a potential phospho-mimicking aspartic acid at position 776 transformed wild type (WT) ATXN1[30Q] into a pathogenic protein. Importantly, while mice expressing ATXN1[30Q]-D776 in Purkinje cells manifested severe ataxia from an early age, i.e. as severe as mice with Purkinje cell expression of ATXN1[82Q], disease in ATXN1[30Q]-D776 mice was not progressive leading to Purkinje cell degeneration, i.e. atrophy of the molecular layer, as was observed in ATXN1[82Q] mice (Fig. 1A and B and [13]). Reasoning that perhaps the failure of Purkinje cell pathology to progress in ATXN1[30Q]D776 mice is due to activation of a neuroprotective pathway(s), it was hypothesized that activation of a protective pathway might elevate expression of critical gene(s) in ATXN1[30Q]D776 cerebellar RNA relative to WT and ATXN1[82Q] cerebellar RNA. An RNA-seq analysis revealed that CCKs expression was much higher in ATXN1[30Q]D776 compared to ATXN1[82Q] cerebella [12]. Moreover, absence of either CCK (Fig. 1B) or CCK1R in ATXN1[30Q]D776 mice [12] resulted in a Purkinje cell disease that is as progressive as in ATXN1[82Q] transgenic mice as assessed by atrophy of the molecular layer and loss of Purkinje cells.

Elevated CCK expression is protective against progressive Purkinje cell atrophy in ATXN1[30Q]D776 mice. A Four-day trial of accelerating Rotarod performance of ATXN1 transgenic mice at 20 weeks of age. Both ATXN1[30Q]-D776 (n = 3) mice and ATXN1[82]-S776 (n = 4) at 20 weeks of age were compared to age-matched WT/FVB (n = 3) (p = 0.04 and p = 0.03) ± SEM. B Cerebellar molecular layer thickness in ATXN1 transgenic mice at 20 weeks of age and 1 year of age. N’s are indicated inside the bars for each genotype. ***p < 0.001
Cholecystokinin (CCK), originally discovered in the gastrointestinal tract and typically associated with regulation of food intake and satiation, is one of the most abundant neuropeptides in the brain [14]. Within the cerebellum, CCK is predominately if not exclusively expressed by Purkinje cells [15, 16], a point further supported by ISH data from the Allen Brain Atlas [https://portal.brain-map.org/]. Cck effects are mediated by two G-protein coupled receptors [14], CCK1R and CCK2R (first designated as Cckar and Cckbr), and CCK1R is expressed in the adult mouse cerebellum in a Purkinje cell-enriched manner (Allen Brain Atlas). Based on the Purkinje cell protective effects of CCK in ATXN1[30Q]D776 mice, we speculated that activation of CCK1R would be protective to Purkinje cells expressing ATXN1 with an expanded polyglutamine tract.
CCK1R Activation in ATXN1[82Q] and ATXN2[127] Transgenic Mice: Restoration of Purkinje Cell Homeostasis and mTORC1 Signaling
A71623, a CCK1R tetrapeptide agonist, is highly selective for CCK1R [17]. In rodents, peripheral administration elicits CNS-mediated behavioral effects [18, 19]. To examine further the protective capability of CCK1R activation in SCA Purkinje cells, we assessed the ability of this CCK1R agonist to improve Purkinje cell function in transgenic mouse models of SCA1 and SCA2 [20]. In these transgenic mice, the expanded ATXNs were expressed specifically in Purkinje cells using the Pcp2 regulatory region [21, 22].
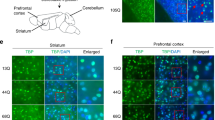
Administration of the CCK1R-selective agonist A71623 mitigates motor performance deficits in both Pcp2-ATXN1[82Q] and Pcp2-ATXN2[127Q] mice (Fig. 2). Consistent with A71623 improving Purkinje cell function, A71623 also increased expression of calbindin, a molecular marker of Purkinje cell health [23], in Pcp2-ATXN1[82Q] and Pcp2-ATXN2[127Q] mice. These results provide direct evidence that activation of CCK1Rs is protective to Purkinje cells in SCA1 and SCA2.

CCK1R agonist A71623 improves motor performance in ATXN1[82Q] and ATXN2[127Q] mice. Mice were tested for motor performance using the bar cross at 3 weeks of age. Then either 0.02 mg/kg/day A71623 or vehicle (20 mM PBS) was given and mice were retested on bar cross at 11 weeks of age. A Time to cross and the number of foot slips on the 10 mm round balance beam for ATXN1[28Q]. B Time to cross and the number of foot slips on the 10 mm round balance beam for ATXN2[127Q]. N’s are indicated inside each bar for each genotype/test. Error bars are SEMs. Two-way ANOVA with Tukey post hoc test, *p < 0.05, **p < 0.01, ***p < 0.001
Activation of CCK1R is known to impact a wide variety of signaling pathways [24]. Interestingly, among the pathways affected by CCK1R activation is mTORC1, and mTORC1 signaling contributes to Purkinje cell dysfunction in Atxn1154Q/2Q knock-in mice [25]. The status of mTORC1 was assessed in cerebella from Pcp2-ATXN1[82Q] and Pcp2-ATXN2[127Q] mice by measuring the levels of phosphorylated ribosomal protein S6 (pS6) and phosphorylated translational repressor 4e-bp1 (p4e-bp10), both targets of mTORC1 [26]. Though mTORC1 was altered in both Pcp2-ATXN1[82Q] and Pcp2-ATXN2[127Q] mice, intriguingly, it was decreased in Pcp2-ATXN1[82Q] mice and increased Pcp2-ATXN2[127Q] mice. Yet, the CCK1R agonist A71623 normalized mTORC1 cerebellar signaling in both Pcp2-ATXN1[82Q] and Pcp2-ATXN2[127Q] mice.
Discussion
Data reviewed here support activation of Purkinje cell CCK1Rs as being protective to these neurons in Pcp2-ATXN1[82Q] and Pcp2-ATXN2[127Q] transgenic mice. A critical aspect of the protective effects of CCK1R activation is a subsequent restoration of normal mTORC1 signaling whether mTORC1 signaling is decreased, as in Pcp2-ATXN1[82Q] Purkinje cells, or enhanced, as seen in Pcp2-ATXN2[127Q] Purkinje cells. Characteristically, mTORC1 signaling is decreased in response to cellular stressors [reviewed in 27]. That either depression or enhancement of mTORC1 signaling can underlie Purkinje cell impairment was previously shown in mice where mTORC1 signaling was decreased due to a loss of mTORC1 or in Purkinje cells in which mTORC1 signaling was enhanced due to the absence of the mTORC1 inhibitor TSC1, the function as well as survival of Purkinje cells were adversely affected [28]. It is intriguing to speculate that, as seen with mTORC1 signaling levels in cerebella of Pcp2-ATXN1[82Q] and Pcp2-ATXN2[127Q] transgenic mice, the SCAs might also be categorized according to mTORC1 signaling activity with SCA1 having reduced Purkinje cell mTORC1 signaling and SCA2 showing enhanced Purkinje cell mTORC1 signaling. Importantly, activation of the CCK1R in both instances restored mTORC1 signaling to a normal level. Understanding the mechanism and cellular pathways by which CCK1R activation in Pcp2-ATXN1[82Q] and Pcp2-ATXN2[127Q] transgenic Purkinje cells restores mTORC1 signaling to a normal level is an area of considerable importance regarding the potential of CCK1R activation as a therapeutic target in the SCAs.
We suggest that the results reported in our studies [12, 20], support a model where a CCK/CCK1R/mTORC1 pathway enables Purkinje cells to adapt to stress. As Golgi discovered many years ago, Purkinje cell axon recurrent collaterals form synaptic connections between Purkinje cell [29] that have a role in modulating synchronized Purkinje cell firing [30]. Perhaps these recurrent Purkinje cell synapses also provide the pathway by which the release of CCK peptide by Purkinje cells activates CCK1R/mTORC1 pathway in an autocrine fashion.
In the case of ATXN1[82Q] expressing Purkinje cells, cumulative stress induced by mutant ATXN1 promotes the cleavage of CCK to the octapeptide CCK-8, the natural ligand with the highest affinity for Cck1R [31] that upon secretion binds to and activates CCK1R on Purkinje cells. In addition, expanded ATXN1 reduces CCK expression, thus dampening the ability of Purkinje cells to respond to stress and promoting ATXN1 pathogenesis. Activation of Purkinje cell CCK1R stimulates mTORC1, which we speculate is a critical component by which CCK1R activation dampens the pathogenic effects of expanded ATXN1. Inactivation of mTORC1 induces a progressive loss of Purkinje cells by apoptosis [28]. mTORC1 signaling, in addition to responding to many stresses, is impaired in Atxn1154Q/2Q knock-in mice, and the absence of mTORC1 in Purkinje cells of Atxn1154Q/2Q mice worsens disease [25]. We are intrigued with the report that impaired striatal mTORC1 activity underlies degenerative phenotypes in a Huntington’s disease mouse model brain and activation of mTORC1 alleviates striatal atrophy [32]. Perhaps, manipulation of the CCK-CCK1R pathway might be a therapeutic target for treating neurodegenerative diseases involving other neurons as well as Purkinje cells.
References
Kasumu AW, Bezprozvanny I. Deranged calcium signaling in Purkinje cells and pathogenesis in spinocerebellar ataxia 2 (SCA2) and other ataxias. Cerebellum. 2012;11:630–9. https://doi.org/10.1007/s12311-010-0182-9.
Bettencourt C, Ryten M, Forabosco P, Schorge S, Hersheson J, Hardy J, et al. Insights from cerebellar transcriptomic analysis into the pathogenesis of ataxia. JAMA Neurol. 2014;71:831–9.
Hekman KE, Gomez CM. The autosomal dominant spinocerebellar ataxias: emerging mechanistic themes suggest pervasive Purkinje cell vulnerability. J Neurol Neurosurg Psychiatry. 2015;86:554–61. https://doi.org/10.1136/jnp-2014-308421.
Robinson KJ, Watchon M, Laird AS. Aberrant cerebellar circuitry in the spinocerebellar ataxias. Front Neurosci. 2020;14:707. https://doi.org/10.3389/fnins.2020.00707.
Cendelin J, Cvetanovic M, Gandelma M, Hirai H, Orr HT, Pulst SM, et al. Consensus paper: strengths and weaknesses of animal models of spiinocerebellar ataxias and their clinical implications. Cerebellum. 2021. https://doi.org/10.1007/s12311-021-01311-1.
Zhao L, Longo-Guess C, Harris BS, et al. Protein accumulation and neurodegeneration in the woozy mutant mouse is caused by disruption of SIL1, a cochaperone of BiP. Nat Genet. 2005;37:974–9.
Lee JW, Beebe K, Nangle LA, et al. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–5.
Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9.
Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4.
Howes J, Shimizu Y, Feige MJ, et al. C-terminal mutations destabilize SIL1/BAP and can cause marinesco-sjogren syndrome. J Biol Chem. 2012;287:8552–60.
Paulson HL, Shakkottai V, Clark HB, Orr HT. Polyglutamine spinocerebellar ataxias – from gene identification to potential treatments. Nature Revs Neurosci. 2017;18:613–26.
Ingram M, Wozniak EAL, Duvick L, Yang R, Bergmann P, Carson R, et al. Cerebellar transcriptome profiles of ATXN1 transgenic mice reveal SCA1 disease progression and protection pathways. Neuron. 2016;89:1194–207.
Duvick L, Barnes J, Ebner B, Agrawal S, Andresen M, Lim J, et al. SCA1-like disease in mice expressing wild type ataxin-1 with a serine to aspartic acid replacement at residue 776. Neuron. 2010;67:929–35.
Lee SY, Soltesz I. Cholecystokinin: a multi-functional molecular switch of neuronal circuits. Dev Neurobiol. 2011;71:83–91.
Ron Y, Wang T, Morgan JL. Identification of candidate Purkinje cell-specific markers by gene expression profiling in wild-type and pcd2J mice. Mol Brain Res. 2004;132:128–45.
Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–62.
Lin CW, Shosaki K, Miller TR, Witte DG, Bianchi BR, Wolfram, et al. Characterization for two novel cholecystokinin tetrapeptide (30–33) analogues, A-71623 and A-70874, that exhibit high ptency and selectivity for cholecystokinin-A receptors. Mol Pharmacol. 1991;99:346–51.
Asin KE, Bednarz L, Nikkel AL, Gore PA, Montana WE, Cullen MJ, et al. Behavioral effects of A71623, a highly selective CCK-A agonist tetrapeptide. Am J Physiol. 1992;263:R125–35.
Asin KE, Bednarz L, Nikkel AL, Gore PA, Nadzan AM. A-71623, a selective CCK-A receptor agonist, suppresses food intake in the mouse, dog, and monkey. Pharmacol Biochem Behav. 1992;42:699–704.
Wozniak EA, Chen Z, Paul S, Yang P, Figueroa KP, Firedrich J, et al. Cholecystokinin 1 receptor (Cck1R) activation restores normal mTORc1 signaling and is protective to Purkinje cells of SCA mice. Cell Rep. 2021;37:109831. https://doi.org/10.1016/j.celrep.2021.109831.
Burright EN, Clark HB, Servadio A, Matilla T, Feddersen RM, Yunis S, et al. SCA1 transgenic mice: a model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell. 1995;82:937–48.
Hansen ST, Meera P, Otis TS, Pulst SM. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet. 2013;22:271–83.
Schmidt H, Stiefel KM, Racay P, Schwaller B, Eilers. Mutational analysis of dendritic Ca2+ kinetics in rodent Purkinje cells: role of parvalbumin and calbindin D28k. J Physiol. 2003;551:13–32. https://doi.org/10.1113/jphysiol.2002.035824.
Cawston EE, Miller LJ. Therapeutic potential for novel drugs targeting the type1 cholecystokinin receptor. Br J Pharmacol. 2010;159:1009–21.
Ruegsegger C, Stucki DM, Steiner S, Angliker N, Radecke J, Keller E, et al. Impaired mTORC1-dependent expression of Homer 3 influences SCA1 pathophysiology. Neuron. 2016;89:129–46.
Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84.
Su K-H, Dai C. mTORC1 senses stresses: coupling stress to proteostasis. Bioassays. 2017;39(5):1600268.
Angliker N, Burri M, Zaichuk M, Fritschy J-M, Rüegg MA. smTORC1 and mTORC2 have largely distinct functions in Purkinje cells. Eur J Neurosci. 2015;42:2595–612.
Ramon y Cajal S. Histogie du Système nerveux de I’Homme et des Vertbrés. Paris: Maloine; 1911.
Orduz D, Llano I. Recurrent axon collaterals underlie facilitating synapses between cerebellar Purkinje cells. Proc Natl Acad Sci USA. 2007;104:17831–6.
Dufresne M, Seva C, Fourmy D. Cholecystokinin and gastrin receptors. Physiol Rev. 2006;86:805–47.
Lee JH, Tecedor L, Chen YH, Monteys AM, Sowada MJ, Thompson LM, Davidson BL. Reinstating aberrant mTORC1 activity in Huntington’s disease mice improves disease phenotypes. Neuron. 2015;85:303–15.
Acknowledgements
The work described was supported by NIH/NINDS grant RO1NS 045667.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The author declares patent US 10,973,812 B2, issued 4–13-2021 as relevant to the work in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Orr, H.T. Cholecystokinin Activation of Cholecystokinin 1 Receptors: a Purkinje Cell Neuroprotective Pathway. Cerebellum 22, 756–760 (2023). https://doi.org/10.1007/s12311-022-01428-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-022-01428-x