
Abstract
A hexanucleotide repeat expansion in the C9orf72 gene is the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) with synaptic dysfunction identified as an early pathological hallmark. Although TDP-43 pathology and overt neurodegeneration are largely absent from the cerebellum, the pathological hallmarks of RNA foci and dipeptide repeat protein (DPR) inclusions are most abundant. Here, we present a systematic literature search in the databases of PubMed, Scopus, Embase, Web of Science and Science Direct up until March 5, 2021, which yielded 19,515 publications. Following the exclusion criteria, 72 articles were included having referred to C9orf72, synapses and the cerebellum. Meta-analyses were conducted on studies which reported experimental and control groups with means and standard deviations extracted from figures using the online tool PlotDigitizer. This revealed dendritic defects (P = 0.03), reduced C9orf72 in human patients (P = 0.005) and DPR-related neuronal loss (P = 0.0006) but no neuromuscular junction abnormalities (P = 0.29) or cerebellar neuronal loss (P = 0.23). Our results suggest that dendritic arborisation defects, synaptic gene dysregulation and altered synaptic neurotransmission may drive cerebellar synaptic dysfunction in C9-ALS/FTD. In this review, we discuss how the chronological appearance of the different pathological hallmarks alters synaptic integrity which may have profound implications for disease progression. We conclude that a reduction in C9orf72 protein levels combined with the accumulation of RNA foci and DPRs act synergistically to drive C9 synaptopathy in the cerebellum of C9-ALS/FTD patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The most common genetic cause of both amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) has been proven to be a large hexanucleotide repeat expansion (G4C2)n within intron 1 of C9orf72 (C9) [1, 2]. As a result of the expansion, three pathogenic mechanisms have been proposed as the underlying cause of C9-ALS/FTD: (1) loss of function due to G4C2 repeat expansion leading to downregulation of C9orf72 protein expression; (2) toxic gain of function by recruitment of other RNA-binding proteins into G4C2 RNA foci; and (3) the non-ATG initiated RAN translation of RNA repeats, which results in the production of toxic dipeptide protein repeat (DPRs) [3,4,5].
The cerebellum is home to approximately 80% of all neurons in the human brain, which mediate reciprocal connections with multiple regions throughout the brain and spinal cord [6, 7]. Importantly, Renton et al. (2011) [2] detected the highest expression level of C9orf72 RNA within the cerebellum of neuropathologically normal individuals. This finding is relevant as the cerebellum executes a major role in regulating sensorimotor control and higher order cognitive functions such as gait, coordination and fine balance, as well as spatial memory, apathy and executive control — all of which can be impaired in patients diagnosed with C9-ALS/FTD [8, 9]. However, this dysfunction is ascribed to frontal lobe pathology, and the cerebellum has been largely overlooked as a region of interest in patients with ALS/FTD, despite key findings suggesting the relevance of this brain region. In this review, we want to focus on the roles played by decreased C9orf72 protein, RNA foci and DPRs in displaying different toxic properties in distinct animal and cellular models [10,11,12,13,14,15] and specifically in developing cerebellar synaptic dysfunction.
C9orf72 protein is predominantly localised to the pre-synaptic and post-synaptic compartments in the mouse brain [16, 17]. Xiao et al. (2019) have shown that C9orf72 is present in synapses of the granular layer of the cerebellum when comparing C9-wild type versus C9-knockout animals [17], resulting in the suggestion that it may be involved in synaptic transmission and autophagy [16,17,18,19]. Downregulation of C9orf72 impairs autophagy and may contribute to the accumulation of the transactive response DNA-binding protein 43 kDa (TDP-43) and p62 [18]. However, a distinct characteristic of C9-ALS/FTD is identifiable in the spatial segregation of TDP-43 and p62 proteinaceous inclusions, which are most abundant in the cerebellum [20].
The role of C9 RAN-translated DPRs in synaptic dysfunction was illustrated by Xu and Xu (2018) who induced the expression of different DPRs in Drosophila models [21]. They observed that poly-GR and poly-PR overexpressing flies presented altered synaptic boutons at neuromuscular junctions (NMJs). In contrast, Jensen et al. (2020) [4] observed that poly-GA aggregates are located in neurites and are less mobile at longer length repeats (400 compared to 50 repeats). Moreover, the authors found that poly-GA causes reductions in synaptic vesicle-associated protein 2 (SV2), alters Ca2+ influx and inhibits synaptic vesicle release resulting in earlier iPSC death [4]. In addition, the presence of DPRs has been linked to marked reductions in dendritic spine densities and overall dendritic arborisation in both in vitro and in vivo models [22,23,24].
Indeed, May et al. (2014) [22] have shown that overexpression of poly-GA in primary neuronal cultures caused severe reductions in dendritic arborisation due to the co-aggregation and sequestration of Unc119, a protein also known to suppress axonal arborisation. In another study by Park et al. (2020) [24], the most significant reduction in dendritic branches was associated with the presence of arginine-rich DPRs (PR and GR) in C9orf72 Drosophila neurons. Moreover, Schweizer-Burguete et al. (2015) [23] showed that the overexpression of 48 × GGGGCC repeat RNA (G4C2-48) caused dendritic branching defects and decreased synaptic densities in rodent spinal cord neurons.
Interestingly, there is evidence supporting DPR aggregation in cerebellar tissues of C9-ALS/FTD patients [25,26,27,28,29]. Several studies have documented that poly-GA and poly-GP DPR aggregates predominate in the cerebellum of C9-ALS/FTD patients and may contribute to disease progression [11, 27, 29,30,31,32]. Indeed, Zhang et al. (2014) [28] have shown that in primary mouse neuronal cultures, the overexpression of poly-GA leads to the upregulation of cytoplasmic p62-immunopositive inclusions within the granule cell layer of the cerebellum in the absence of neurodegeneration. Moreover, in vivo green fluorescent protein (GFP) tagged mouse models overexpressing poly-GA (GFP-GA50), demonstrating more severe neuronal cell loss in the Purkinje layer of the cerebellum which were associated to the aggregation and sequestration of HR23 proteins, responsible for normal proteasome degradation and nucleocytoplasmic transport functions [33]. Conversely, there are conflicting studies where, despite detecting significant poly-GA, GP and GR inclusions in the cerebellar tissues of C9orf72 patients, no sign of neurodegeneration in the cerebellum, cognitive decline or clinical phenotypes have been found [30,31,32, 34, 35].
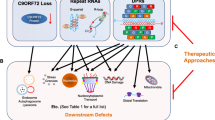
Furthermore, RNA foci are also frequently identified in the molecular and granular cell layers of the cerebellum, where intranuclear foci were significantly larger (~ 500 nm) in comparison to the neocortex (~ 200 nm) in both in vitro and zebrafish models. This has been suggested to be linked to caspase-3-initiated mechanisms of apoptotic neurodegeneration [36]. Interestingly, a more recent clinico-pathological study examining cerebellar and frontal cortical post-mortem tissue from C9orf72 expansion mutation carriers identified the largest RNA foci burden levels in the Purkinje cells of the cerebellum (~ 70%) compared to all other regional tissue types, without any cerebellar neuronal loss [37]. All these results could suggest a synergistic combination of RNA foci and DPR accumulation which could be underlying cerebellar synaptic dysfunction usually overlooked in C9-ALS/FTD patients (overviewed in Fig. 1).

Putative mechanisms underlying synaptic dysfunction in C9orf72-ALS/FTD. A schematic detailing the role of the hexanucleotide expansion, (G4C2)n, of the C9orf72 gene in driving synaptic, axonal and dendritic dysfunction. This operates through the three main pathogenic mechanisms implicated in C9-ALS/FTD which are haploinsufficiency of the C9orf72 protein and the accumulation of RNA foci and dipeptide repeats (DPRs). Abbreviations: p53, tumour protein p53; RAN, repeat-associated non-AUG; Ca2+, calcium ions; mRNP, messenger ribonucleoprotein; RNA, ribonucleic acid
In the present systematic review, we will examine the most recent literature for cerebellar synaptic dysfunction in C9orf72 gene mutation carriers of ALS/FTD. We will discuss alterations in neuronal morphology, including structural and functional changes to synapses, deterioration in dendritic morphology and axonal degeneration. Finally, we will address the role of DPRs and RNA foci and whether these pathological features precede cerebellar neuronal dysfunction during the course of gradual neurodegeneration in the cerebellum of C9-ALS/FTD patients.
Methods
Literature Search
A systematic literature search was conducted in accordance to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines [38]. Original research articles and reviews pertaining to cerebellar synaptic dysfunction in C9-ALS/FTD have been independently searched for in five electronic databases — PubMed, Web of Science (WoS), Scopus, Science Direct and EMBASE. The search was performed by three researchers (NA, JA, AK) on March 5, 2021, using the following search terms and combinations: [“cerebellum” AND “synaptic” AND “C9ORF72”], [“cerebellum” AND “synapsis” AND “C9ORF72”], [“cerebellum” AND “pruning” AND “C9ORF72”], [“cerebellum” AND “dendrites” AND “C9ORF72”], [“cerebellum” AND “neuronal loss” AND “C9ORF72”], [“cerebellum” AND “axonal” AND “C9ORF72”], [“cerebellum” AND “neuron” AND “C9ORF72”], [“cerebellum” AND “C9ORF72” AND “ALS” “cerebellum” AND “C9ORF72” AND “ALS” OR “Amyotrophic lateral sclerosis”], [“cerebellum” AND “C9ORF72” AND “FTD” OR “frontotemporal dementia”], [“synaptic” AND “C9ORF72”], [“dendrites” AND “C9ORF72”], [“axon” AND “C9ORF72”], [“neuronal loss” AND “C9ORF72”]; [“neuronal degeneration” AND “C9ORF72”], [“neuron” AND “C9ORF72” AND “ALS” OR “amyotrophic lateral sclerosis”] and [“neuron” AND “C9ORF72” AND “FTD” OR “Frontotemporal dementia”]. No chronological, language or methodological filters have been imposed on the search engines, and all resulting data sets were exported and compiled in an Excel document. The search strategy was further broadened to include screening of references cited in relevant review articles.
Study Selection
Following the removal of duplicates, all remaining articles had their titles and abstracts screened for eligibility. Epidemiological studies and articles which did not specifically pertain to C9orf72 mutation in ALS-FTD were deemed ineligible. After the initial screening phase, full texts of selected studies were retrieved and reviewed in detail against the inclusion criteria. In order for a study to be included in the systematic review, it had to (i) show clear evidence of either synaptic dysfunction or findings relating to C9orf72 protein/ DPR aggregates/RNA foci, (ii) employ genetic models of C9orf72 mutation and/ or C9-ALS/FTD patient samples and (iii) examine cerebellum tissue or present findings which can be extrapolated to cerebellar synaptic pathology.
Meta-Analysis
A continuous random effects model with a standard mean difference was employed to conduct the meta-analysis. Publications that reported (i) dendritic arborisation defects, (ii) NMJ abnormalities, (iii) alterations in neurite length, (iv) reductions in C9orf72 protein, (v) cerebellar neuronal loss and (vi) DPR-related neuronal loss underwent methodological quality assessment performed by two independent researchers to minimise the risk of bias. Studies were excluded from meta-analysis for not reporting the mean, standard deviation (SD) or sample size such as Zhang et al. (2014) [28] and lack of quantitative analysis such as Lee et al. (2017) [39]. Additionally, G4C2related neuronal loss was not statistically assessed due to several factors (RNA foci, DPRs, reduced C9orf72) having a potential role in neuronal loss. Significance played no role in the selection process, with studies reporting null findings included by the experimenters. Authors of the relevant publications were not contacted directly regarding the raw data sets. Instead, numerical data was extracted directly from the figures using the online data extractor tool PlotDigitizer. Information regarding the figures used to calculate the different outcomes of meta-analysis is summarised in Table 1. Means, standard deviations and sample sizes were entered into Review Manager [40] which automatically calculated standard mean difference (SMD), confidence intervals (CIs), heterogeneity and overall effect size using a random effects model. Studies were weighted in the final analysis based on the precision of their data as determined by confidence intervals, with greater weights usually indicative of larger sample sizes.
Results
The searches conducted in PubMed, Scopus, Web of Science (WoS), EMBASE and Science Direct electronic databases yielded 1489, 2561, 1664, 3144 and 10,293 articles, respectively, reaching a total of 19,515 publications, of which 16,754 were identified as duplicates and removed from the data set. The titles and abstracts of the remaining 2397 articles were screened for eligibility, with 2292 publications deemed to fall outside the scope of the systematic review and excluded. Full texts of the final 105 articles were retrieved, read in full and carefully assessed against the inclusion criteria, with 70 studies deemed eligible for inclusion in the systematic review. Additionally, two relevant studies have been identified through cross-reference screening of relevant literature, giving rise to a total of 72 studies included in our analysis (see Fig. 2).

Inclusion of articles by Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram
General Characteristics of Selected Studies
The studies selected for inclusion in the systematic review were published between 2011 and 2021 (n = 72). Of the papers included, approximately half used C9orf72 mutation-positive subjects (n = 49) whilst the remaining papers attempted to recapitulate C9-ALS/FTD pathology in either in vitro models (n = 28) or in vivo models (n = 30). The characteristics of all selected studies, including the methodology and main findings reported, are summarised in Tables 2, 3 and 4 and in Fig. 3.

Frequency graphs illustrating the characteristics of selected studies. (A) Shows the number of studies that report synaptic dysfunction. (B) Illustrates the number of studies which report ALS/FTD pathology in C9-patients. (C) Shows the number of different animal models used in in vivo experiments. (D) Shows the number of different in vitro models used in the selected studies. (E) illustrates the pathomechanism type assessed in different animal models. (F) Illustrates the number of DPR constructs introduced to different animal models. Abbreviations: DPRs, dipeptide repeat proteins; NMJ, neuromuscular junction; cDNA, circular DNA
In total, 23 studies which specifically referenced the synapse were found, although two of these reported no changes (Table 2). The most frequent finding was that of dendritic arborisation defects (n = 8). Moreover, studies reported dysregulation in synaptic genes (n = 2), synaptic neurotransmission (n = 2) and NMJ morphological changes such as blebbing and loss of synaptic boutons (n = 5). Three studies focused on the interaction of C9orf72 with synaptic proteins, such as the Rab family of GTPases, and two other studies reported axonal degeneration (n = 1) and axonal projection impairments (n = 1). Of the remaining 50 papers, 32 discussed changes in the cerebellum (Table 3) and the other 18 were relevant publications to C9orf72 pathology (Table 4).
All human patient data was derived from ALS/FTD cases with a confirmed C9 mutation. The majority of human studies employed histological and/ or molecular analysis of post-mortem tissue (n = 45). Publications analysing post-mortem tissue of C9-ALS/FTD cases most commonly reported (i) reduced levels of C9orf72 proteins (n = 5), (ii) DPR aggregates/ toxicity (n = 24), (iii) abundant RNA foci (n = 10) and (iv) brain region-specific neuronal loss (n = 5). There were also case study reports (n = 2) demonstrating a link between C9 mutation and cerebellar pathology (cerebellar ataxia and pure cerebellar syndrome) and a large-scale screening clinical study (n = 1).
The methodologies used to recapitulate C9-ALS/FTD pathology in vivo and in vitro can be broadly divided into two categories: (i) insertion of G4C2 repeat expansions of varying length and (ii) expression of DPRs in the absence of G4C2 repeats. In all selected studies, in vitro data was complemented by post-mortem human data and/or in vivo data. Investigating the contribution of RNA repeats, studies utilised sense and antisense (G4C2)n expression vectors (n = 6) which were used to transfect cell culture lines and study the formation of RNA foci and DPRs. In vitro transfection of DPRs was even more frequently used (n = 13). Alternatively, ten studies used C9orf72-ALS/FTD patient iPSC-derived neurons. In contrast, a model composed of 100 synthetic cDNA encoded repeats of the five main DPRs — GA, PA, PR, GP and GR — was used to study the effect of those DPRs on transfected cells and primary neuronal cultures.
Transgenic mouse lines were the most commonly used in vivo (n = 17) with models generated to contain G4C2 repeats (n = 3), express DPR proteins (n = 9) or knockout the C9orf72 gene (n = 5). G4C2 repeats and DPR models were created by means of AAV viral injections. Viral transduction was achieved in the CNS through different promotors such as the cyan fluorescent protein (CFP)-GA149 line which expressed DPRs under the control of the Thy1 promotor, ensuring neuron-specific expression. Alternatively, DPR-Nestin lines were generated to drive ubiquitous CNS expression. Of the transgenic mouse lines used to study DPRs, poly-GA (n = 4) and poly-PR (n = 4) were the most common models followed by poly-GR (n = 1). Drosophila models were also frequent throughout the studies (n = 11). Transgenic fly lines were generated using traditional crossing methods to investigate DPRs (n = 7) and G4C2 repeat-mediated (n = 6) pathology. Drosophila lines expressing poly-GR (n = 6) were the most common, followed by poly-PR (n = 5), poly-PA (n = 3) and poly-GA (n = 2). Zebrafish models (n = 3) were generated to study the effect of reduced C9orf72 protein expression by injection of antisense oligonucleotides (n = 1), transient expression of DPRs (n = 1) and (G4C2)n repeats (n = 1). Additionally, a C. elegans model expressing 50 repeats of -GA, -PA, -GR or -PR DPRs (n = 1) and two chick embryo models expressing DPRs and G4C2 repeats were used.
Meta-Analyses
Of the included papers, meta-analysis was conducted on dendritic arborisation defects (n = 8) with this being refined to dendritic abnormalities (n = 5) and neurite length (n = 3) as two separate analyses and NMJ abnormalities (n = 5). Furthermore, reductions in C9orf72 protein (n = 4), cerebellar- and DPR-related neuronal loss were also conducted (n = 4 and n = 5, respectively). When analysing articles, we also found that many studies reported results that would fit into the previously mentioned categorisations; however, frequently, the data reported was visual (i.e. immunofluorescent imaging) without any quantitative data supporting gain/loss or no effect of each analysis and was therefore excluded.
We found that in all meta-analyses (Figs. 4 and 5), the studies were highly heterogenous (I2 > 75%; P ≤ 0.001), most likely a result of different species, repeat lengths, DPR models and other variables changing in each study. Nevertheless, significant dendritic abnormalities were seen in C9orf72 models of disease (P = 0.03) as well as reductions in C9orf72 protein in human patients (P = 0.005) and DPR-related neuronal loss (P = 0.0006). Whereas NMJ abnormalities and cerebellar neuronal loss failed to reach significance (P = 0.29 and P = 0.23, respectively). Moreover, neurite length data was extracted with the intent to analyse; however, the required study size (n = 3) was not reached. Therefore, we have included the already extracted data values in Table 5 to be used in future meta-analyses.

Meta-analysis using a random effects model of selected studies relating to synaptic deficits. (A) Shows the meta-analysis for dendritic defects assessing reductions in arborisations such as crossings and branchpoints (P = 0.03). (B) Shows the meta-analysis for neuromuscular junction (NMJs) abnormalities assessing synaptic bouton counts and fractured NMJs (P = 0.29). (C) Shows the meta-analysis for reductions of C9orf72 protein in human patients using frontal cortex and cerebellar samples (P = 0.005). All studies were highly heterogenous (I2 ≥ 84%; P ≤ 0.0004). The figure was generated using the RevMan 5.4 software. Abbreviations: SD, standard deviation; CI, confidence interval

Meta-analysis using a random effects model of selected studies relating to neuronal loss in C9orf72. (A) Shows the meta-analysis of neuronal loss in the cerebellum as a result of G4C2 repeats and DPR models (P = 0.23). (B) Shows the meta-analysis of DPR-related neuronal loss using animal models transfected with DPR constructs (P = 0.0006). Studies were highly heterogenous (I2 \(\ge\) 78%; P ≤ 0.001). The figure was generated using RevMan 5.4. Abbreviations: SD, standard deviation; CI, confidence interval
Discussion
Synaptic dysfunction is a common feature in neurodegenerative disease which represents an early disease event taking place before the development of neuronal degeneration and loss [5, 21, 41,42,43,44,45]. Freibaum et al. (2015) [42] assessed the impact of C9orf72 repeat pathology in Drosophila larvae, showing not only a dramatic loss in synapse structure, with severe reductions found at presynaptic active zones [42, 43], but also a significant reduction in synaptic bouton counts [42, 44] and synaptic quantal content [43, 44].
Interestingly, a meta-analysis conducted to assess the state of NMJs (Fig. 4B) found no significant alterations in bouton counts and NMJ integrity despite the reports from Freibaum et al. (2015) [32, 42]. This may be explained by Xu and Xu [21] finding increased synaptic bouton counts whilst the remaining studies found the opposite. Given that an increase in synaptic bouton count could be a result of excitotoxic mechanisms which Xu and Xu themselves report, this may then skew the meta-analysis, as this statistical test is not sensitive to phenomena in which both an increase and decrease in synaptic boutons may represent pathological changes. Indeed, the presence of excitotoxic mechanisms has been reported by other studies [4, 39] in the early stages of the disease. Devlin et al. (2014) [41] demonstrated that C9orf72 patient iPSC-derived motor neurons (MNs) revealed hyperexcitability at early stages in culture followed by a progressive loss of action potential output, a finding previously reported in other animal models of ALS [46, 47]. This initial phase of increased activity has been suggested to trigger a cascade of excitotoxic disease mechanisms involving pathological changes in Ca2+ handling [48, 49], accumulation of intracellular Ca2+ and the eventual activation of cell death pathways. Other early events observed by the authors were the loss of synaptic activity in C9orf72-iPSC-derived MNs which could be reflecting a general loss of action potential generation in culture; however, due to the evidence of loss and dysfunction of synapses in ALS [50,51,52,53,54,55], specific deficits in synaptic transmission might also contribute to reductions in synaptic activity recorded from patient iPSC-derived MNs. Indeed, Jensen et al. (2020) [4] found that the presence of poly-GA results in hyperexcitability, through Ca2+ depolarisation, of neurons followed by a reduction in SV2 — mirroring Devlin et al.’s [41] results of the initial appearance of excitotoxic mechanisms followed by a loss of synaptic activity, or in this case, an inability to unload the synaptic vesicles properly. Moreover, the animal model used by Jensen et al. [4] exclusively expressed poly-GA in the spinal cord, brainstem and cerebellum before any motor neuron loss was observed. This resulted in time-dependent and poly-GA-dependent gait and behavioural deficits [4], possibly suggesting that SV2 and neuronal excitability are more widespread than just MNs as Devlin et al. [41] reported and may include any neuron which harbours poly-GA expression. Devlin et al. also demonstrated changes in ionic conductance which could indicate that early dysfunction or loss of ion channels may contribute to the initiation of downstream degenerative pathways that ultimately lead to MN loss in ALS [41]. This ‘functional loss’ of neurons may render the motor system, including the cerebellum, inactive before neurodegeneration occurs. Both papers highlight the importance of understanding synaptic pathology as this may precede and promote neuronal death. Therefore, they present viable therapeutic options to prevent neurodegeneration from occurring rather than attempting to rescue already dying tissues.
Moreover, the dysregulation in synaptic transmission in the motor system was also corroborated by Hao et al. (2019) [56]. In their study, they expressed 28 × poly-PR (GFP-PR28) under the control of the neuronal Thy1 promoter, and they found that heterozygous mice developed deficiency of motor performance at 6 months of age. These motor deficits were accompanied by cerebellar poly-PR inclusions together with increased activation of astrocytes and microglia in the cerebellum and spinal cord, whereas only a few astrocytes in the motor cortex and no gliosis in the hippocampus were observed in these animals. Moreover, the GFP-PR28 heterozygous mice showed atrophy of the cerebral cortex and loss of Purkinje cells in the cerebellum and motor neurons in the spinal cord. These results are in line with the greater cerebellar atrophy showed by Gendron et al. (2013) [27] in C9orf72-FTD patients, highlighting the direct relationship between the C9orf72 mutation and cerebellar and motor defects. Moreover, other subtypes of ALS such as SOD1 and MAPT have shown cerebellar pathology [57], revealing the relevance of this brain region in ALS pathology. Finally, we wanted to note that although cerebellar ataxia is not typically observed in other types of FTD [58,59,60], Fogel et al. (2012) [56] have found a case where cerebellar ataxia was identified in C9orf72-ALS/FTD patients.
Understanding the Role of DPRs in C9-Cerebellar Pathology
Moreover, although the evidence has pointed out that the expression of DPRs is one of the causative factors for C9orf72-ALS/FTD based on the identification of DPRs in patient brains [12, 26, 29, 33, 61,62,63,64,65,66,67,68] and expression of DPRs without hexanucleotide-repeat RNA in animal models [33, 64], it is unknown whether DPR localisation correlates to neuronal loss. In order to elucidate this, Zhang et al. (2019) [69] generated a mouse model through viral infection of AAV1 GFP-PR50. This resulted in motor dysfunction and cognitive deficits, with reduced brain weight and an age-dependent loss of poly-PR expressing Purkinje cells and cortical neurons at 3 months of age, suggesting that poly-PR expression caused cell-autonomous neuron death. These pathological hallmarks were also accompanied by increased astrogliosis and microgliosis in the cortex and cerebellum of GFP-PR50 animals. Moreover, a poly-GR100 AAV-infected animal model also showed cortical thinning, hippocampal cell loss and cerebellar Purkinje cell loss from 1.5 to 6 months of age, whereas no spinal cord neuron loss was seen. These changes were parallel to increased astrogliosis and microgliosis from 1.5 months in GFP-GR100 mice and locomotor impairment [65]. Both studies not only highlight the impact of arginine enriched DPRs on the cerebellum and motor function, but also the correlation between DPR length and onset of pathogenesis, with shorter lengths of poly-PR having a later onset of age-related deficits [56, 69].
Indeed, our meta-analysis provides evidence linking DPRs with neuronal loss in C9-ALS/FTD (Fig. 5B). However, the effect DPRs have in the cerebellum specifically is difficult to assess due to multiple brain regions being pooled for this meta-analysis in order to reach an appropriate study number. In fact, when we assessed neuronal loss in the cerebellum, regardless of DPR involvement, we found no significant reduction in the Purkinje cells (Fig. 5A). This may be due to the fact that different areas in the cerebellum may be more prone to degeneration such as the spinocerebellum compared to the lateral cerebellar hemispheres [70]. Therefore, it is important for studies to report which specific subregion was used to carry out quantification to allow for more accurate pooling of data which may lead to more meaningful conclusions from a meta-analysis in the future.
Another important finding in these studies is that different DPRs interfere with different cell processes. The gene ontology (GO) analyses after RNA sequencing in the GFP-PR28 heterozygous mice model used by Hao et al. (2019) [56] revealed dysregulation of synaptic transmission-related genes, specifically downregulated genes included calcium ion-regulated exocytosis of neurotransmitters, intracellular signal transduction and neurotransmitter secretion. These results point out that transmission across chemical synapses is a major pathway implicated in PR28 cerebellar pathology. They have also found that two genes (Rims3, Doc2b) related to synaptic function were downregulated in the cerebellum of 2-month-old heterozygous mice. Moreover, upregulation of ER-stress genes (Chac1 and Atf5) has been found in the cerebellum of 2-month- and 5- month-old heterozygous mice, suggesting that ER stress and synaptic dysfunction in the cerebellum are early events in poly-PR expressing neurons.
However, the GO analyses in the GFP-GR100 mice reported by Zhang et al. (2018) [65] did not show synaptic-related differences but ribosomal and protein translational changes. These results are opposite to the studies where the accumulation of arginine-rich DPRs (GR and PR) in the nuclei of many cell types [11, 63, 71, 72] have suggested that they could be the cause of the dendritic defects observed, which is thought to precede neuronal cell death in ALS-FTD [24]. Park et al. (2020), using a C9orf72 Drosophila model, proved that arginine-rich DPRs led to the most significant reduction in dendritic branches and reduced the number of Golgi outposts in dendrites, which are organelles with a role in the cytoarchitecture of neuronal dendrites, leading to defective vesicular trafficking in dendrites. Moreover, Swaminathan et al. (2018) show that [73] poly-GR100 was capable of reducing motor neuron length in a zebrafish model [74]. Despite the methodological heterogeneity, our meta-analysis found significant alterations in dendritic branching, further giving evidence that synaptic dysfunction is a feature of C9-ALS/FTD (Fig. 4A). These results encourage further research into the cerebellum where early pathological features are detected. Most importantly, these studies compile evidence that the accumulation of different DPRs in different brain regions during the progression of the disease could explain the dynamics contributing to the neurodegeneration seen in C9orf72-ALS/FTD patients.
The Relevance of DPR Dynamics in C9-Cerebellar Pathology
A clear example of the need to study DPR dynamics during the cerebellar progression of the disease is represented by Yamakawa et al. (2015) in the brains of patients with C9orf72-ALS/FTD [75]. The authors found that the five DPRs (poly-GA, poly-GP, poly-GR, poly-PR and poly-PA) are deposited in the granular neurons of the cerebellum as reported by others [25, 76]. Further to this, Mackenzie et al. (2015) found that poly-GA inclusions (sense transcript) were by far the most abundant, followed by poly-GP (sense and antisense transcripts) and poly-GR inclusions (sense transcript) with only rare poly-PR and poly-PA inclusions (antisense transcripts) throughout the premotor frontal cortex, lower spinal cord MNs and cerebellum [26]. These data, as well as early-onset DPR inclusions in animal models and their inherent toxicity, illustrate the importance of studying the accumulation of brain DPRs in a time- and cell-dependent fashion and may explain why some poly-DPR pathologies are rare in post-mortem brain tissues from C9orf72-ALS/FTD patients, which reflect the end stage of disease and may likely have neuronal death obscuring the mechanisms through which DPRs are causing toxicity.
The toxicity of DPRs may also be related to and depend on each other. Zhang et al. (2018) [65] confirmed a previous report by Yang et al. (2015) [77] that poly-GR only aggregates in cells that have insoluble poly-GA aggregates and remains diffuse without the presence of poly-GA. Interestingly, Yang et al. found that poly-GA recruiting poly-GR to aggregates was found to prevent certain measures of toxicity and restore defective Notch signalling, suggesting that poly-GA, which readily aggregates, may provide some defence mechanism against toxicity [77]. However, it was revealed that poly-GR was still able to interact with ribosomal/mitochondrial targets when aggregated [65]. The ability to do so may impact the dynamics within the cell such that aggregated poly-GR interacting and binding to targets may impair nearby reactions due to steric hindrance.
Moreover, poly-GA’s ability to alter DPR chemistry is not limited to poly-GR. Darling et al. (2019) co-expressed poly-GA with poly-PR and interestingly found that high levels of poly-GA in relation to poly-PR — ratios of 10:1 and 5:1 — localised poly-PR to the cytoplasm, with the nucleus being the typical aggregate locale of poly-PR [78]. Additionally, it was found that co-expression of poly-GA50 reduced aberrant phosphorylation in the unfolded protein response — a characteristic of poly-PR aggregates — preventing potential triggering of an apoptotic event through the PERK pathway. However, there is some controversy regarding the roles of DPR synergy in causing toxicity as Lee et al. (2017) found that poly-GA did not co-localise frequently with poly-GR and -PR [39]. Although this could be explained by poly-GR and -PR being much rarer in the brain in general, they demonstrated that poly-GA could sequester poly-GP and -PA, but, in their model, sequestration of poly-PA with poly-GA actually prevented GA-mediated toxicity and not GA-mediated reduction of toxicity of the other DPRs as seen by Zhang and Yang (2018; 2015) [65, 77]. Nevertheless, the ability for DPRs to interact with one another and alter their toxicity suggests that structures which express all variants of DPRs such as the cerebellum will likely have different alterations to metabolic pathways which could explain differences from cortical and motor neuron degeneration along the life span of C9orf72 patients [39, 65, 75, 77].
Pathways Mediating DPRs Toxicity in C9-Cerebellar Pathology
Another important hallmark of the cerebellum in C9orf72 mutation carriers is the presence of TDP-43-negative and p62-positive neuronal cytoplasmic inclusions [22, 25, 79, 80] which have been shown to be associated with poly-GA, GP, GR, PA and PR [11, 22, 30, 31, 61, 81]. In this regard, Mann (2013) observed a correlation between p62 inclusions and poly-GA and poly-GP in the cerebellum [31]. Moreover, in the study, a correlation between poly-GA and -GP, together with poly-GR and -PR, was also demonstrated. Although the role of poly-PR was recently established by Maor-Nof et al. (2021) [82] and Far and Shorter (2021) [83] as a remodeller of the neuronal epigenome to promote proapoptotic p53 activity involving PUMA, the inconsistency of its presence in all expansion bearers of the Mann study (2013) [31] casts doubt into its relevance to cerebellar pathology. Despite this, Maor-Nof’s study [82] proposed for the first time a relationship between poly-PR and axonal degeneration that could be rescued by p53 reduction, increasing survival of C9orf72-ALS/FTD patient-induced pluripotent stem cell (iPSC)-derived motor neurons. Moreover, we cannot rule out that poly-PR and its inconsistent presence may be a result of the toxicity it exerts on the neuron, such that all neurons which are PR positive die earlier than neurons which lack PR inclusions or have a low poly-PR burden.
Furthermore, Lopez-Gonzalez et al. also found that poly-GR increases p53 levels in neurons of C9orf72 patients, and axonal degeneration may be a unique pathology associated with arginine-DPRs [84]. Mori et al. (2013b) demonstrated a rostro-caudal gradient of poly-GR inclusions being abundant in all neocortical areas, hippocampus and cerebellum, with moderate abundance in subcortical nuclei and low abundance in the brain stem and spinal cord which may highlight areas particularly susceptible to poly-GR-mediated degeneration [85]. However, in these studies, the contribution of these DPR inclusions to cerebellar synaptic function and the pathways potentially involved has been scarcely studied. Despite this, these results shed light on the relationship between DPRs and downstream degenerative cascades that could be targeted at early stages in specific brain regions. Moreover, p53 cerebellar reduction in relation to poly-PR and poly-GR accumulation in C9orf72 ALS/FTD-patient remains unclear, but further research may highlight potential therapies which could be applicable to other diseases such as spinal muscular atrophy and Purkinje cell degeneration [86, 87].
In a poly-GA mouse model, LaClair et al. (2020) demonstrated that poly-GA has a greater propensity to aggregate in the cerebellum which may drive toxicity seen in these mice [81]. These results are also supported by the increased number of inclusions found in the molecular and granular layers of the cerebellum of C9orf72 mutation cases by Mori et al. (2013b), whereas inclusions were rarely found in the Purkinje layer [85]. Moreover, LaClair et al. (2020) [81] have shown that poly-GA promotes interferon responses in C9orf72 disease and contributes to TDP-43 abnormalities and neuron loss selectively in disease-relevant regions.
An important study from May et al. (2014) [22] showed that poly-GA co-localised with Unc119, a transport factor previously linked to neuromuscular and axonal function, in the cerebellum. In the study, it was demonstrated that similar to poly-GA expression, Unc119 knockdown inhibits dendritic branching and causes neurotoxicity, suggesting that poly-GA expression may be the driving force for Unc119 aggregation. Interestingly, those who present with C9orf72-FTD have higher aggregated levels of poly-GA and Unc119 in the cerebellum than those of C9orf72-ALS. However, Schludi et al. (2017) [88] have shown that despite eliciting behavioural deficits and inflammation, no neuronal loss was seen in mouse models expressing poly-GA, suggesting that poly-GA may be instrumental in protein sequestration [89] and inflammation of ALS but not end-stage neuronal death. Jensen et al. (2020) [4] further support this intermediary role of poly-GA in reporting aberrant synaptic unloading and motor deficits without overt neuronal death, as we have previously discussed.
The Role of DPR Solubility in C9-Cerebellar Pathology
Despite the overwhelming evidence of DPR aggregates in C9-ALS/FTD, it is unclear whether binding DPRs to insoluble aggregates causes toxicity or whether the soluble form of DPRs is toxic and aggregation is a defence mechanism [30, 90]. In this regard, Quaegebeur et al. (2020) demonstrated differing solubility profiles of DPRs across human brain tissue [91]. Notably, soluble DPRs tend to be less abundant in areas associated with clinical pathologies, such as the frontal cortex in FTD. Interestingly, the cerebellum has significantly different dynamics from the other cortices measured. Despite the aggregate load of poly-GA and -GR being comparable to the frontal and temporal cortices, insoluble poly-GP was highest in the cerebellum. Moreover, the soluble forms of poly-GA and -GP are at much greater concentrations in the cerebellum than other regions, with poly-GR showing significant variability between all cortices. With Quaegebeur et al. (2020) reporting that reductions in soluble fractions were associated with disease severity [91], one would likely presume that this is because they are aggregating. Therefore, it is surprising that the cerebellum, which has higher levels of soluble and insoluble DPRs, shows less neurodegeneration when compared to the frontal cortex. This may indicate a mechanism by which the toxicity of DPR aggregation is in some way mitigated in the cerebellum and is seen as ‘spared’ in C9orf72-ALS/FTD. These regional differences in DPR solubility could be pointing out the selective vulnerability of different brain regions to DPRs, underlined by different degradation pathways along the brain. Furthermore, this may mean the cerebellum may present as the most useful structure to investigate the effect of synaptopathy and of the macro-mechanisms of reduced C9orf72 protein and RNA foci due to the mitigation of DPR-dependent toxicity as well as investigating solubility mechanisms and its regulation.
Reduced C9orf72 Protein Show Toxicity Through Downstream Effects
As a consequence of C9orf72 repeat expansions, C9orf72 protein levels are reduced, and the resulting loss of function is one of the key patho-mechanisms of the disease. As we have previously mentioned, the cerebellum is a brain region known to express high levels of C9orf72 mRNA [2], and differences in cerebellar transcript levels between C9orf72 mutations carriers and controls [92, 93] have also been found. Frick et al. (2018) confirmed previous reports of reduced C9orf72 protein levels in the cerebellum of C9orf72 mutation carriers with no association to clinical phenotypes (ALS, ALS/FTD or FTD), age at onset and disease duration [16]. The authors explained this finding by a strong positive correlation between the presence of neurodegeneration/cell death and C9orf72 levels in cerebellar regions. Similar results in post-mortem tissues were reported by Tan et al. (2016) [70] where no neuronal loss was identified in the cerebellum. However, opposite results were found by Chew et al. (2015) in which mouse models of (G4C2)66 showed loss of Purkinje cells [94]. Similar results were found by Liu et al. (2016) in a BAC mouse model of C9orf72 [95], so Frick’s [16] results might be potentially explained by protein degradation due to post-mortem delay in human samples. Moreover, Saberi et al. (2017) [96] and Waite et al. (2014) [93] found reduced C9orf72 protein expression in frontal areas but not in the cerebellum. Nevertheless, performing a meta-analysis (Fig. 4C) which combined frontal and cerebellar brain regions found that C9orf72 levels are significantly reduced in human patients at the time of autopsy.
It is also important to note that the mechanisms by which reduced protein levels contribute to C9orf72 disease pathogenesis are still unknown and may not be causing neurodegeneration per se. This is evident with reductions in C9orf72 being present in brain regions affected by neurodegeneration and those spared from it. Moreover, the lack of clinical phenotypes in C9orf72 knockout mice seems to support this view [97,98,99,100]. However, Frick et al. (2018) identified C9orf72 protein to be localised presynaptically and able to interact with members of the RAB3 protein family, suggestive of a role for C9orf72 in regulating stress vesicles function by potentially acting as guanine nucleotide exchange factors (GEFs) for specific Rab GTPases (Rabs) such as RAB3 [16]. These results could support a role for cerebellar C9orf72 in regulating synaptic vesicle function.
Another factor that could be contributing to contradictory results regarding the precise functions and properties of C9orf72 protein is the lack of specific antibodies [101, 102]. In this regard, it is important to mention that C9orf72 generates three transcripts through alternative splicing that encode 2 protein isoforms: a long isoform of approximately 54 kDa (termed C9-L), corresponding to variants 2 (V2) and 3 (V3) and a short isoform of approximately 24 kDa (termed C9-S), corresponding to variant 1 (V1) (overviewed in Fig. 6) [103]. Haploinsufficiency was initially suggested as a disease mechanism owing to the decreased abundance of V2 and V3 transcripts in C9orf72-ALS cases, leaving the contribution of C9-S to the disease unknown. Xiao et al. (2015) reported two antibodies capable of detecting both C9 isoforms [104]. In their study, C9-L showed diffuse labelling in the cytoplasm of cerebellar Purkinje cells, with a striking labelling of numerous speckles that were observed in both the neuronal perikarya and dendritic processes in C9orf72 carriers and non-carriers. In contrast, the C9-S antibody gave a very specific labelling of the nuclear membrane. These data showed that C9-L and C9-S have different subcellular localisations in Purkinje cells and suggest that C9orf72 proteins could play a role in the disruption of the nucleocytoplasmic transport. Davidson et al. (2018) later expanded on this to include commercial antibodies, and whilst some could recapitulate the staining of C9-L, none showed similar staining to C9-S which remains elusive [105].

Splice variants of human C9orf72 mRNA. An overview of the exons included in the long and short isoforms of C9orf72 and their subcellular localisation. Abbreviations: aa, amino acids; kDa, kilodalton; RNA, ribonucleic acid; V1/2/3, variant 1/2/3
Recent evidence suggests that C9orf72 protein complexes with p62 and may lead to pathology through inclusion formation [106]. P62-positive inclusions remain a hallmark of C9-ALS/FTD; however, little research has been conducted into the side effects of the aggregated protein, and there remains a large gap in the current literature [22, 25, 79, 80]. Having reduced C9orf72 protein, p62 accumulates with additional symmetrical methylated arginine proteins ordinarily used in the complex with C9orf72 [106]. Bieniek et al. (2013) suggest that aggregation of p62 means there is less to bind to tau resulting in build-up of hyperphosphorylated tau and axonopathy [107]. Moreover, as we have previously discussed, the co-localisation of p62 to DPRs, specifically poly-GA which sequesters Unc119 [22], highlights the complex and potentially synergistic effects of both reduction in C9orf72 protein levels and DPR expression in dendritic and axonopathy and the pathogenesis of C9-ALS/FTD [11, 22, 30, 31, 81, 85].
RNA foci
Finally, we also want to discuss the role of intranuclear neuronal RNA foci containing G4C2 repeats in cerebellar ALS and FTD tissues with RNA foci accumulation being reported by several authors [27, 36, 37, 108]. Indeed, Lee et al. (2013) have found discrete intranuclear neuronal RNA foci where larger RNA foci were found in the cerebellum (500 nm) compared to the cortex (200 nm), which were most frequent in neurons adjacent to Purkinje cells [36]. Also, the authors observed co-localisation between the foci and hnRNP-H, suggesting that sequestration of hnRNP-H itself, other RNA-binding protein and multiple RNA transcripts could be leading to significant dysregulation of RNA processing and toxicity in the cerebellum. Similar results were found by Cooper-Knock et al. (2014) where co-localisation of RNA foci and hnRNP in cerebellar granule cells were found [109]. More recently, Mehta et al. (2020) found evidence of abundant RNA foci in all cell types of the cerebellum without concomitant TDP-43 pathology in C9orf72 post-mortem tissue whilst using a high-resolution modified in situ hybridisation technique, BaseScope™ [110]. Moreover, an AAV9-mediated expression of (G4C2)102 repeats in mice leads to increased number of RNA foci in the Purkinje cell layer of the cerebellum at 12 months after AAV delivery with increased apoptotic markers and infrequent TDP-43 aggregates [111]. These results are opposite to the correlation identified between antisense RNA foci and TDP-43 pathology in motor neurons of C9orf72 patients [112], where the authors suggested that RNA foci may be a cause of TDP-43 inclusions.
Neuritic RNA foci are not a commonly discussed feature of C9orf72, usually focusing on intranuclear inclusions. However, Schweizer Burguete et al. (2015) found that approximately 80% of neurons with intranuclear inclusions also presented neuritic foci [23]. Most importantly, neuritic RNA foci were found to reduce primary dendritic branching by up to 50% which was not a result of branching capability being hampered. Indeed, early fly larvae exhibited normal branching morphologies; however, upon body growth within the same instar stage, these dendrites failed to extend to cover the increased brain expanse, thereby indicating that neuritic foci disrupt the dendrite’s ability to extend its branches during growth and may indicate early-stage synaptopathies in C9orf72 carriers during development. Moreover, they found that neuritic-localised foci were the only RNA foci to elicit such branching defects with somatic or nuclear inclusions not impacting dendritic arborisation. Supporting this, foci were bidirectionally transported throughout the cell through mRNP transport vesicles, and this was found to directly impact morphological features of the dendrites. In neurons with a knockdown of FMRP (fragile X mental retardation protein) and other transport-associated proteins, dendritic pathology was attenuated as foci could no longer move from proximal–somatic areas to neuritic–distal locales of the neuron. The concomitance of nuclear foci and neuritic foci suggests a role in the cerebellum which has high foci pathology; however, the exact abundance of neuritic foci is yet to be explored in these areas and therefore may help elucidate the state of dendritic processes and ultimately, synaptic dysfunction, within the cerebellum. Indeed, special attention should be given to understanding the dynamics of differentially localised RNA foci in relation to cerebellar synaptic dysfunction and other deleterious effects their localisation may result in.
Conclusion
Collectively, the data assembled through this review provides clear evidence that RNA foci and proteinaceous inclusions contribute to synaptic deficits and cerebellar neurodegeneration and should be considered characteristic features of C9-ALS/FTD. This review highlights the relevance of the cerebellum in understanding C9orf72 pathology and may act as a unique structure to understand synaptic pathology which, to date, has been largely neglected. Special attention should be given to cerebellar pathology not only at early stages, but also throughout the course of the disease, which could shed light on some assumptions regarding the combined actions of reductions in C9orf72 protein, RNA foci and DPRs as contributing factors to C9 synaptopathy.
References
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56. https://doi.org/10.1016/j.neuron.2011.09.011.
Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68. https://doi.org/10.1016/j.neuron.2011.09.010.
Gendron TF, Petrucelli L. Disease mechanisms of C9ORF72 repeat expansions. Cold Spring Harb Perspect Med. 2018;8(4). https://doi.org/10.1101/cshperspect.a024224.
Jensen BK, Schuldi MH, McAvoy K, Russell KA, Boehringer A, Curran BM, et al. Synaptic dysfunction induced by glycine-alanine dipeptides in C9orf72-ALS/FTD is rescued by SV2 replenishment. EMBO molecular medicine. 2020;12(5):e10722. https://doi.org/10.15252/emmm.201910722.
Starr A, Sattler R. Synaptic dysfunction and altered excitability in C9ORF72 ALS/FTD. Brain Res. 2018;1693(Pt A):98–108. https://doi.org/10.1016/j.brainres.2018.02.011.
Middleton FA, Strick PL. Cerebellar projections to the prefrontal cortex of the primate. J Neurosci. 2001;21(2):700–12.
O’Reilly JX, Beckmann CF, Tomassini V, Ramnani N, Johansen-Berg H. Distinct and overlapping functional zones in the cerebellum defined by resting state functional connectivity. Cereb Cortex. 2010;20(4):953–65. https://doi.org/10.1093/cercor/bhp157.
Prell T, Grosskreutz J. The involvement of the cerebellum in amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis & frontotemporal degeneration. 2013;14(7–8):507–15. https://doi.org/10.3109/21678421.2013.812661.
Tan RH, Devenney E, Dobson-Stone C, Kwok JB, Hodges JR, Kiernan MC, et al. Cerebellar integrity in the amyotrophic lateral sclerosis-frontotemporal dementia continuum. PLoS ONE. 2014;9(8): e105632. https://doi.org/10.1371/journal.pone.0105632.
Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639–46. https://doi.org/10.1016/j.neuron.2013.02.004.
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science (New York, NY). 2013;339(6125):1335–8. https://doi.org/10.1126/science.1232927.
Zu T, Liu Y, Bañez-Coronel M, Reid T, Pletnikova O, Lewis J, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA. 2013;110(51):E4968–77. https://doi.org/10.1073/pnas.1315438110.
Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345(6201):1192–4. https://doi.org/10.1126/science.1256800.
Haeusler AR, Donnelly CJ, Rothstein JD. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci. 2016;17(6):383–95. https://doi.org/10.1038/nrn.2016.38.
Lee YB, Baskaran P, Gomez-Deza J, Chen HJ, Nishimura AL, Smith BN, et al. C9orf72 poly GA RAN-translated protein plays a key role in amyotrophic lateral sclerosis via aggregation and toxicity. Hum Mol Genet. 2020. https://doi.org/10.1093/hmg/ddaa181.
Frick P, Sellier C, Mackenzie IRA, Cheng CY, Tahraoui-Bories J, Martinat C, et al. Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol Commun. 2018;6(1):72. https://doi.org/10.1186/s40478-018-0579-0.
Xiao S, McKeever PM, Lau A, Robertson J. Synaptic localization of C9orf72 regulates post-synaptic glutamate receptor 1 levels. Acta Neuropathol Commun. 2019;7(1):161. https://doi.org/10.1186/s40478-019-0812-5.
Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb-Cheynel I, Oulad-Abdelghani M, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016;35(12):1276–97. https://doi.org/10.15252/embj.201593350.
Yang M, Liang C, Swaminathan K, Herrlinger S, Lai F, Shiekhattar R, et al. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv. 2016;2(9): e1601167. https://doi.org/10.1126/sciadv.1601167.
Troakes C, Maekawa S, Wijesekera L, Rogelj B, Siklós L, Bell C, et al. An MND/ALS phenotype associated with C9orf72 repeat expansion: abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology: official journal of the Japanese Society of Neuropathology. 2012;32(5):505–14. https://doi.org/10.1111/j.1440-1789.2011.01286.x.
Xu W, Xu J. C9orf72 dipeptide repeats cause selective neurodegeneration and cell-autonomous excitotoxicity in Drosophila glutamatergic neurons. J Neurosci. 2018;38(35):7741–52. https://doi.org/10.1523/jneurosci.0908-18.2018.
May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, et al. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta Neuropathol. 2014;128(4):485–503. https://doi.org/10.1007/s00401-014-1329-4.
Burguete AS, Almeida S, Gao FB, Kalb R, Akins MR, Bonini NM. GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. eLife. 2015;4:e08881. https://doi.org/10.7554/eLife.08881.
Park JH, Chung CG, Seo J, Lee BH, Lee YS, Kweon JH, et al. C9orf72-associated arginine-rich dipeptide repeat proteins reduce the number of golgi outposts and dendritic branches in Drosophila neurons. Mol Cells. 2020;43(9):821–30. https://doi.org/10.14348/molcells.2020.0130.
Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122(6):691–702. https://doi.org/10.1007/s00401-011-0911-2.
Mackenzie IR, Frick P, Grässer FA, Gendron TF, Petrucelli L, Cashman NR, et al. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 2015;130(6):845–61. https://doi.org/10.1007/s00401-015-1476-2.
Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126(6):829–44. https://doi.org/10.1007/s00401-013-1192-8.
Zhang YJ, Jansen-West K, Xu YF, Gendron TF, Bieniek KF, Lin WL, et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014;128(4):505–24. https://doi.org/10.1007/s00401-014-1336-5.
Gendron TF, van Blitterswijk M, Bieniek KF, Daughrity LM, Jiang J, Rush BK, et al. Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers. Acta Neuropathol. 2015;130(4):559–73. https://doi.org/10.1007/s00401-015-1474-4.
Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K, et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 2013;126(6):859–79. https://doi.org/10.1007/s00401-013-1181-y.
Mann DM, Rollinson S, Robinson A, Bennion Callister J, Thompson JC, Snowden JS, et al. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun. 2013;1:68. https://doi.org/10.1186/2051-5960-1-68.
Schludi MH, May S, Grasser FA, Rentzsch K, Kremmer E, Kupper C, et al. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol. 2015;130(4):537–55. https://doi.org/10.1007/s00401-015-1450-z.
Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu YF, et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci. 2016;19(5):668–77. https://doi.org/10.1038/nn.4272.
Davidson YS, Robinson AC, Snowden JS, Mann DM. Pathological assessments for the presence of hexanucleotide repeat expansions in C9ORF72 in Alzheimer’s disease. Acta Neuropathol Commun. 2013;1:50. https://doi.org/10.1186/2051-5960-1-50.
Zhou Q, Lehmer C, Michaelsen M, Mori K, Alterauge D, Baumjohann D, et al. Antibodies inhibit transmission and aggregation of C9orf72 poly-GA dipeptide repeat proteins. EMBO molecular medicine. 2017;9(5):687–702. https://doi.org/10.15252/emmm.201607054.
Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Stalekar M, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5(5):1178–86. https://doi.org/10.1016/j.celrep.2013.10.049.
DeJesus-Hernandez M, Finch NA, Wang X, Gendron TF, Bieniek KF, Heckman MG, et al. In-depth clinico-pathological examination of RNA foci in a large cohort of C9ORF72 expansion carriers. Acta Neuropathol. 2017;134(2):255–69. https://doi.org/10.1007/s00401-017-1725-7.
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ (Clinical research ed). 2009;339: b2535. https://doi.org/10.1136/bmj.b2535.
Lee YB, Baskaran P, Gomez-Deza J, Chen HJ, Nishimura AL, Smith BN, et al. C9orf72 poly GA RAN-translated protein plays a key role in amyotrophic lateral sclerosis via aggregation and toxicity. Hum Mol Genet. 2017;26(24):4765–77. https://doi.org/10.1093/hmg/ddx350.
Review Manager (RevMan). The Cochrane Collaboration; 2020.
Devlin AC, Burr K, Borooah S, Foster JD, Cleary EM, Geti I, et al. Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat Commun. 2015;6:5999. https://doi.org/10.1038/ncomms6999.
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525(7567):129–33. https://doi.org/10.1038/nature14974.
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525(7567):56–61. https://doi.org/10.1038/nature14973.
Perry S, Han Y, Das A, Dickman D. Homeostatic plasticity can be induced and expressed to restore synaptic strength at neuromuscular junctions undergoing ALS-related degeneration. Hum Mol Genet. 2017;26(21):4153–67. https://doi.org/10.1093/hmg/ddx304.
Sareen D, O'Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5(208):208ra149. https://doi.org/10.1126/scitranslmed.3007529.
Fuchs A, Kutterer S, Mühling T, Duda J, Schütz B, Liss B, et al. Selective mitochondrial Ca2+ uptake deficit in disease endstage vulnerable motoneurons of the SOD1G93A mouse model of amyotrophic lateral sclerosis. J Physiol. 2013;591(10):2723–45. https://doi.org/10.1113/jphysiol.2012.247981.
Delestrée N, Manuel M, Iglesias C, Elbasiouny SM, Heckman CJ, Zytnicki D. Adult spinal motoneurones are not hyperexcitable in a mouse model of inherited amyotrophic lateral sclerosis. J Physiol. 2014;592(7):1687–703. https://doi.org/10.1113/jphysiol.2013.265843.
von Lewinski F, Fuchs J, Vanselow BK, Keller BU. Low Ca2+ buffering in hypoglossal motoneurons of mutant SOD1 (G93A) mice. Neurosci Lett. 2008;445(3):224–8. https://doi.org/10.1016/j.neulet.2008.08.084.
Jaiswal MK, Keller BU. Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol Pharmacol. 2009;75(3):478–89. https://doi.org/10.1124/mol.108.050831.
Matsumoto S, Goto S, Kusaka H, Ito H, Imai T. Synaptic pathology of spinal anterior horn cells in amyotrophic lateral sclerosis: an immunohistochemical study. J Neurol Sci. 1994;125(2):180–5. https://doi.org/10.1016/0022-510x(94)90032-9.
Sasaki S, Maruyama S. Synapse loss in anterior horn neurons in amyotrophic lateral sclerosis. Acta Neuropathol. 1994;88(3):222–7. https://doi.org/10.1007/bf00293397.
Ince PG, Slade J, Chinnery RM, McKenzie J, Royston C, Roberts GW, et al. Quantitative study of synaptophysin immunoreactivity of cerebral cortex and spinal cord in motor neuron disease. J Neuropathol Exp Neurol. 1995;54(5):673–9. https://doi.org/10.1097/00005072-199509000-00009.
Jiang M, Schuster JE, Fu R, Siddique T, Heckman CJ. Progressive changes in synaptic inputs to motoneurons in adult sacral spinal cord of a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2009;29(48):15031–8. https://doi.org/10.1523/jneurosci.0574-09.2009.
Chang Q, Martin LJ. Glycinergic innervation of motoneurons is deficient in amyotrophic lateral sclerosis mice: a quantitative confocal analysis. Am J Pathol. 2009;174(2):574–85. https://doi.org/10.2353/ajpath.2009.080557.
Schütz B. Imbalanced excitatory to inhibitory synaptic input precedes motor neuron degeneration in an animal model of amyotrophic lateral sclerosis. Neurobiol Dis. 2005;20(1):131–40. https://doi.org/10.1016/j.nbd.2005.02.006.
Hao Z, Liu L, Tao Z, Wang R, Ren H, Sun H, et al. Motor dysfunction and neurodegeneration in a C9orf72 mouse line expressing poly-PR. Nat Commun. 2019;10(1):2906. https://doi.org/10.1038/s41467-019-10956-w.
King A, Maekawa S, Bodi I, Troakes C, Al-Sarraj S. Ubiquitinated, p62 immunopositive cerebellar cortical neuronal inclusions are evident across the spectrum of TDP-43 proteinopathies but are only rarely additionally immunopositive for phosphorylation-dependent TDP-43. Neuropathology: official journal of the Japanese Society of Neuropathology. 2011;31(3):239–49. https://doi.org/10.1111/j.1440-1789.2010.01171.x.
Goldman JS, Quinzii C, Dunning-Broadbent J, Waters C, Mitsumoto H, Brannagan TH 3rd, et al. Multiple system atrophy and amyotrophic lateral sclerosis in a family with hexanucleotide repeat expansions in C9orf72. JAMA Neurol. 2014;71(6):771–4. https://doi.org/10.1001/jamaneurol.2013.5762.
Fogel BL, Pribadi M, Pi S, Perlman SL, Geschwind DH, Coppola G. C9ORF72 expansion is not a significant cause of sporadic spinocerebellar ataxia. Movement disorders: official journal of the Movement Disorder Society. 2012;27(14):1832–3. https://doi.org/10.1002/mds.25245.
Corcia P, Vourc’h P, Guennoc AM, Del Mar AM, Blasco H, Andres C, et al. Pure cerebellar ataxia linked to large C9orf72 repeat expansion. Amyotrophic lateral sclerosis & frontotemporal degeneration. 2016;17(3–4):301–3. https://doi.org/10.3109/21678421.2015.1113298.
Mori K, Lammich S, Mackenzie IR, Forné I, Zilow S, Kretzschmar H, et al. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 2013;125(3):413–23. https://doi.org/10.1007/s00401-013-1088-7.
Tao Z, Wang H, Xia Q, Li K, Li K, Jiang X, et al. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum Mol Genet. 2015;24(9):2426–41. https://doi.org/10.1093/hmg/ddv005.
Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, et al. C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell. 2016;167(3):774-88.e17. https://doi.org/10.1016/j.cell.2016.10.002.
Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science (New York, NY). 2014;345(6201):1192–4. https://doi.org/10.1126/science.1256800.
Zhang YJ, Gendron TF, Ebbert MTW, O’Raw AD, Yue M, Jansen-West K, et al. Poly(GR) impairs protein translation and stress granule dynamics in C9orf72-associated frontotemporal dementia and amyotrophic lateral sclerosis. Nat Med. 2018;24(8):1136–42. https://doi.org/10.1038/s41591-018-0071-1.
Murray ME, DeJesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 2011;122(6):673–90. https://doi.org/10.1007/s00401-011-0907-y.
King A, Al-Sarraj S, Troakes C, Smith BN, Maekawa S, Iovino M, et al. Mixed tau, TDP-43 and p62 pathology in FTLD associated with a C9ORF72 repeat expansion and p.Ala239Thr MAPT (tau) variant. Acta Neuropathol. 2013;125(2):303–10. https://doi.org/10.1007/s00401-012-1050-0.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–34. https://doi.org/10.1016/j.cell.2010.02.016.
Zhang YJ, Guo L, Gonzales PK, Gendron TF, Wu Y, Jansen-West K, et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science (New York, NY). 2019;363(6428). https://doi.org/10.1126/science.aav2606.
Tan RH, Kril JJ, McGinley C, Hassani M, Masuda-Suzukake M, Hasegawa M, et al. Cerebellar neuronal loss in amyotrophic lateral sclerosis cases with ATXN2 intermediate repeat expansions. Ann Neurol. 2016;79(2):295–305. https://doi.org/10.1002/ana.24565.
Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, et al. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science (New York, NY). 2014;345(6201):1139–45. https://doi.org/10.1126/science.1254917.
Rudich P, Snoznik C, Watkins SC, Monaghan J, Pandey UB, Lamitina ST. Nuclear localized C9orf72-associated arginine-containing dipeptides exhibit age-dependent toxicity in C. elegans. Human molecular genetics. 2017;26(24):4916–28. https://doi.org/10.1093/hmg/ddx372.
Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84(6):1213–25. https://doi.org/10.1016/j.neuron.2014.12.010.
Swaminathan A, Bouffard M, Liao M, Ryan S, Callister JB, Pickering-Brown SM, et al. Expression of C9orf72-related dipeptides impairs motor function in a vertebrate model. Hum Mol Genet. 2018;27(10):1754–62. https://doi.org/10.1093/hmg/ddy083.
Yamakawa M, Ito D, Honda T, Kubo K, Noda M, Nakajima K, et al. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum Mol Genet. 2015;24(6):1630–45. https://doi.org/10.1093/hmg/ddu576.
Pikkarainen M, Hartikainen P, Alafuzoff I. Ubiquitinated p62-positive, TDP-43-negative inclusions in cerebellum in frontotemporal lobar degeneration with TAR DNA binding protein 43. Neuropathology: official journal of the Japanese Society of Neuropathology. 2010;30(2):197–9. https://doi.org/10.1111/j.1440-1789.2009.01043.x.
Yang D, Abdallah A, Li Z, Lu Y, Almeida S, Gao FB. FTD/ALS-associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta Neuropathol. 2015;130(4):525–35. https://doi.org/10.1007/s00401-015-1448-6.
Darling AL, Breydo L, Rivas EG, Gebru NT, Zheng D, Baker JD, et al. Repeated repeat problems: combinatorial effect of C9orf72-derived dipeptide repeat proteins. Int J Biol Macromol. 2019;127:136–45. https://doi.org/10.1016/j.ijbiomac.2019.01.035.
Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain: a journal of neurology. 2012;135(Pt 3):693–708. https://doi.org/10.1093/brain/awr355.
Mahoney CJ, Beck J, Rohrer JD, Lashley T, Mok K, Shakespeare T, et al. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain: a journal of neurology. 2012;135(Pt 3):736–50. https://doi.org/10.1093/brain/awr361.
LaClair KD, Zhou Q, Michaelsen M, Wefers B, Brill MS, Janjic A, et al. Congenic expression of poly-GA but not poly-PR in mice triggers selective neuron loss and interferon responses found in C9orf72 ALS. Acta Neuropathol. 2020;140(2):121–42. https://doi.org/10.1007/s00401-020-02176-0.
Maor-Nof M, Shipony Z, Lopez-Gonzalez R, Nakayama L, Zhang YJ, Couthouis J, et al. p53 is a central regulator driving neurodegeneration caused by C9orf72 poly(PR). Cell. 2021;184(3):689-708.e20. https://doi.org/10.1016/j.cell.2020.12.025.
Fare CM, Shorter J. Open access: a role for p53 in c9ALS/FTD? trends in genetics: TIG. 2021. https://doi.org/10.1016/j.tig.2021.01.008.
Lopez-Gonzalez R, Yang D, Pribadi M, Kim TS, Krishnan G, Choi SY, et al. Partial inhibition of the overactivated Ku80-dependent DNA repair pathway rescues neurodegeneration in C9ORF72-ALS/FTD. Proc Natl Acad Sci USA. 2019;116(19):9628–33. https://doi.org/10.1073/pnas.1901313116.
Mori K, Arzberger T, Grässer FA, Gijselinck I, May S, Rentzsch K, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013;126(6):881–93. https://doi.org/10.1007/s00401-013-1189-3.
Simon CM, Dai Y, Van Alstyne M, Koutsioumpa C, Pagiazitis JG, Chalif JI, et al. Converging mechanisms of p53 activation drive motor neuron degeneration in spinal muscular atrophy. Cell Rep. 2017;21(13):3767–80. https://doi.org/10.1016/j.celrep.2017.12.003.
Baltanás FC, Berciano MT, Tapia O, Narcis JO, Lafarga V, Díaz D, et al. Nucleolin reorganization and nucleolar stress in Purkinje cells of mutant PCD mice. Neurobiol Dis. 2019;127:312–22. https://doi.org/10.1016/j.nbd.2019.03.017.
Schludi MH, Becker L, Garrett L, Gendron TF, Zhou Q, Schreiber F, et al. Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol. 2017;134(2):241–54. https://doi.org/10.1007/s00401-017-1711-0.
Davidson YS, Flood L, Robinson AC, Nihei Y, Mori K, Rollinson S, et al. Heterogeneous ribonuclear protein A3 (hnRNP A3) is present in dipeptide repeat protein containing inclusions in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9orf72 gene. Acta Neuropathol Commun. 2017;5(1):31. https://doi.org/10.1186/s40478-017-0437-5.
Freibaum BD, Taylor JP. The role of dipeptide repeats in C9ORF72-related ALS-FTD. Front Mol Neurosci. 2017;10:35. https://doi.org/10.3389/fnmol.2017.00035.
Quaegebeur A, Glaria I, Lashley T, Isaacs AM. Soluble and insoluble dipeptide repeat protein measurements in C9orf72-frontotemporal dementia brains show regional differential solubility and correlation of poly-GR with clinical severity. Acta Neuropathol Commun. 2020;8(1):184. https://doi.org/10.1186/s40478-020-01036-y.
van Blitterswijk M, Gendron TF, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, et al. Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol. 2015;130(6):863–76. https://doi.org/10.1007/s00401-015-1480-6.
Waite AJ, Baumer D, East S, Neal J, Morris HR, Ansorge O, et al. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014;35(7):1779 e5- e13. https://doi.org/10.1016/j.neurobiolaging.2014.01.016.
Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science (New York, NY). 2015;348(6239):1151–4. https://doi.org/10.1126/science.aaa9344.
Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, et al. C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron. 2016;90(3):521–34. https://doi.org/10.1016/j.neuron.2016.04.005.
Saberi S, Stauffer JE, Jiang J, Garcia SD, Taylor AE, Schulte D, et al. Sense-encoded poly-GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co-localize with TDP-43 in dendrites of repeat-expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol. 2018;135(3):459–74. https://doi.org/10.1007/s00401-017-1793-8.
Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016;6:23204. https://doi.org/10.1038/srep23204.
Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sá R, et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Annals of neurology. 2015;78(3):426–38. https://doi.org/10.1002/ana.24453.
O’Rourke JG, Bogdanik L, Muhammad A, Gendron TF, Kim KJ, Austin A, et al. C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron. 2015;88(5):892–901. https://doi.org/10.1016/j.neuron.2015.10.027.
Sudria-Lopez E, Koppers M, de Wit M, van der Meer C, Westeneng HJ, Zundel CA, et al. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol. 2016;132(1):145–7. https://doi.org/10.1007/s00401-016-1581-x.
Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics (Oxford, England). 2013;29(4):499–503. https://doi.org/10.1093/bioinformatics/bts725.
Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet. 2014;23(13):3579–95. https://doi.org/10.1093/hmg/ddu068.
Belzil VV, Bauer PO, Prudencio M, Gendron TF, Stetler CT, Yan IK, et al. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013;126(6):895–905. https://doi.org/10.1007/s00401-013-1199-1.
Xiao S, MacNair L, McGoldrick P, McKeever PM, McLean JR, Zhang M, et al. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol. 2015;78(4):568–83. https://doi.org/10.1002/ana.24469.
Davidson YS, Robinson AC, Rollinson S, Pickering-Brown S, Xiao S, Robertson J, et al. Immunohistochemical detection of C9orf72 protein in frontotemporal lobar degeneration and motor neurone disease: patterns of immunostaining and an evaluation of commercial antibodies. Amyotrophic lateral sclerosis & frontotemporal degeneration. 2018;19(1–2):102–11. https://doi.org/10.1080/21678421.2017.1359304.
Chitiprolu M, Jagow C, Tremblay V, Bondy-Chorney E, Paris G, Savard A, et al. A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat Commun. 2018;9(1):2794. https://doi.org/10.1038/s41467-018-05273-7.
Bieniek KF, Murray ME, Rutherford NJ, Castanedes-Casey M, DeJesus-Hernandez M, Liesinger AM, et al. Tau pathology in frontotemporal lobar degeneration with C9ORF72 hexanucleotide repeat expansion. Acta Neuropathol. 2013;125(2):289–302. https://doi.org/10.1007/s00401-012-1048-7.
Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, et al. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013;126(6):845–57. https://doi.org/10.1007/s00401-013-1200-z.
Cooper-Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D, et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain: a journal of neurology. 2014;137(Pt 7):2040–51. https://doi.org/10.1093/brain/awu120.
Mehta AR, Selvaraj BT, Barton SK, McDade K, Abrahams S, Chandran S, et al. Improved detection of RNA foci in C9orf72 amyotrophic lateral sclerosis post-mortem tissue using BaseScope™ shows a lack of association with cognitive dysfunction. Brain communications. 2020;2(1):fcaa009. https://doi.org/10.1093/braincomms/fcaa009.
Herranz-Martin S, Chandran J, Lewis K, Mulcahy P, Higginbottom A, Walker C, et al. Viral delivery of C9orf72 hexanucleotide repeat expansions in mice leads to repeat-length-dependent neuropathology and behavioural deficits. Dis Model Mech. 2017;10(7):859–68. https://doi.org/10.1242/dmm.029892.
Cooper-Knock J, Higginbottom A, Stopford MJ, Highley JR, Ince PG, Wharton SB, et al. Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol. 2015;130(1):63–75. https://doi.org/10.1007/s00401-015-1429-9.
Acknowledgements
We would like to thank George Popescu-Craiova for his help in tabulating, sorting and removing duplicate articles during the systematic search of papers.
Funding
This work was supported by the Ministry of Science, Innovation and Universities (PSI2017-83893-R and PID2020-117259RB-I00), the Ministry of Economy and Business (PSI2017-90806-REDT) (Spain) and the UK Dementia Research Institute which receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK. The funders had no role in the study design or data collection. We would like to thank the Alzheimer’s Research UK, Dementia Research Institute (DRI), King’s College London Network Centre and AINDACE foundation for their technical and human support.
Author information
Authors and Affiliations
Contributions
N.A. and C.T.T.: research project: (a) conception, (b) organisation and (c) execution. N.A., C.T.T., A.K., J.A. and C.S.: manuscript: (a) writing of the first draft, (b) review and critique and (c) review.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaliszewska, A., Allison, J., Col, TT. et al. Elucidating the Role of Cerebellar Synaptic Dysfunction in C9orf72-ALS/FTD — a Systematic Review and Meta-Analysis. Cerebellum 21, 681–714 (2022). https://doi.org/10.1007/s12311-021-01320-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-021-01320-0