
Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD) is becoming an increasingly pressing global health challenge, with increasing mortality rates showing an upward trend. Two million deaths occur annually from cirrhosis and liver cancer together each year. Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), key effectors of the Hippo signaling pathway, critically regulate tissue homeostasis and disease progression in the liver. While initial studies have shown that YAP expression is normally restricted to cholangiocytes in healthy livers, the activation of YAP/TAZ is observed in other hepatic cells during chronic liver disease. The disease-driven dysregulation of YAP/TAZ appears to be a critical element in the MASLD progression, contributing to hepatocyte dysfunction, inflammation, and fibrosis. In this study, we focused on the complex roles of YAP/TAZ in MASLD and explored how the YAP/TAZ dysregulation of YAP/TAZ drives steatosis, inflammation, fibrosis, and cirrhosis. Finally, the cell-type-specific functions of YAP/TAZ in different types of hepatic cells, such as hepatocytes, hepatic stellate cells, hepatic macrophages, and biliary epithelial cells are discussed, highlighting the multifaceted impact of YAP/TAZ on liver physiology and pathology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) are key effectors of the Hippo signaling pathway and play vital roles in regulating cell fate decisions, tissue growth, regeneration, and metabolism cell proliferation (Maglic et al. 2018; Koo et al. 2020; Daoud et al. 2021). With a strong evolutionary conservation, their roles were initially studied in Drosophila homolog Yki. A landmark report revealed that YAP overexpression in mice leads to dramatic growth in liver size by up to six times and robust development of HCC (Dong et al. 2007). This striking phenotype has attracted substantial scientific interest, highlighting the potential roles of YAP and TAZ in mammalian tissue homeostasis. Specifically, as liver disease progresses, YAP/TAZ levels increase as hepatocytes undergo continuous death and regeneration, which is thought to be related to the disease progression. Since then, YAP/TAZ has been strongly implicated in a wide range of diseases, including metabolic dysfunction-associated steatotic liver disease (MASLD), as discussed in this review.
YAP and TAZ share highly homologous sequences but have some structural variances (Plouffe et al. 2018). YAP is larger than TAZ, and while both contain WW domains that mediate protein–protein interactions, their transcriptional activation domains and binding capacities can differ (Reggiani et al. 2021). Structurally, the TEAD-binding domain of YAP comprises two α-helices connected by a loop containing the PXXΦP motif, whereas TAZ lacks the motif (Chen et al. 2010; Reggiani et al. 2021). They also show some functional differences as well. For example, YAP knockout mice are not viable, showing embryo lethality at embryonic day 8.5 due to defective yolk sac vasculogenesis and embryonic axis development (Morin-Kensicki et al. 2006). In contrast, TAZ knockout mice are viable and fertile albeit they develop renal and lung abnormalities (Hossain et al. 2007; Makita et al. 2008).
In the liver, the Hippo pathway regulates substantial hepatocyte proliferation, differentiation, and survival (Lee et al. 2018). When Hippo signaling is active, its core kinases, namely mammalian sterile 20-like kinase 1 and 2 (MST1/2) and mitogen-activated protein kinase kinase kinase kinase (MAP4Ks), activate large tumor suppressor kinases 1 and 2 (LATS1/2) and induce the phosphorylation of YAP/TAZ at multiple sites (Plouffe et al. 2018). This phosphorylation forces cytoplasmic retention and degradation of YAP/TAZ, thereby preventing its transcriptional activity (Vassilev et al. 2001). Conversely, when Hippo signaling is suppressed, unphosphorylated YAP/TAZ translocates to the nucleus. They interact with the transcription enhancer factor-associated domain (TEAD) family of transcription factors and other transcription partners to regulate the expression of genes crucial for cell proliferation, survival, and metabolic processes (Huh et al. 2019; Jin et al. 2023). Examples of key YAP/TAZ target genes include the cysteine-rich angiogenic inducer 61 (CYR61) and connective tissue growth factor (CTGF), which are classically known mediators of hepatic inflammation and fibrosis. Therefore, understanding the interplay between YAP/TAZ dysregulation and cellular dysfunction in diverse liver cell types is crucial for the development of novel therapeutic strategies to target complex diseases.
YAP/TAZ dysregulation in liver diseases
MASLD is defined as a progressive fatty liver disease, developing from simple steatosis to hepatitis, fibrosis, and cirrhosis which eventually causes liver cancer, that meets two of the following metabolic risk factors: obesity, type 2 diabetes, and hyperlipidemia (Eslam et al. 2020). Hepatic steatosis caused by insulin resistance and excess fatty acids can be enhanced by genetic mutations, making it vulnerable to steatohepatitis (Farese et al. 2012). Insulin resistance promotes fat synthesis in the liver and inhibits fat decomposition, causing fatty liver disease and increasing the expression of inflammatory cytokines, causing liver damage and death (Reyes-Gordillo et al. 2017; Chen et al. 2017). In its inflammatory phase called metabolic dysfunction-associated steatohepatitis (MASH), when fat accumulation in hepatocytes becomes severe, structural changes and functional damage to liver tissue occur, leading to fibrosis, and continued fibrosis leads to cirrhosis (Friedman 2024). Additionally, oxidative stress, inflammation, and fibrosis can lead to cirrhosis and hepatocellular carcinoma, which can lead to death from liver disease (Abu Rmilah et al. 2019). MASLD presents a substantial global health challenge, with increasing mortality rates. Two million deaths occur annually from cirrhosis and liver cancer together each year (Asrani et al. 2019). It was not until this year that resmetirom (marketed as Rezdiffra) was approved by the United States Food and Drug Administration drug for the treatment of MASH. It is indicated for use only in relatively mild cases of the F2 stage fibrosis which have not yet progressed to cirrhosis, suggesting that there remains a vast and unmet clinical need (Harrison et al. 2024).
In the last decade, it has become evident that the aberrant regulation of the Hippo signaling pathway is linked to various aspects of hepatic dysfunction, including changes in the proportion of hepatic cells, altered regeneration capacity, parenchymal death, and inflammation. While initial studies have shown that YAP expression is normally restricted to cholangiocytes in the healthy liver, robust activation of YAP/TAZ was observed in hepatocytes after partial hepatectomy, which is necessary for normal liver regeneration, although not essential (Lu et al. 2018). Subsequent discoveries have demonstrated a substantial increase in YAP/TAZ activity in hepatocytes during acute liver injury, as well as in liver fibrosis (Machado et al. 2015; Wang et al. 2016; Zhang et al. 2016; Kwon et al. 2024). This disease-driven dysregulation of YAP/TAZ appears to be critical for MASLD progression, contributing to hepatocyte dysfunction, inflammation, and fibrosis (Fig. 1).

Regulation of YAP/TAZ during MASLD progression in patients and experimental animal models. YAP/TAZ activity during different stages of MASLD. YAP/TAZ expression or activity observed in patients (blue box) and mouse models to mimic human disease (green box) are shown. In physiological conditions, YAP expression is mostly restricted to cholangiocytes in both humans and mice (yellow box). APAP acetaminophen; CCA cholangiocarcinoma; CCl4 carbon tetrachloride; CDAHFD choline-deficient amino acid-defined high-fat diet; DEN diethylnitrosamine; FPC high-fructose high-palmitate and high-cholesterol diet; HCC hepatocellular carcinoma; HFD high-fat diet; LPS lipopolysaccharide; MCD methionine- and choline-deficient diet; MyrAKT O/E Hepatocytic overexpression of myristoylated AKT
Steatosis and steatohepatitis
Steatosis refers to the abnormal accumulation of fat within the liver, typically characterized by fat content exceeding 5% of the liver weight (Nassir et al. 2015). Steatosis commonly arises from metabolic disorders associated with alcohol consumption, obesity, diabetes, and other related conditions. Although the simple accumulation of fat is not regarded as a disease, excess lipids may cause lipotoxicity and induce inflammation, leading to steatohepatitis (Peng et al. 2020). In humans, analysis of liver biopsies from patients with MASH often reveals increased nuclear localization of YAP/TAZ in hepatocytes compared to healthy liver tissues (Mooring et al. 2020; Salloum et al. 2021). Mouse models fed a high-fat diet (HFD) or choline-deficient, amino acid-defined high-fat diet also showed that YAP activation was accompanied by the development of steatosis and steatohepatitis (Salloum et al. 2021). In addition, mice fed a cholesterol-rich, MASH-inducing diet showed increased TAZ expression in hepatocytes (Wang et al. 2020).
Hepatocyte stress is one of the most prominent causes of hepatic YAP/TAZ activation in MASLD. Although direct clinical studies on acute liver injury are less common, elevated YAP expression and nuclear localization have been observed in patients with acute liver failure (Hyun et al. 2019), suggesting that acute hepatocyte damage can trigger substantial YAP/TAZ dysregulation in humans. Studies using animal models have provided strong evidence for YAP/TAZ activation following hepatocyte injury. A recent study has reported aberrant YAP activation in an animal model of acetaminophen-induced liver injury (Kwon et al. 2024). This study revealed that mitochondrial stress activates YAP/TAZ via superoxide-mediated oxidation and activation of Ras homolog family member A (RhoA), a potent activator of YAP/TAZ. Consistent observations have been reported in another mouse model that utilized a single intraperitoneal injection of carbon tetrachloride (CCl4), confirming the contribution of oxidative stress to YAP activation (Mannaerts et al. 2015; Verboven et al. 2021). Another study found that pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNFα) can directly modulate hepatocyte YAP activity in a dose-dependent manner, promoting either survival (low TNFα) or death (high TNFα)(Zhao et al. 2020). This highlights a context-dependent regulatory mechanism that adds complexity to the interplay between YAP/TAZ and inflammatory signaling. Notably, high cholesterol levels, which are a characteristic of MASLD, stabilize TAZ. In hepatocytes, cholesterol-induced activation of TAZ is linked to lipid dysregulation and MASLD pathogenesis (Wang et al. 2020). Excessive cholesterol triggers the inositol trisphosphate receptor (IP3R)-calcium-RhoA pathway to activate TAZ.
Fibrosis and cirrhosis
Liver fibrosis is characterized by the accumulation of extracellular matrix (ECM) proteins. Although it is a regenerative process against hepatocyte loss, excessive ECM production causes liver fibrosis, which can disrupt liver architecture, impair organ function, and alter intrahepatic blood flow. Cirrhosis is an end-stage liver fibrosis characterized by severe architectural distortion and impaired liver function. Accumulating evidence strongly links YAP/TAZ upregulation to the progression of fibrosis and the development of cirrhosis (Machado et al. 2015; Chen et al. 2018; Wang et al. 2020; Zhao et al. 2023). Analysis of liver tissues from patients with fibrosis and cirrhosis repetitively confirmed an increased YAP/TAZ activity compared to healthy livers throughout multiple cohorts. Moreover, the extent of YAP activity is frequently correlated with the severity of fibrosis (Machado et al. 2015; Salloum et al. 2021). Mouse models of liver fibrosis using a methionine and choline-deficient diet, high-fructose, high-palmitate, and high-cholesterol (FPC) diets, or repetitive intraperitoneal injections of CCl4 all show increased YAP/TAZ activity accompanied by fibrous scarring and ECM accumulation (Wang et al. 2016; Salloum et al. 2021; Du et al. 2023). In addition to YAP/TAZ activation in hepatocytes upon chronic liver injury, damaged hepatocytes release signals, such as the cytokine Indian Hedgehog (Ihh) and the morphogen Sonic Hedgehog (Shh), and activate YAP/TAZ signaling in hepatic stellate cells (HSCs) as well (Machado et al. 2015; Wang et al. 2016). Increased production of Ihh and Shh has been observed in both human and mouse livers with fibrosis, which was attributed to the enhanced activity of YAP/TAZ in HSCs. Notably, the production of Ihh was shown to be induced by YAP or TAZ in hepatocytes, suggesting its role in intercellular communication.
Increased stiffness of the fibrotic ECM is one of the mechanisms of YAP/TAZ activation owing to their distinct mechanosensitivity (Dupont et al. 2011). A stiff ECM prompts YAP/TAZ nuclear localization and upregulation of the YAP/TAZ target gene, indicating the role of YAP/TAZ as a mechanosensor. Inhibition of YAP reduces the mechanosensitive spontaneous transdifferentiation of HSCs (Mannaerts et al. 2015). YAP/TAZ activation and subsequent HSC transdifferentiation were strongly correlated with alterations in the physical stiffness of the surrounding matrix, as demonstrated using a light-induced secondary crosslinking hydrogel matrix. Excess accumulation of specific ECM proteins such as collagen I and IV is accompanied by liver fibrosis progression. Therefore, changes in ECM composition may also regulate YAP/TAZ through integrin-mediated signaling (Caliari et al. 2016).
Primary liver cancers
Primary liver cancers refer to cancers that occur in the liver itself, which include HCC and cholangiocarcinoma (CCA). MASLD is a major risk factor, with cirrhosis being the most substantial (Llovet et al. 2021; Ebrahimi et al. 2023). There are several features of MASLD that contribute to the development of cancer. In MASLD, persistent inflammatory signals, endoplasmic reticulum stress, and fatty acid overload induce cell death and DNA damage, which promote the formation of malignant cells. Accumulation of DNA damage from repeated cell death and regeneration can lead to cancerous transformation of liver cells (Matchett et al. 2024). In addition, oxidative stress, or metabolic abnormalities per se can also interfere with physiological regulation of proliferation and tumor suppression, either by alteration of signaling pathways or genetic mutation (Brahma et al. 2021). These changes play an important role in the early stages of primary liver cancer, closely linking MASLD to liver carcinogenesis (Llovet et al. 2021).
Over 60% of human HCC cases exhibit elevated YAP/TAZ expression and nuclear localization, along with increased expression of their target genes. This overexpression correlates with poor prognosis in HCC patients (Zender et al. 2006; Xu et al. 2009; Han et al. 2014; Sohn et al. 2016). Similarly, CCAs display increased YAP/TAZ expression and activity compared with normal bile duct cells (Li et al. 2012; Sugihara et al. 2019). Despite the frequent amplification of YAP/TAZ activity in liver cancers, mutations in the Hippo pathway are rare in HCC and CCA. Therefore, YAP/TAZ signaling may not be the driver of tumor initiation. Nonetheless, unrestrained YAP/TAZ activity is a common feature of many cancers and strongly supports proliferation, invasion, and drug resistance (Guo et al. 2015; Zhou et al. 2019; Cho et al. 2021; Gao et al. 2021; Guegan et al. 2022). Notably, increased nuclear localization of YAP was also observed in normal hepatocytes near liver tumors. Peritumoral activation of YAP/TAZ restrains tumor growth; however, the exact mechanism is unclear (Moya et al. 2019). Similarly, YAP/TAZ has been implicated in the activation of cancer-associated fibroblasts (CAFs) (Calvo et al. 2013). Although this has not been demonstrated in liver cancers, it is probable that hepatic CAFs and HSCs surrounding tumors also have high YAP activity.
Oncogenic signaling pathways found in liver cancers, such as growth factor receptor signaling, Wnt, and Notch, have been reported to promote YAP/TAZ activity to synergize cell proliferation and survival following anticancer treatment (Tschaharganeh et al. 2013; Kim et al. 2017; Moon et al. 2022). For example, synergistic activation of Myc/β-catenin, and YAP/TAZ boosts liver cancer cell proliferation and drug resistance (Bisso et al. 2020). Among the specific upstream inputs in liver cancers, one study suggested that COP9 signalosome subunit 6 (CSN6), a member of the constitutive photomorphogenic 9 (COP9) protein complex implicated in tumorigenesis, drives YAP activation in HCC (Li et al. 2024). In the diethylnitrosamine plus CCl4 mouse model, CSN6 antagonizes speckle-type POZ protein (SPOP) and stabilizes hydroxymethylglutaryl-CoA synthase 1 (HMGCS1), resulting in YAP activation through the mevalonate pathway. Hydrodynamic transfection of Myr-Akt with TAZS89A, which increases TAZ activity, accelerates iCCA development via the neurogenic locus notch homolog protein 2 (Notch2) (Cigliano et al. 2022). Pre-existing cirrhosis or advanced fibrosis promotes tumorigenesis in liver cancer (Chakraborty et al. 2017; Roy et al. 2023). In addition to oncogenic signaling pathways, altered matrix stiffness is another major contributor to YAP/TAZ activation, promoting cell proliferation and invasiveness (Yang et al. 2020; Deng et al. 2022). Notably, livers with steatohepatitis that are not considered stiff can promote tumor incidence through altered viscoelasticity, which also causes mechanosensitive YAP/TAZ activation (Fan et al. 2024).
Cell-type specific roles of YAP/TAZ in liver disease
While YAP/TAZ are primarily expressed in BECs in normal liver, their distribution is observed to spread throughout all major cell types in the liver during the progression of MASLD. As liver disease progression is orchestrated by different cells in the liver, dissecting the cell type-specific role of YAP/TAZ is crucial to develop effective treatment and management strategies for MASLD.
Hepatocytes
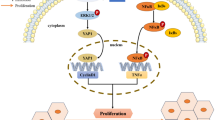
Hepatocytes account for 80% of the liver mass and are involved in the major functions of the liver, such as protein synthesis, metabolism of stored carbohydrates, synthesis of cholesterol and bile juice, and detoxification and excretion of substances (Fabris et al. 2019). YAP/TAZ activity affects hepatocyte proliferation, lipid metabolism, inflammation, and fibrosis, demonstrating its multifaceted effect on liver pathophysiology (Fig. 2). The contribution of YAP to liver homeostasis is represented by the significant hepatomegaly observed in mice with hepatocyte-specific overexpression of active mutant YAP (Dong et al. 2007). After massive tissue loss, YAP/TAZ is activated in a controlled and transient manner in the remaining hepatocytes (Lu et al. 2018). This accelerates hepatocyte proliferation, as demonstrated in mouse models, where partial hepatectomy leads to the rapid nuclear localization of YAP and the subsequent induction of target genes involved in cell proliferation (Wu et al. 2013). Conversely, the deletion of YAP/TAZ in hepatocytes leads to an impaired rate of tissue regeneration. The pharmacological inhibition or genetic deletion of MST1/2 therefore enhances liver regeneration, highlighting the pharmacological value of modulating the Hippo pathway (Lu et al. 2018). Interestingly, however, the mice lacking YAP/TAZ are still able to fully repopulate the liver, implying the existence of additional compensatory machineries (Verboven et al. 2021). Although promoting hepatocyte proliferation is generally beneficial for repopulating tissues, the precise consequences of YAP/TAZ activity depend on the overall health of the liver microenvironment. For example, sustained or excessive YAP overexpression in an injured liver may paradoxically lead to the elimination of proliferating hepatocytes as part of the injury response (Miyamura et al. 2017).

Cell type-specific functions promoted by YAP/TAZ. Upon activation, YAP/TAZ exerts distinct cellular effects in both parenchymal and non-parenchymal cells that comprise the liver. The boxes show the YAP/TAZ-dependent functions that have been identified in the indicated cell type to date
YAP/TAZ dysregulation in hepatocytes plays a multifaceted role in driving steatosis and inflammation, thereby contributing to the progression of MASLD. A key mechanism contributing to steatosis involves the direct regulation of sterol regulatory element-binding proteins (SREBPs) in hepatocytes, leading to increased lipid synthesis and accumulation (Wang et al. 2016). The components of the upstream Hippo pathway also affect this process. For example, inhibition of MST1/2 or LATS1/2 enhances SREBP activity and promotes fatty liver disease (Aylon et al. 2016; Lu et al. 2018). Furthermore, MST1 influences lipid metabolism by promoting the expression of Sirt1, a negative regulator of lipid synthesis (Zhou et al. 2019). Although specific investigations into the link between YAP/TAZ and inflammation in MASLD are warranted, insights from general liver injury models suggest that YAP dysregulation in hepatocytes promotes the production of pro-inflammatory cytokines that exacerbate the disease (Mooring et al. 2020). For example, acute hepatocyte injury caused by intraperitoneal injection of CCl4 induces CYR61 expression through YAP/TAZ to promote pro-inflammatory monocyte infiltration associated with steatohepatitis (Noguchi et al. 2018).
The dysregulation of YAP/TAZ in hepatocytes also contributes to the development of fibrosis. Hepatocyte-specific YAP-knockout mice are resistant to the development of liver fibrosis induced by chronic CCl4 administration (Mooring et al. 2020). In the same study, deletion of both YAP and TAZ did not further inhibit the development of fibrosis, indicating that YAP, but not TAZ, is the primary contributor. Another study has reported that elevated TAZ activity induces the production of Ihh, a potent activator of HSCs, and exaggerates diet-induced fibrosis driven by an FPC diet (Wang et al. 2016). A separate study by the same group demonstrated that hepatocyte-specific N-acetylgalactosamine-conjugated siRNA targeting TAZ was sufficient to ameliorate diet-induced liver fibrosis in mice (Wang et al. 2019). Further studies are warranted to determine how YAP and TAZ differentially contribute to MASLD in humans.
Hepatic stellate cells
HSCs reside in the space of Disse, a region between the liver sinusoidal endothelial cells and hepatocytes (Wake 1971). In the normal liver, HSCs maintain a quiescent state, retaining retinyl esters, representing up to 10% of all liver-resident cells. Lineage-tracing studies have identified HSCs as the primary cause of liver fibrosis, except for cholestatic injury (Mederacke et al. 2013; Iwaisako et al. 2014). Upon liver injury, these cells transdifferentiate into myofibroblast-like cells. Activated HSCs secrete ECM proteins and pro-inflammatory cytokines with enhanced proliferative, migratory, and contractile phenotypes (Tsuchida and Friedman 2017). Accumulating evidence implicates YAP/TAZ as crucial modulators of HSC activation. Transcriptomic analysis has revealed that the upregulation of YAP target genes is an early hallmark of activation, occurring both in cell culture models and in vivo during liver injury (Mannaerts et al. 2015; De Smet et al. 2021). During liver injury, multiple stimuli converge to promote nuclear translocation and transcriptional activity of YAP/TAZ in HSCs. These factors regulate the expression of genes crucial for HSC proliferation (e.g., CTGF and CYR61) and survival (e.g., BIRC5) along with the production of ECM components, such as collagen I and alpha-smooth muscle actin (α-SMA). Mechanistically, stiffening of the ECM, which occurs in fibrotic livers, efficiently promotes YAP/TAZ nuclear localization in HSCs (Caliari et al. 2016). YAP/TAZ also forms an autocrine feed-forward loop in HSCs. For instance, YAP/TAZ can promote transforming growth factor-beta (TGF-β) signaling, which further reinforces YAP/TAZ activity, as demonstrated by enhanced SMAD2/3 phosphorylation upon YAP/TAZ overexpression (Haak et al. 2019). Most importantly, the necessity of YAP/TAZ in the pro-fibrotic function of HSCs has been demonstrated in studies where pharmacological inhibition with verteporfin attenuated HSC activation and collagen production in vitro and reduced liver fibrosis in mouse models (Mannaerts et al. 2015; Martin et al. 2016; Du et al. 2018; Liu et al. 2019).
YAP/TAZ also plays a crucial role in regulating the fate of HSCs. Specifically, YAP enhances the sensitivity to ferroptosis while conferring resistance to senescence. Silencing of YAP and TAZ renders HSCs susceptible to ferroptosis and promotes senescence. Notably, HSC-specific YAP knockout in the mouse leads to an increase in senescent HSCs and offers protection against liver fibrosis, as evidenced by reduced expression of αSMA and decreased collagen accumulation (Du et al. 2023). Collectively, these findings highlight the multifaceted role of YAP/TAZ in HSCs and their potential as therapeutic targets in liver fibrosis.
Hepatic macrophages
Hepatic macrophages comprise resident macrophages known as Kupffer cells (KCs), and macrophages derived from circulating monocytes (Wen et al. 2021). Hepatic macrophages play a crucial role in maintaining liver homeostasis and repair. Although there is little evidence regarding the role of YAP/TAZ in hepatic macrophages, several reports have suggested their regulatory potential (Thomann et al. 2021). In a MASH mouse model, YAP expression in KCs promoted M1 polarization and increased the production of inflammatory cytokines (Song et al. 2020). In addition, the deletion of YAP in KC, HFD, and lipopolysaccharide (LPS) treatment resulted in the inhibition of M1 polarization and an increase in the M2 proportion in the HFD plus LPS model of NASH in mice. However, its role in KCs seems to be restricted to the regulation of the inflammatory response and not to hepatic metabolism, since it does not alter hepatic lipid accumulation (Song et al. 2020). Another study showed that increased YAP expression in KC promotes the secretion of inflammatory cytokines, leading to liver damage (Liu et al. 2024). This demonstrated that YAP plays an important role in the regulation of liver injury and inflammation through interactions between hepatic cells. Furthermore, YAP/TAZ interplay with other signaling pathways, notably Notch1 and TGF-β, in regulating macrophage activation and function in the liver milieu (Feng et al. 2018; Yang et al. 2023). Such crosstalk may contribute to fine-tuning the balance between the pro- and anti-inflammatory responses.
Biliary epithelial cells
BECs, also known as cholangiocytes, line the bile ducts in the liver and play a critical role in liver function and pathology (O'Hara et al. 2013). Physiologically, BECs regulate bile acid composition and flow (Hrncir and Gracz 2023). Disruption in bile acid homeostasis upon malfunction of BECs can contribute to liver injury and inflammation, which are central to the pathogenesis of MASLD (Chiang and Ferrell 2018). Excess bile acids exert direct cytotoxic effects that can exacerbate hepatocyte injury in steatohepatitis and influence lipid metabolism. This inflammatory response can further drive hepatocyte damage and fibrosis, which are hallmarks of steatohepatitis. Moreover, in conditions where chronic inflammation is present, activated BECs secrete profibrogenic cytokines and growth factors that promote the activation of hepatic stellate cells, ultimately leading to fibrosis (Poulsen et al. 2022).
Enforced expression of S127-YAP or deletion of NF2 causes hepatocyte dedifferentiation into Sox9- or CK19-expressing BEC-like cells. This process is dependent on Notch signaling. Notably, dedifferentiated cells were able to re-differentiate into hepatocytes when the expression of S127A-YAP was reversed, indicating a role for YAP in hepatic cell plasticity (Yimlamai et al. 2014). BECs are epithelial cells that surround the biliary tubules and transport bile from the liver to the small intestine (Tabibian et al. 2013). Recent studies have demonstrated the significance of YAP/TAZ in the regulation of BECs proliferation and maintenance. For instance, deletion of YAP in adult liver cells results in bile duct paucity and delayed regeneration or hepatocyte necrosis (Airik et al. 2020; Verboven et al. 2021). The role of YAP in early biliary development is important because YAP is required for the differentiation of the second layer of BECs (Molina et al. 2021, 2022). Notably, TAZ overexpression promotes the conversion of lipid-rich hepatocytes into fully malignant BECs in combination with AKT (Cigliano et al. 2022). YAP has been reported to be a key feature of CCA, and YAP activity is associated with a poor overall survival rate (Pei et al. 2015; Sugimachi et al. 2017; Toth et al. 2021).
Drug candidates targeting YAP/TAZ in the liver
The discovery and development of small-molecule inhibitors and activators targeting the YAP/TAZ signaling pathway have emerged as a promising strategy in cancer research. A previous study reported the discovery of a small molecule inhibitor called verteporfin, the most widely used to date, which effectively suppressed YAP/TAZ signaling and was confirmed to inhibit YAP-induced liver overgrowth (Liu-Chittenden et al. 2012). Pharmacological inhibition of YAP by verteporfin enhances the sensitivity of cancer cells to sorafenib and overcomes tumorigenesis (Sun et al. 2021). The combination of sorafenib and verteporfin substantially inhibits HCC growth. CA3 is another small molecule that inhibits YAP and has been proposed as an anticancer agent against HCC (Han et al. 2022). Enhanced expression of cyclin-dependent kinase 6 (CDK6) has been proposed as one of the main causes of lenvatinib resistance in HCC (Leung et al. 2023; Jing et al. 2023). A recent study showed that CA3 effectively inhibits CDK6 expression, demonstrating the potential of YAP/TAZ inhibitors in combination therapy for HCC (Leung et al. 2023). As a common mechanism, these inhibitors prevent the interaction between YAP/TAZ and TEAD, thereby inhibiting YAP/TAZ-mediated transcription (Table 1). In addition, GalNAc-siTAZ can be another viable therapeutic given the current acceptance of nucleic acid medicines. Conjugation of a GalNAc ligand targeting the asialoglycoprotein receptor with chemically modified siRNA resulted in robust and durable RNAi-mediated TAZ silencing specifically in hepatocytes (Debacker et al. 2020). Systemic injection of GalNAc-siTAZ reduced liver inflammation, hepatocyte damage, liver fibrosis, and profibrotic mediator expression compared to the GalNAc-control group (Wang et al. 2023).
Therapeutic modulation of YAP/TAZ is expected to require a delicate balance. The challenge lies in inhibiting the pathological amplification of YAP/TAZ signaling in liver cells without impairing the regenerative capacity of the liver or stimulating regeneration without promoting tumorigenesis. The development of dual modulators that can inhibit or activate YAP/TAZ signaling in a context-dependent manner may provide more viable therapeutic options. While both inhibitors and activators have the potential to contribute substantially to the management of liver tumors and other complications, such as MASLD, the discovery and optimization of such modulators of YAP/TAZ signaling represents a promising frontier in the treatment of liver disease.
Conclusion
The intricate interplay between YAP/TAZ and liver dysfunction emphasizes their significance in MASLD. Based on their regulation of lipid metabolism, inflammation, and fibrosis, and their impact on different types of cells in the liver, YAP/TAZ have emerged as central players governing diverse aspects of liver homeostasis and disease progression. In MASLD, aberrant activation of YAP/TAZ is intricately linked to hepatocyte stress, steatosis, inflammation, and fibrosis, representing a critical nexus throughout disease progression. Furthermore, their dysregulation contributes to the development and progression of MASLD and primary liver cancer. Understanding the cell type-specific roles of YAP and TAZ in different liver cell populations, including hepatocytes, HSCs, hepatic macrophages, and BECs, is crucial to deciphering their diverse functions in liver physiology and pathology.
It is important to note that this review should be interpreted with caution. In particular, the experimental models using mice may not fully mimic the human condition of MASLD, potentially limiting the applicability of the findings to clinical settings. The complex interplay of (epi)genetic and environmental factors in clinical settings may introduce unaccounted variables that could influence the outcomes that cannot be explained by the currently used experimental models. In addition, given the incomplete understanding of the mechanism by which YAP/TAZ are redistributed and activated in different hepatic cells during MASLD, it is inadvisable to rely on a single general mechanism for YAP/TAZ regulation. In a similar context, considering the importance of the interplay between YAP/TAZ and MASLD progression, identification of their upstream input(s) will be the next aim for developing therapeutic modulators. For instance, studies on the signaling pathways linked to YAP/TAZ activity have identified the mechanical properties of the extracellular matrix as an emerging target. Consequently, the regulation of the mechanosensitive activation of YAP/TAZ may be an important research topic for understanding the differences between healthy and diseased tissues in MASLD.
In summary, the elucidation of YAP/TAZ-mediated molecular mechanisms and their intricate involvement in liver pathophysiology not only enhances our understanding of liver diseases but also paves the way for the development of innovative therapeutic strategies aimed at addressing unmet clinical needs in the field of hepatology.
References
Abu Rmilah A, Zhou W, Nelson E, Lin L, Amiot B, Nyberg SL (2019) Understanding the marvels behind liver regeneration. Wiley Interdiscip Rev Dev Biol 8:e340. https://doi.org/10.1002/wdev.340
Airik M, Schuler M, Mccourt B, Weiss AC, Herdman N, Ludtke TH, Widmeier E, Stolz DB, Nejak-Bowen KN, Yimlamai D, Wu YL, Kispert A, Airik R, Hildebrandt F (2020) Loss of Anks6 leads to YAP deficiency and liver abnormalities. Hum Mol Genet 29:3064–3080. https://doi.org/10.1093/hmg/ddaa197
Asrani SK, Devarbhavi H, Eaton J, Kamath PS (2019) Burden of liver diseases in the world. J Hepatol 70:151–171. https://doi.org/10.1016/j.jhep.2018.09.014
Aylon Y, Gershoni A, Rotkopf R, Biton IE, Porat Z, Koh AP, Sun X, Lee Y, Fiel MI, Hoshida Y, Friedman SL, Johnson RL, Oren M (2016) The LATS2 tumor suppressor inhibits SREBP and suppresses hepatic cholesterol accumulation. Genes Dev 30:786–797. https://doi.org/10.1101/gad.274167.115
Bisso A, Filipuzzi M, Gamarra Figueroa GP, Brumana G, Biagioni F, Doni M, Ceccotti G, Tanaskovic N, Morelli MJ, Pendino V, Chiacchiera F, Pasini D, Olivero D, Campaner S, Sabo A, Amati B (2020) Cooperation between MYC and beta-Catenin in liver tumorigenesis requires Yap/Taz. Hepatology 72:1430–1443. https://doi.org/10.1002/hep.31120
Brahma MK, Gilglioni EH, Zhou L, Trepo E, Chen P, Gurzov EN (2021) Oxidative stress in obesity-associated hepatocellular carcinoma: sources signaling and therapeutic challenges. Oncogene 40:5155–5167. https://doi.org/10.1038/s41388-021-01950-y
Caliari SR, Perepelyuk M, Cosgrove BD, Tsai SJ, Lee GY, Mauck RL, Wells RG, Burdick JA (2016) Stiffening hydrogels for investigating the dynamics of hepatic stellate cell mechanotransduction during myofibroblast activation. Sci Rep 6:21387. https://doi.org/10.1038/srep21387
Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E (2013) Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 15:637–646. https://doi.org/10.1038/ncb2756
Chakraborty S, Njah K, Pobbati AV, Lim YB, Raju A, Lakshmanan M, Tergaonkar V, Lim CT, Hong W (2017) Agrin as a mechanotransduction signal regulating YAP through the hippo pathway. Cell Rep 18:2464–2479. https://doi.org/10.1016/j.celrep.2017.02.041
Chen L, Chan SW, Zhang X, Walsh M, Lim CJ, Hong W, Song H (2010) Structural basis of YAP recognition by TEAD4 in the hippo pathway. Genes Dev 24:290–300. https://doi.org/10.1101/gad.1865310
Chen Z, Yu R, Xiong Y, Du F, Zhu S (2017) A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis 16:203. https://doi.org/10.1186/s12944-017-0572-9
Chen P, Luo Q, Huang C, Gao Q, Li L, Chen J, Chen B, Liu W, Zeng W, Chen Z (2018) Pathogenesis of non-alcoholic fatty liver disease mediated by YAP. Hepatol Int 12:26–36. https://doi.org/10.1007/s12072-017-9841-y
Chiang JYL, Ferrell JM (2018) Bile acid metabolism in liver pathobiology. Gene Expr 18:71–87. https://doi.org/10.3727/105221618X15156018385515
Cho K, Ro SW, Lee HW, Moon H, Han S, Kim HR, Ahn SH, Park JY, Kim DY (2021) YAP/TAZ suppress drug penetration into hepatocellular carcinoma through stromal activation. Hepatology 74:2605–2621. https://doi.org/10.1002/hep.32000
Cigliano A, Zhang S, Ribback S, Steinmann S, Sini M, Ament CE, Utpatel K, Song X, Wang J, Pilo MG, Berger F, Wang H, Tao J, Li X, Pes GM, Mancarella S, Giannelli G, Dombrowski F, Evert M, Calvisi DF, Chen X, Evert K (2022) The Hippo pathway effector TAZ induces intrahepatic cholangiocarcinoma in mice and is ubiquitously activated in the human disease. J Exp Clin Cancer Res 41:192. https://doi.org/10.1186/s13046-022-02394-2
Daoud F, Holmberg J, Alajbegovic A, Grossi M, Rippe C, Sward K, Albinsson S (2021) Inducible deletion of YAP and TAZ in adult mouse smooth muscle causes rapid and lethal colonic pseudo-obstruction. Cell Mol Gastroenterol Hepatol 11:623–637. https://doi.org/10.1016/j.jcmgh.2020.09.014
De Smet V, Eysackers N, Merens V, Kazemzadeh Dastjerd M, Halder G, Verhulst S, Mannaerts I, Van Grunsven LA (2021) Initiation of hepatic stellate cell activation extends into chronic liver disease. Cell Death Dis 12:1110. https://doi.org/10.1038/s41419-021-04377-1
Debacker AJ, Voutila J, Catley M, Blakey D, Habib N (2020) Delivery of oligonucleotides to the liver with GalNAc: from research to registered therapeutic drug. Mol Ther 28:1759–1771. https://doi.org/10.1016/j.ymthe.2020.06.015
Deng B, Zhao Z, Kong W, Han C, Shen X, Zhou C (2022) Biological role of matrix stiffness in tumor growth and treatment. J Transl Med 20:540. https://doi.org/10.1186/s12967-022-03768-y
Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D (2007) Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130:1120–1133. https://doi.org/10.1016/j.cell.2007.07.019
Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M, Dalton GD, Thelen E, Rizi BS, Jung Y, Diehl AM (2018) Hedgehog-YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology 154(1465–1479):e13. https://doi.org/10.1053/j.gastro.2017.12.022
Du K, Maeso-Diaz R, Oh SH, Wang E, Chen T, Pan C, Xiang K, Dutta RK, Wang XF, Chi JT, Diehl AM (2023) Targeting YAP-mediated HSC death susceptibility and senescence for treatment of liver fibrosis. Hepatology 77:1998–2015. https://doi.org/10.1097/HEP.0000000000000326
Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S (2011) Role of YAP/TAZ in mechanotransduction. Nature 474:179–183. https://doi.org/10.1038/nature10137
Ebrahimi F, Hagstrom H, Sun J, Bergman D, Shang Y, Yang W, Roelstraete B, Ludvigsson JF (2023) Familial coaggregation of MASLD with hepatocellular carcinoma and adverse liver outcomes: nationwide multigenerational cohort study. J Hepatol 79:1374–1384. https://doi.org/10.1016/j.jhep.2023.08.018
Eslam M, Sanyal AJ, George J & International Consensus P (2020) MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 158(1999–2014):e1. https://doi.org/10.1053/j.gastro.2019.11.312
Fabris G, Dumortier O, Pisani DF, Gautier N, Van Obberghen E (2019) Amino acid-induced regulation of hepatocyte growth: possible role of Drosha. Cell Death Dis 10:566. https://doi.org/10.1038/s41419-019-1779-7
Fan W, Adebowale K, Vancza L, Li Y, Rabbi MF, Kunimoto K, Chen D, Mozes G, Chiu DK, Li Y, Tao J, Wei Y, Adeniji N, Brunsing RL, Dhanasekaran R, Singhi A, Geller D, Lo SH, Hodgson L, Engleman EG, Charville GW, Charu V, Monga SP, Kim T, Wells RG, Chaudhuri O, Torok NJ (2024) Matrix viscoelasticity promotes liver cancer progression in the pre-cirrhotic liver. Nature 626:635–642. https://doi.org/10.1038/s41586-023-06991-9
Farese RV Jr, Zechner R, Newgard CB, Walther TC (2012) The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab 15:570–573. https://doi.org/10.1016/j.cmet.2012.03.004
Feng Y, Liang Y, Zhu X, Wang M, Gui Y, Lu Q, Gu M, Xue X, Sun X, He W, Yang J, Johnson RL, Dai C (2018) The signaling protein Wnt5a promotes TGFbeta1-mediated macrophage polarization and kidney fibrosis by inducing the transcriptional regulators Yap/Taz. J Biol Chem 293:19290–19302. https://doi.org/10.1074/jbc.RA118.005457
Friedman SL (2024) Hepatic fibrosis and cancer: the silent threats of metabolic syndrome. Diabetes Metab J 48:161–169. https://doi.org/10.4093/dmj.2023.0240
Gao R, Kalathur RKR, Coto-Llerena M, Ercan C, Buechel D, Shuang S, Piscuoglio S, Dill MT, Camargo FD, Christofori G, Tang F (2021) YAP/TAZ and ATF4 drive resistance to Sorafenib in hepatocellular carcinoma by preventing ferroptosis. EMBO Mol Med 13:e14351. https://doi.org/10.15252/emmm.202114351
Guegan JP, Lapouge M, Voisin L, Saba-El-Leil MK, Tanguay PL, Levesque K, Bregeon J, Mes-Masson AM, Lamarre D, Haibe-Kains B, Trinh VQ, Soucy G, Bilodeau M, Meloche S (2022) Signaling by the tyrosine kinase Yes promotes liver cancer development. Sci Signal 15:ebj4743. https://doi.org/10.1126/scisignal.abj4743
Guo Y, Pan Q, Zhang J, Xu X, Liu X, Wang Q, Yi R, Xie X, Yao L, Liu W, Shen L (2015) Functional and clinical evidence that TAZ is a candidate oncogene in hepatocellular carcinoma. J Cell Biochem 116:2465–2475. https://doi.org/10.1002/jcb.25117
Haak AJ, Kostallari E, Sicard D, Ligresti G, Choi KM, Caporarello N, Jones DL, Tan Q, Meridew J, Diaz Espinosa AM, Aravamudhan A, Maiers JL, Britt RD Jr, Roden AC, Pabelick CM, Prakash YS, Nouraie SM, Li X, Zhang Y, Kass DJ, Lagares D, Tager AM, Varelas X, Shah VH, Tschumperlin DJ (2019) Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aau6296
Han SX, Bai E, Jin GH, He CC, Guo XJ, Wang LJ, Li M, Ying X, Zhu Q (2014) Expression and clinical significance of YAP TAZ and AREG in hepatocellular carcinoma. J Immunol Res 2014:261365. https://doi.org/10.1155/2014/261365
Han S, Lim JY, Cho K, Lee HW, Park JY, Ro SW, Kim KS, Seo HR, Kim DY (2022) Anti-cancer effects of YAP inhibitor (CA3) in combination with Sorafenib against hepatocellular carcinoma (HCC) in patient-derived multicellular tumor spheroid models (MCTS). Cancers (Basel). https://doi.org/10.3390/cancers14112733
Harrison SA, Bedossa P, Guy CD, Schattenberg JM, Loomba R, Taub R, Labriola D, Moussa SE, Neff GW, Rinella ME, Anstee QM, Abdelmalek MF, Younossi Z, Baum SJ, Francque S, Charlton MR, Newsome PN, Lanthier N, Schiefke I, Mangia A, Pericas JM, Patil R, Sanyal AJ, Noureddin M, Bansal MB, Alkhouri N, Castera L, Rudraraju M, Ratziu V, Investigators M-N (2024) A phase 3 randomized controlled trial of resmetirom in NASH with liver fibrosis. N Engl J Med 390:497–509. https://doi.org/10.1056/NEJMoa2309000
Hossain Z, Ali SM, Ko HL, Xu J, Ng CP, Guo K, Qi Z, Ponniah S, Hong W, Hunziker W (2007) Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc Natl Acad Sci USA 104:1631–1636. https://doi.org/10.1073/pnas.0605266104
Hrncir HR, Gracz AD (2023) Cellular and transcriptional heterogeneity in the intrahepatic biliary epithelium. Gastro Hep Adv 2:108–120. https://doi.org/10.1016/j.gastha.2022.07.015
Huh HD, Kim DH, Jeong HS, Park HW (2019) Regulation of TEAD transcription factors in cancer biology. Cells. https://doi.org/10.3390/cells8060600
Hyun J, Oh SH, Premont RT, Guy CD, Berg CL, Diehl AM (2019) Dysregulated activation of fetal liver programme in acute liver failure. Gut 68:1076–1087. https://doi.org/10.1136/gutjnl-2018-317603
Iwaisako K, Jiang C, Zhang M, Cong M, Moore-Morris TJ, Park TJ, Liu X, Xu J, Wang P, Paik YH, Meng F, Asagiri M, Murray LA, Hofmann AF, Iida T, Glass CK, Brenner DA, Kisseleva T (2014) Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci USA 111:E3297–E3305. https://doi.org/10.1073/pnas.1400062111
Jin C, Luo Y, Liang Z, Li X, Kolat D, Zhao L, Xiong W (2023) Crucial role of the transcription factors family activator protein 2 in cancer: current clue and views. J Transl Med 21:371. https://doi.org/10.1186/s12967-023-04189-1
Jing F, Li X, Jiang H, Sun J, Guo Q (2023) Combating drug resistance in hepatocellular carcinoma: no awareness today no action tomorrow. Biomed Pharmacother 167:115561. https://doi.org/10.1016/j.biopha.2023.115561
Kim W, Khan SK, Gvozdenovic-Jeremic J, Kim Y, Dahlman J, Kim H, Park O, Ishitani T, Jho EH, Gao B, Yang Y (2017) Hippo signaling interactions with Wnt/beta-catenin and Notch signaling repress liver tumorigenesis. J Clin Invest 127:137–152. https://doi.org/10.1172/JCI88486
Koo JH, Plouffe SW, Meng Z, Lee DH, Yang D, Lim DS, Wang CY, Guan KL (2020) Induction of AP-1 by YAP/TAZ contributes to cell proliferation and organ growth. Genes Dev 34:72–86. https://doi.org/10.1101/gad.331546.119
Kwon A, Lee NY, Yu JH, Choi MG, Park J, Koo JH (2024) Mitochondrial stress activates YAP/TAZ through RhoA oxidation to promote liver injury. Cell Death Dis 15:51. https://doi.org/10.1038/s41419-024-06448-5
Lee YA, Noon LA, Akat KM, Ybanez MD, Lee TF, Berres ML, Fujiwara N, Goossens N, Chou HI, Parvin-Nejad FP, Khambu B, Kramer EGM, Gordon R, Pfleger C, Germain D, John GR, Campbell KN, Yue Z, Yin XM, Cuervo AM, Czaja MJ, Fiel MI, Hoshida Y, Friedman SL (2018) Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nat Commun 9:4962. https://doi.org/10.1038/s41467-018-07338-z
Leung CON, Yang Y, Leung RWH, So KKH, Guo HJ, Lei MML, Muliawan GK, Gao Y, Yu QQ, Yun JP, Ma S, Zhao Q, Lee TKW (2023) Broad-spectrum kinome profiling identifies CDK6 upregulation as a driver of lenvatinib resistance in hepatocellular carcinoma. Nat Commun 14:6699. https://doi.org/10.1038/s41467-023-42360-w
Li H, Wolfe A, Septer S, Edwards G, Zhong X, Abdulkarim AB, Ranganathan S, Apte U (2012) Deregulation of Hippo kinase signalling in human hepatic malignancies. Liver Int 32:38–47. https://doi.org/10.1111/j.1478-3231.2011.02646.x
Li K, Zhang J, Lyu H, Yang J, Wei W, Wang Y, Luo H, Zhang Y, Jiang X, Yi H, Wang M, Zhang C, Wu K, Xiao L, Wen W, Xu H, Li G, Wan Y, Yang F, Yang R, Fu X, Qin B, Zhou Z, Zhang H, Lee MH (2024) CSN6-SPOP-HMGCS1 axis promotes hepatocellular carcinoma progression via YAP1 activation. Adv Sci (Weinh). https://doi.org/10.1002/advs.202306827
Liu Y, Lu T, Zhang C, Xu J, Xue Z, Busuttil RW, Xu N, Xia Q, Kupiec-Weglinski JW, Ji H (2019) Activation of YAP attenuates hepatic damage and fibrosis in liver ischemia-reperfusion injury. J Hepatol 71:719–730. https://doi.org/10.1016/j.jhep.2019.05.029
Liu K, Wehling L, Wan S, Weiler SME, Toth M, Ibberson D, Marhenke S, Ali A, Lam M, Guo T, Pinna F, Pedrini F, Damle-Vartak A, Dropmann A, Rose F, Colucci S, Cheng W, Bissinger M, Schmitt J, Birner P, Poth T, Angel P, Dooley S, Muckenthaler MU, Longerich T, Vogel A, Heikenwalder M, Schirmacher P, Breuhahn K (2024) Dynamic YAP expression in the non-parenchymal liver cell compartment controls heterologous cell communication. Cell Mol Life Sci 81:115. https://doi.org/10.1007/s00018-024-05126-1
Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D (2012) Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev 26:1300–1305. https://doi.org/10.1101/gad.192856.112
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J, Finn RS (2021) Hepatocellular carcinoma. Nat Rev Dis Primers 7:6. https://doi.org/10.1038/s41572-020-00240-3
Lu L, Finegold MJ, Johnson RL (2018) Hippo pathway coactivators Yap and Taz are required to coordinate mammalian liver regeneration. Exp Mol Med 50:e423. https://doi.org/10.1038/emm.2017.205
Machado MV, Michelotti GA, Pereira TA, Xie G, Premont R, Cortez-Pinto H, Diehl AM (2015) Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease. J Hepatol 63:962–970. https://doi.org/10.1016/j.jhep.2015.05.031
Maglic D, Schlegelmilch K, Dost AF, Panero R, Dill MT, Calogero RA, Camargo FD (2018) YAP-TEAD signaling promotes basal cell carcinoma development via a c-JUN/AP1 axis. EMBO J. https://doi.org/10.15252/embj.201798642
Makita R, Uchijima Y, Nishiyama K, Amano T, Chen Q, Takeuchi T, Mitani A, Nagase T, Yatomi Y, Aburatani H, Nakagawa O, Small EV, Cobo-Stark P, Igarashi P, Murakami M, Tominaga J, Sato T, Asano T, Kurihara Y, Kurihara H (2008) Multiple renal cysts urinary concentration defects and pulmonary emphysematous changes in mice lacking TAZ. Am J Physiol Renal Physiol 294:F542–F553. https://doi.org/10.1152/ajprenal.00201.2007
Mannaerts I, Leite SB, Verhulst S, Claerhout S, Eysackers N, Thoen LF, Hoorens A, Reynaert H, Halder G, Van Grunsven LA (2015) The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol 63:679–688. https://doi.org/10.1016/j.jhep.2015.04.011
Martin K, Pritchett J, Llewellyn J, Mullan AF, Athwal VS, Dobie R, Harvey E, Zeef L, Farrow S, Streuli C, Henderson NC, Friedman SL, Hanley NA, Piper Hanley K (2016) PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat Commun 7:12502. https://doi.org/10.1038/ncomms12502
Matchett KP, Paris J, Teichmann SA, Henderson NC (2024) Spatial genomics: mapping human steatotic liver disease. Nat Rev Gastroenterol Hepatol. https://doi.org/10.1038/s41575-024-00915-2
Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, Pradere JP, Schwabe RF (2013) Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 4:2823. https://doi.org/10.1038/ncomms3823
Miyamura N, Hata S, Itoh T, Tanaka M, Nishio M, Itoh M, Ogawa Y, Terai S, Sakaida I, Suzuki A, Miyajima A, Nishina H (2017) Erratum: YAP determines the cell fate of injured mouse hepatocytes in vivo. Nat Commun 8:16146. https://doi.org/10.1038/ncomms16146
Molina LM, Zhu J, Li Q, Pradhan-Sundd T, Krutsenko Y, Sayed K, Jenkins N, Vats R, Bhushan B, Ko S, Hu S, Poddar M, Singh S, Tao J, Sundd P, Singhi A, Watkins S, Ma X, Benos PV, Feranchak A, Michalopoulos G, Nejak-Bowen K, Watson A, Bell A, Monga SP (2021) Compensatory hepatic adaptation accompanies permanent absence of intrahepatic biliary network due to YAP1 loss in liver progenitors. Cell Rep 36:109310. https://doi.org/10.1016/j.celrep.2021.109310
Molina L, Nejak-Bowen K, Monga SP (2022) Role of YAP1 signaling in biliary development repair and disease. Semin Liver Dis 42:17–33. https://doi.org/10.1055/s-0041-1742277
Moon H, Park H, Chae MJ, Choi HJ, Kim DY, Ro SW (2022) Activated TAZ induces liver cancer in collaboration with EGFR/HER2 signaling pathways. BMC Cancer 22:423. https://doi.org/10.1186/s12885-022-09516-1
Mooring M, Fowl BH, Lum SZC, Liu Y, Yao K, Softic S, Kirchner R, Bernstein A, Singhi AD, Jay DG, Kahn CR, Camargo FD, Yimlamai D (2020) Hepatocyte stress increases expression of Yes-associated protein and transcriptional coactivator With PDZ-binding Motif in hepatocytes to promote parenchymal inflammation and fibrosis. Hepatology 71:1813–1830. https://doi.org/10.1002/hep.30928
Morin-Kensicki EM, Boone BN, Howell M, Stonebraker JR, Teed J, Alb JG, Magnuson TR, O’neal W & Milgram SL, (2006) Defects in yolk sac vasculogenesis chorioallantoic fusion and embryonic axis elongation in mice with targeted disruption of Yap65. Mol Cell Biol 26:77–87. https://doi.org/10.1128/MCB.26.1.77-87.2006
Moya IM, Castaldo SA, Van Den Mooter L, Soheily S, Sansores-Garcia L, Jacobs J, Mannaerts I, Xie J, Verboven E, Hillen H, Alguero-Nadal A, Karaman R, Van Haele M, Kowalczyk W, De Waegeneer M, Verhulst S, Karras P, Van Huffel L, Zender L, Marine JC, Roskams T, Johnson R, Aerts S, Van Grunsven LA, Halder G (2019) Peritumoral activation of the Hippo pathway effectors YAP and TAZ suppresses liver cancer in mice. Science 366:1029–1034. https://doi.org/10.1126/science.aaw9886
Nassir F, Rector RS, Hammoud GM, Ibdah JA (2015) Pathogenesis and prevention of hepatic steatosis. Gastroenterol Hepatol (N Y) 11:167–175
Noguchi S, Saito A, Nagase T (2018) YAP/TAZ signaling as a molecular link between fibrosis and cancer. Int J Mol Sci. https://doi.org/10.3390/ijms19113674
O’hara SP, Tabibian JH, Splinter PL, Larusso NF, (2013) The dynamic biliary epithelia: molecules pathways and disease. J Hepatol 58:575–582. https://doi.org/10.1016/j.jhep.2012.10.011
Pei T, Li Y, Wang J, Wang H, Liang Y, Shi H, Sun B, Yin D, Sun J, Song R, Pan S, Sun Y, Jiang H, Zheng T, Liu L (2015) YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget 6:17206–20. https://doi.org/10.18632/oncotarget.4043
Peng C, Stewart AG, Woodman OL, Ritchie RH, Qin CX (2020) Non-alcoholic steatohepatitis: a review of its mechanism models and medical treatments. Front Pharmacol 11:603926. https://doi.org/10.3389/fphar.2020.603926
Plouffe SW, Lin KC, Moore JL 3rd, Tan FE, Ma S, Ye Z, Qiu Y, Ren B, Guan KL (2018) The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. J Biol Chem 293:11230–11240. https://doi.org/10.1074/jbc.RA118.002715
Poulsen KL, Cajigas-Du Ross CK, Chaney JK, Nagy LE (2022) Role of the chemokine system in liver fibrosis: a narrative review. Dig Med Res. https://doi.org/10.21037/dmr-21-87
Reggiani F, Gobbi G, Ciarrocchi A, Sancisi V (2021) YAP and TAZ are not identical twins. Trends Biochem Sci 46:154–168. https://doi.org/10.1016/j.tibs.2020.08.012
Reyes-Gordillo K, Shah R, Muriel P (2017) Oxidative stress and inflammation in hepatic diseases: current and future therapy. Oxid Med Cell Longev 2017:3140673. https://doi.org/10.1155/2017/3140673
Roy AM, Iyer R, Chakraborty S (2023) The extracellular matrix in hepatocellular carcinoma: mechanisms and therapeutic vulnerability. Cell Rep Med 4:101170. https://doi.org/10.1016/j.xcrm.2023.101170
Salloum S, Jeyarajan AJ, Kruger AJ, Holmes JA, Shao T, Sojoodi M, Kim MH, Zhuo Z, Shroff SG, Kassa A, Corey KE, Khan SK, Lin W, Alatrakchi N, EaK S, Chung RT (2021) Fatty acids activate the transcriptional coactivator YAP1 to promote liver fibrosis via p38 mitogen-activated protein kinase. Cell Mol Gastroenterol Hepatol 12:1297–1310. https://doi.org/10.1016/j.jcmgh.2021.06.003
Sohn BH, Shim JJ, Kim SB, Jang KY, Kim SM, Kim JH, Hwang JE, Jang HJ, Lee HS, Kim SC, Jeong W, Kim SS, Park ES, Heo J, Kim YJ, Kim DG, Leem SH, Kaseb A, Hassan MM, Cha M, Chu IS, Johnson RL, Park YY, Lee JS (2016) Inactivation of Hippo pathway is significantly associated with poor prognosis in hepatocellular carcinoma. Clin Cancer Res 22:1256–1264. https://doi.org/10.1158/1078-0432.CCR-15-1447
Song K, Kwon H, Han C, Chen W, Zhang J, Ma W, Dash S, Gandhi CR, Wu T (2020) Yes-associated protein in Kupffer cells enhances the production of proinflammatory cytokines and promotes the development of nonalcoholic steatohepatitis. Hepatology 72:72–87. https://doi.org/10.1002/hep.30990
Sugihara T, Isomoto H, Gores G, Smoot R (2019) YAP and the Hippo pathway in cholangiocarcinoma. J Gastroenterol 54:485–491. https://doi.org/10.1007/s00535-019-01563-z
Sugimachi K, Nishio M, Aishima S, Kuroda Y, Iguchi T, Komatsu H, Hirata H, Sakimura S, Eguchi H, Bekki Y, Takenaka K, Maehara Y, Suzuki A, Mimori K (2017) Altered expression of Hippo signaling pathway molecules in intrahepatic cholangiocarcinoma. Oncology 93:67–74. https://doi.org/10.1159/000463390
Sun T, Mao W, Peng H, Wang Q, Jiao L (2021) YAP promotes sorafenib resistance in hepatocellular carcinoma by upregulating survivin. Cell Oncol (dordr) 44:689–699. https://doi.org/10.1007/s13402-021-00595-z
Tabibian JH, Masyuk AI, Masyuk TV, O’hara SP & Larusso NF, (2013) Physiology of cholangiocytes. Compr Physiol 3:541–565. https://doi.org/10.1002/cphy.c120019
Thomann S, Weiler SME, Wei T, Sticht C, De La Torre C, Toth M, Rose F, Tang Y, Ritz T, Ball C, Glimm H, Ryschich E, Schirmacher P, Breuhahn K (2021) YAP-induced Ccl2 expression is associated with a switch in hepatic macrophage identity and vascular remodelling in liver cancer. Liver Int 41:3011–3023. https://doi.org/10.1111/liv.15048
Toth M, Wehling L, Thiess L, Rose F, Schmitt J, Weiler SME, Sticht C, De La Torre C, Rausch M, Albrecht T, Grabe N, Duwe L, Andersen JB, Kohler BC, Springfeld C, Mehrabi A, Kulu Y, Schirmacher P, Roessler S, Goeppert B, Breuhahn K (2021) Co-expression of YAP and TAZ associates with chromosomal instability in human cholangiocarcinoma. BMC Cancer 21:1079. https://doi.org/10.1186/s12885-021-08794-5
Tschaharganeh DF, Chen X, Latzko P, Malz M, Gaida MM, Felix K, Ladu S, Singer S, Pinna F, Gretz N, Sticht C, Tomasi ML, Delogu S, Evert M, Fan B, Ribback S, Jiang L, Brozzetti S, Bergmann F, Dombrowski F, Schirmacher P, Calvisi DF, Breuhahn K (2013) Yes-associated protein up-regulates Jagged-1 and activates the Notch pathway in human hepatocellular carcinoma. Gastroenterology 144(1530–1542):e12. https://doi.org/10.1053/j.gastro.2013.02.009
Tsuchida T, Friedman SL (2017) Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 14:397–411. https://doi.org/10.1038/nrgastro.2017.38
Vassilev A, Kaneko KJ, Shu H, Zhao Y, Depamphilis ML (2001) TEAD/TEF transcription factors utilize the activation domain of YAP65 a Src/Yes-associated protein localized in the cytoplasm. Genes Dev 15:1229–1241. https://doi.org/10.1101/gad.888601
Verboven E, Moya IM, Sansores-Garcia L, Xie J, Hillen H, Kowalczyk W, Vella G, Verhulst S, Castaldo SA, Alguero-Nadal A, Romanelli L, Mercader-Celma C, Souza NA, Soheily S, Van Huffel L, Van Brussel T, Lambrechts D, Roskams T, Lemaigre FP, Bergers G, Van Grunsven LA, Halder G (2021) Regeneration defects in Yap and Taz mutant mouse livers are caused by bile duct disruption and cholestasis. Gastroenterology 160:847–862. https://doi.org/10.1053/j.gastro.2020.10.035
Wake K (1971) “Sternzellen” in the liver: perisinusoidal cells with special reference to storage of vitamin A. Am J Anat 132:429–462. https://doi.org/10.1002/aja.1001320404
Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, Masia R, Chung RT, Lefkowitch JH, Schwabe RF, Tabas I (2016) Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab 24:848–862. https://doi.org/10.1016/j.cmet.2016.09.016
Wang X, Sommerfeld MR, Jahn-Hofmann K, Cai B, Filliol A, Remotti HE, Schwabe RF, Kannt A, Tabas I (2019) A therapeutic silencing RNA targeting hepatocyte TAZ prevents and reverses fibrosis in nonalcoholic steatohepatitis in mice. Hepatol Commun 3:1221–1234. https://doi.org/10.1002/hep4.1405
Wang X, Cai B, Yang X, Sonubi OO, Zheng Z, Ramakrishnan R, Shi H, Valenti L, Pajvani UB, Sandhu J, Infante RE, Radhakrishnan A, Covey DF, Guan KL, Buck J, Levin LR, Tontonoz P, Schwabe RF, Tabas I (2020) Cholesterol stabilizes TAZ in hepatocytes to promote experimental non-alcoholic steatohepatitis. Cell Metab 31(969–986):e7. https://doi.org/10.1016/j.cmet.2020.03.010
Wang X, Moore MP, Shi H, Miyata Y, Donnelly SK, Radiloff DR, Tabas I (2023) Hepatocyte-targeted siTAZ therapy lowers liver fibrosis in NASH diet-fed chimeric mice with hepatocyte-humanized livers. Mol Ther Methods Clin Dev 31:101165. https://doi.org/10.1016/j.omtm.2023.101165
Wen Y, Lambrecht J, Ju C, Tacke F (2021) Hepatic macrophages in liver homeostasis and diseases-diversity plasticity and therapeutic opportunities. Cell Mol Immunol 18:45–56. https://doi.org/10.1038/s41423-020-00558-8
Wu H, Xiao Y, Zhang S, Ji S, Wei L, Fan F, Geng J, Tian J, Sun X, Qin F, Jin C, Lin J, Yin ZY, Zhang T, Luo L, Li Y, Song S, Lin SC, Deng X, Camargo F, Avruch J, Chen L, Zhou D (2013) The Ets transcription factor GABP is a component of the hippo pathway essential for growth and antioxidant defense. Cell Rep 3:1663–1677. https://doi.org/10.1016/j.celrep.2013.04.020
Xu MZ, Yao TJ, Lee NP, Ng IO, Chan YT, Zender L, Lowe SW, Poon RT, Luk JM (2009) Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer 115:4576–4585. https://doi.org/10.1002/cncr.24495
Yang N, Chen T, Wang L, Liu R, Niu Y, Sun L, Yao B, Wang Y, Yang W, Liu Q, Tu K, Liu Z (2020) CXCR4 mediates matrix stiffness-induced downregulation of UBTD1 driving hepatocellular carcinoma progression via YAP signaling pathway. Theranostics 10:5790–5801. https://doi.org/10.7150/thno.44789
Yang Y, Ni M, Zong R, Yu M, Sun Y, Li J, Chen P, Li C (2023) Targeting Notch1-YAP circuit reprograms macrophage polarization and alleviates acute liver injury in mice. Cell Mol Gastroenterol Hepatol 15:1085–1104. https://doi.org/10.1016/j.jcmgh.2023.01.002
Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, Cahan P, Stanger BZ, Camargo FD (2014) Hippo pathway activity influences liver cell fate. Cell 157:1324–1338. https://doi.org/10.1016/j.cell.2014.03.060
Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, Mu D, Lucito R, Powers S, Lowe SW (2006) Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 125:1253–1267. https://doi.org/10.1016/j.cell.2006.05.030
Zhang K, Chang Y, Shi Z, Han X, Han Y, Yao Q, Hu Z, Cui H, Zheng L, Han T, Hong W (2016) omega-3 PUFAs ameliorate liver fibrosis and inhibit hepatic stellate cells proliferation and activation by promoting YAP/TAZ degradation. Sci Rep 6:30029. https://doi.org/10.1038/srep30029
Zhao S, Jiang J, Jing Y, Liu W, Yang X, Hou X, Gao L, Wei L (2020) The concentration of tumor necrosis factor-alpha determines its protective or damaging effect on liver injury by regulating Yap activity. Cell Death Dis 11:70. https://doi.org/10.1038/s41419-020-2264-z
Zhao Y, Wang H, He T, Ma B, Chen G, Tzeng C (2023) Knockdown of Yap attenuates TAA-induced hepatic fibrosis by interaction with hedgehog signals. J Cell Commun Signal 17:1335–1354. https://doi.org/10.1007/s12079-023-00775-6
Zhou Y, Wang Y, Zhou W, Chen T, Wu Q, Chutturghoon VK, Lin B, Geng L, Yang Z, Zhou L, Zheng S (2019) YAP promotes multi-drug resistance and inhibits autophagy-related cell death in hepatocellular carcinoma via the RAC1-ROS-mTOR pathway. Cancer Cell Int 19:179. https://doi.org/10.1186/s12935-019-0898-7
Funding
Open Access funding enabled and organized by Seoul National University. This work was supported by the National Research Foundation of Korea grants (2021R1C1C1013323) and RS-2024-00348340) and the Korea Basic Science Institute (National Research Facilities and Equipment Center) grant (RS-2024-00398668) funded by the Korea government (MSIT), as well as by the Creative-Pioneering Researchers Program from Seoul National University. NYL was supported by the Basic Science Research Program funded by the Korean Ministry of Education (RS-2023-00271224).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, N.Y., Choi, M.G., Lee, E.J. et al. Interplay between YAP/TAZ and metabolic dysfunction-associated steatotic liver disease progression. Arch. Pharm. Res. 47, 558–570 (2024). https://doi.org/10.1007/s12272-024-01501-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-024-01501-5