
Abstract
This study aimed to identify possible pathogenic genes in a 90-member family with a rare combination of multiple neurodegenerative disease phenotypes, which has not been depicted by the known neurodegenerative disease. We performed physical and neurological examinations with International Rating Scales to assess signs of ataxia, Parkinsonism, and cognitive function, as well as brain magnetic resonance imaging scans with seven sequences. We searched for co-segregations of abnormal repeat-expansion loci, pathogenic variants in known spinocerebellar ataxia-related genes, and novel rare mutations via whole-genome sequencing and linkage analysis. A rare co-segregating missense mutation in the CARS gene was validated by Sanger sequencing and the aminoacylation activity of mutant CARS was measured by spectrophotometric assay. This pedigree presented novel late-onset core characteristics including cerebellar ataxia, Parkinsonism, and pyramidal signs in all nine affected members. Brain magnetic resonance imaging showed cerebellar/pons atrophy, pontine-midline linear hyperintensity, decreased rCBF in the bilateral basal ganglia and cerebellar dentate nucleus, and hypo-intensities of the cerebellar dentate nuclei, basal ganglia, mesencephalic red nuclei, and substantia nigra, all of which suggested neurodegeneration. Whole-genome sequencing identified a novel pathogenic heterozygous mutation (E795V) in the CARS gene, meanwhile, exhibited none of the known repeat-expansions or point mutations in pathogenic genes. Remarkably, this CARS mutation causes a 20% decrease in aminoacylation activity to charge tRNACys with L-cysteine in protein synthesis compared with that of the wild type. All family members carrying a heterozygous mutation CARS (E795V) had the same clinical manifestations and neuropathological changes of Parkinsonism and spinocerebellar-ataxia. These findings identify novel pathogenesis of Parkinsonism-spinocerebellar ataxia and provide insights into its genetic architecture.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The autosomal dominant spinocerebellar ataxias (SCAs) are a clinically, pathologically, and genetically heterogeneous group of neurodegenerative disorders principally characterized by progressive cerebellar ataxia [1]. SCAs usually onset in middle age and present with extensive phenotypic variations that overlap with other motor/sensory neuropathology [2], such as parkinsonism, dystonia, chorea, and myoclonus [3], revealing more than 40 different phenotypic subtypes of SCAs [4]. Movement disorders are frequent in many of the various SCA subtypes and are even predominantly expressed in some SCA subtypes [3]. When combined with cerebellar ataxia, the occurrence of multisystem atrophy (MSA-C) makes differential clinical diagnoses particularly challenging. Most SCAs are caused by dynamic DNA-repeat expansions in specific genes [5], whereas other SCA subtypes are caused by point mutations [6]. Although the SCA subtypes exhibit complex phenotypic overlap, each SCA subtype corresponds to a specific causative gene [5, 6], indicating that genetic factors play a crucial role in the development of these subtypes. Here, we characterized a comprehensive spectrum of disease phenotypes in a 90-member Han Chinese family and identified a heterozygous mutation in cysteinyl-tRNA synthetase (CARS; E795V mutation) that significantly co-segregates in this pedigree. Moreover, the functional impact of mutation in CARS aminoacylation activity was further elucidated.
Methods
Participants and Clinical Evaluations
The first proband (IV-15) was evaluated at the Memory and Movement Disorder Center of Peking Union Medical College Hospital (PUMCH). In her family with a pedigree composed of 90 people from six generations, 17 individuals developed symptoms similar to those of the proband, while six died of illness and two refused any further examinations. Surveys were conducted at either a local hospital or within each individual’s home. Three professors of neurology collected the family and medical histories of each individual, performed physical and neurological examinations, and assessed signs of ataxia via the International Cooperative Ataxia Rating Scale (ICARS), signs of parkinsonism via the Unified Parkinson's Disease Rating Scale (UPDRS) part III, and cognitive function via a PUMCH-FHS neuropsychological test battery (Fig. S1). Brain magnetic resonance imaging (MRI) scans, via a GE Signa 3·0-T MRI scanner at PUMCH, were performed in four members with symptoms and in one member at the presymptomatic stage. MRI sequences consisted of sagittal three-dimensional (3D), T1-weighted, axial T2-weighted (1·0-mm isotropy), T2*-weighted, fluid-attenuated inversion recovery (FLAIR), susceptibility-weighted imaging (SWI), and arterial spin-labeling (ASL) images. Moreover, MRI scans of the neck and thoracic spinal cord, as well as electrophysiological examinations (i.e., sensor and motor nerve conduction velocities, somatosensory-evoked potentials, and auditory-evoked potentials), were also conducted for the first proband (IV-15).
Whole-Genome Sequencing and Variants Calling
Whole-genome sequencing was used for identifying genetic variants, including single nucleotide variants (SNVs), short insertions-deletions (INDELs), short tandem repeats (STRs), copy number variants (CNVs), and structure variants (SVs). An average depth coverage of 66 × raw sequencing reads was generated by the BGISEQ-500 platform. Reads were filtered by SOAPnuke [7] and aligned to the human reference genome (hg19) by Burrows-Wheeler aligner [8] (version 0.7.15). Aligned reads were stored in cram format and the alignment summaries were calculated by samtools [9] (Table S1).
SNVs and INDELs were detected by the Genome Analysis Tool Kit [10] (version 3.6). Variants with any of the following quality control criticisms were excluded: (1) read depth < 4. (2) map quality < 55; (3) variant quality < 30; (4) genotype quality < 30. SNVs and INDELs were annotated by VEP [11] to predict the functional impact. Minor allele frequency (MAF) of variants were accessed in the gnomAD [12] database and two additional Chinese databases: ChinaMAP [13] and PGG.Han [14]. Variants with MAF > 1% in gnomAD [12] were excluded. The MAF cutoff threshold for rare variants was set at 1% by considering that deleterious alleles causing aging-related diseases may not be purged from the population before onset age. Patients with pathogenic mutation can survive for a long time and pass the mutant allele to their offspring, resulting in a low frequency and/or rare variant in the population.
Repeat-expansion calling was performed by expansionHunter [15]. The chromosomal locations, repeat motifs, diseases, and normal repeat-expansion sizes for 11 repeat-expansion loci of SCAs [5, 6, 16, 17] are listed in Table S2. A recently reported [18] repeat-expansion locus of SCA27B (FGF14 [GAA]) was validated by Sanger sequencing. CNVs calling was performed by CNVnator [19]. CNVs with any of the quality control criticisms suggested by the author were excluded: (1) Q-value > 0.05; (2) fraction of reads with zero map quality > 0.5; (3) fraction of gap > 0. CNVs coordinates of all samples with 1 bp overlap were merged together by bedtools [20]. SVs calling was performed by LUMPY [21] (v.0.2.13) and SVs genotyping was performed by SVTyper [22] (v.0.1.4). SVs with any of the quality control criticisms employed from Abel’s study [23] were excluded: (1) The proportion of split-read and paired-end read counts < 10%; (2) mean sample quality < 150; (3) the deletion size < the insert size of sequencing library estimated by SVTyper; (4) deletion copy-number estimated by CNVnator [19] > 0.5 or duplication copy-number < 1.5.
Linkage Analysis and Bioinformatic Analysis
To identify the pathological mutation, we performed linkage analysis via Merlin [24] software. We found three novel rare mutations co-segregated with the disease phenotype of sequencing individuals (four affected and three unaffected members) and no co-segregations of repeat-expansion, CNVs, SVs, and known pathogenic point mutations of SCA, Parkinson's disease [25, 26] (PD) and Multiple system atrophy [27] (MSA) reported by ClinVar database [28]. Sanger sequencing was used to validate the three mutations and confirm the co-segregations in the pedigree (all nine affected and seven unaffected members). Full-likelihood Bayes factor (BF) of co-segregation was calculated by segregated R-package to quantify the pathogenic evidence [29]. SIFT [30], PolyPhen-2 [31], M-CAP [32], and MutationTaster [33] were used to predict the functional impact of the mutations. Homologous sequence analysis was used to investigate the conservation of the CARS mutation across multiple species. I-TASSER [34] was used to model the structural consequence of the CARS protein altered by mutation.
CARS Expression in Mouse Brain and Spinal Cord
To indicate whether the CARS gene is dosage sensitive, we employed a method developed in our previous study [35]. We collected 37 aminoacyl-tRNA synthetase genes in the human genome and calculated the standard deviation (SD) of gene expression across 156,049 mononuclear transcriptomes of mice brains and spinal cords (GSE110823) [36] via the formula: \(SD=\sqrt{\frac{\sum_{i}^{n}{({C}_{i}-\overline{C })}^{2}}{n-1}}\), in which \(C\) refers to fold change, \(i\) refers to the \(i\) th cell, and \(n\) refers to the total number of cells. The fold change was calculated by the expression in one cell divided by the mean expression of all cells: \({C}_{i}={E}_{i}/\overline{E }\), in which \(E\) refers to the expression in one cell, \(\overline{E }\) refers to the mean expression, \(i\) refers to the \(i\) th cell. We compared the SD of the CARS gene with loss-of-function tolerant genes [37] and dosage-sensitive genes [38]. Mann-Whitney U-test was used to indicate the significance of low SD values of aminoacyl-tRNA synthetase genes compared with loss-of-function tolerant genes and dosage-sensitive genes.
CARS Immunohistochemistry
Paraffin-embedded temporal cortex sections (obtained from Netherlands Brain Bank) from a 92-year-old control male were hydrated, rinsed in phosphate-buffered saline (PBS) for 10 min, and treated with 1.5% hydrogen peroxide in PBS for 1 hour at 37℃ to quench endogenous peroxidase activity. After being washed in PBS containing 0.5% triton X-100 (PBST) (3*5 min), the sections were treated with microwaves (700 W) in 0.05 mol/L citrate-buffered saline (pH 6.0) for 2×10 min for antigen retrieval. Subsequently, the sections were washed in PBST (3×5 min) and incubated in 5% normal goat serum (Vector Laboratories, Burlingame, CA) in PBST for 1 hour at 37℃ to block nonspecific staining. Then the sections were incubated with primary antiserum of rabbit anti-CARS (Novus biological, NBP1-86624, at 1:200 dilution) in PBST containing 5% normal goat serum for 24 hours at 4℃. Amplification was performed with biotinylated goat anti-rabbit IgG (1:200; Vector Laboratories, USA) and avidin-biotin-peroxidase complex (1:200; Vector Laboratories). Finally, the immune complex was visualized by incubation with 0.05% 3, 3’-diaminobenzidine (Sigma-Aldrich) in PBST containing 0.03% H2O2 for 10 min. The sections were mounted in Tris–HCl buffer containing 0.5% gelatin and dried overnight at room temperature. Photographs were collected using an Olympus BX52 microscope (Olympus, Japan).
Aminoacylation Activity of Mutant CARS
Quantification of aminoacylation of tRNA is used to measure the ability of mutant CARS to catalyze the attachment of L-cystine to tRNAcys (Fig. S5). Wild-type (WT) CARS and a known mutant CARS (c.2061) that have been reported to modify the aminoacylation activity [39] were used for negative and positive control. A high throughput spectrophotometric assay for quantitative measurement is designed and performed in a transparent 96-well plate [40]. Each 100μl aminoacylation reaction containing 30 mmol/L HEPES (pH7.5), 30 mmol/L NaCl, 30 mmol/L KCl, 40 mmol/L MgCl2, 1 mmol/L DTT, 200 μmol/L ATP, 2 U/mL PPiase, 55 μg/mL miRNA and enzyme was incubated at 37℃ for an hour. In aminoacylation, pyrophosphate was released in ATP catabolism, and catalyzed to inorganic phosphate by PPiase. EDTA was used to terminate the aminoacylation and a 25 μL malachite green dye was added to each reaction and then the reaction was incubated at room temperature for 10 minutes. After that, the absorbance value of the chromogenic complex of inorganic phosphorus and malachite green dye was measured by a multimode microplate reader at 620 nm and its standard curve was estimated.
Results
Clinical Characteristics
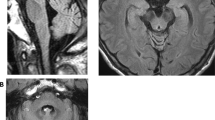
Among all 30 available participants, nine affected members were identified from four families of offspring in the third or fourth generation of this pedigree (Fig. 1A). The specific clinical features are shown in Table 1. The onset age ranged from 42–62 years and the duration of the disease varied from 1–25 years. All nine affected members presented core characteristics, namely difficulty in walking as an early feature, slow progression, followed by varying degrees of cerebellar ataxia, parkinsonism, and pyramidal signs (except IV-12, one-year course, no pyramidal signs). The severities of these three core clinical manifestations were closely related to the course of the disease. All patients over the eight-year course were confined to wheelchairs. The non-core features consisted of a variety of neurodegenerative manifestations. Among them, peripheral neuropathy and vertical gaze dysfunction were only found in the posterity's family D (III-6, III-7). Cognitive impairment, stridor, and cold hands were only found in the posterity's family C (IV-3, IV-5). Erectile dysfunction was found only in the posterity's family B (IV-13), and facial grimacing was found in the posterity's families A and C (IV-5, IV-12, IV-10). Four of the nine affected members underwent brain MRI scans, and all showed varying degrees of atrophy in the cerebellar cortex, vermis, and pons (Table 1, Fig. 2, Fig. S2). The axial T2-weighted images showed pontine-midline linear hyperintensities, similar to MSA-C in two affected members (IV-10, IV-15). SWI revealed hypo-intensities of the cerebellar dentate nuclei, basal ganglia, mesencephalic red nuclei, and substantia nigra (SN) in one affected member (IV-10), suggesting iron accumulation. ASL images showed decreased regional cerebral blood flow (rCBF) in the bilateral basal ganglia and the cerebellar dentate nucleus, followed by further deterioration over two years (IV-15), whereas there was reduction only in the dentate nuclei for the member at the pre-symptomatic stage (IV-10). Moreover, the following were all normal in the first proband (IV-15): cervical and thoracic spinal cord MRI scans; electrophysiological examinations of sensor- and motor-nerve conduction velocities; somatosensory-evoked potentials; and auditory-evoked potentials. These findings excluded cervical and thoracic spinal cord demyelination, peripheral neuropathy, deep sensory disorder, and auditory nerve damage.

A heterozygous CARS mutation causes a rare neurodegenerative disease in a 90-member family. The first proband (IV-15) was evaluated at the Memory and Movement Disorder Center at Peking Union Medical College Hospital (PUMCH). Three neurologists examined all 30 available participants in this pedigree and identified nine affected members and 21 clinically unaffected members. Four affected members (III-7, IV-5, IV-12, and IV-15) and three age-matched unaffected members (III-5, III-9, and IV-1) were used for whole-genome sequencing. A significant co-segregated rare mutation, CARS: p.Glu795Val, was identified in whole-genome-sequencing samples and was confirmed, via Sanger sequencing, in five additional affected members (III-6, 75 years old; IV-4, 63 years old; IV-10, 52 years old; IV-11, 50 years old; IV-13, 52 years old) and four additional unaffected members who were at or close to the onset age of disease (IV-6, 52 years old; IV-7, 48 years old; IV-18, 52 years old; IV-25, 38 years old). All 30 members (except for IV-26 and V-24 with failure of Sanger sequencing) with available DNA are marked with (+) or without (–) the CARS mutation (A), and their genotypes are marked by the red arrows in the CARS sequence chromatograms (B).

Magnetic resonance imaging in the brain. Brain MRI scans were performed on four clinically affected family members (IV-3, IV-10, IV-13, and, IV-15), and one asymptomatic member carrying the CARS mutation (IV-23). A Marked atrophy of the pons, vermis, and B cerebellar cortex is shown in MRI scans from member IV-3; C Atrophy of the pons, vermis, and D cerebellar cortex with a pontine-midline linear hyperintensity on T2WI is shown for member IV-10. E, F A normal iron-induced SWI hypo-signal in the dentate nuclei and basal ganglia is shown in MRI scans from member IV-10. G, H Mild atrophy of the pons, vermis, and cerebellar cortex with lacunas in the pons is shown in MRI scans from member IV-13. I–K An iron-induced SWI hypo-intensity in the dentate nuclei, red nuclei, SN, and basal ganglia is shown in MRI scans from member IV-13. L Marked atrophy of the vermis, pons, and L–N diffuse cerebellar atrophy with N a pontine-midline linear hyperintensity on T2WI is shown in MRI scans from member IV-15. O Decreased regional cerebral blood flow (rCBF) in the bilateral basal ganglia (from IV-15) and P in the cerebellar dentate nucleus, Q with further deterioration over two years, is shown in MRI scans from member IV-15. R Decreased rCBF in the cerebellar dentate nucleus (from member IV-10). S Decreased rCBF in the left putamen and the head of the caudate nucleus (from V-23), T but not in the cerebellar dentate nucleus.
Pathogenic Variant Identification
To identify the genetic cause of disease in this pedigree, we selected four affected members (III-7, IV-5, IV-12, and IV-15) and three age-matched unaffected members (III-5, 79 years old; III-9, 65 years old; IV-1, 72 years old) to perform whole-genome sequencing (Table S1). All seven members were identified to exhibit normal ranges of expression levels in 11 repeat-expansion loci of SCAs (Table S2) and did not carry any reported pathogenic mutations (SNVs and INDELs) related to SCAs, indicating that known pathogenic mutations were not the cause of disease in this pedigree. The normal repeat number of two loci (SCA17: TBP[CAG]; SCA37: DAB1[ATTTC]) caused similar phenotypes and a recently reported locus (SCA27B: FGF14[GAA]) was validated by Sanger sequencing in the proband (IV-15) (Fig. S3). To identify the novel genetic cause, we screened all the variants—including SNVs, INDELs, CNVs, and SVs—and then found three rare missense mutations (Table 2) co-segregated in this pedigree (Merlin: LOD=2.9). We performed Sanger sequencing for the three candidate mutations in all 30 members with available DNA (Fig. 1B). A CARS heterozygous mutation (c.2384A>T; p.Glu795Val; E795V) was detected in all nine affected members, whereas no such mutation was detected in any of the seven unaffected members who were at or close to the onset-age of disease (III-5, 79 years old; III-9, 65 years old; IV-1, 72 years old; IV-6, 52 years old; IV-7, 48 years old; IV-18, 52 years old; IV-25, 38 years old). ERVFRD-1 mutation and OR51G2 mutation were not detected in affected member IV-13 (Fig. S4). We calculate the LOD score and BF of co-segregation in nine affected and seven unaffected members via Merlin and segregated R-package. Notably, a LOD score of 4.96 indicated the CARS mutation is significantly co-segregated with the disease status in this pedigree. A BF score of 4631 is strong evidence that suggests CARS mutation is pathogenicity. We screened the SVs and CNVs in the E795V neighboring region and found none of the SVs/CNVs at the CARS gene, which is consistent with the rare event of CARS SVs/CNVs in the gnomAD v4 database. These results indicated CARS E795V is the cause of the disease.
Specifically, four currently unaffected members (IV-14, 45 years old; IV-23, 46 years old; V-6, 42 years old; V-23, 22 years old) carried this pathogenic mutation. V-23 had undertaken brain MRI scans and showed preclinical features, but the other three unaffected members refused to undergo MRIs or further examinations.
Functional Impact
CARS mutation is novel (0) in the Chinese population of the ChinaMAP database, and rare (0.006) in Han-Chinese of PGG. Han database and (0.008) in East Asian of gnomAD-v3 [12] database (Table 2). This rare mutation was predicted to have a harmful effect on gene function (Table 2), be conservative across multi-species (Fig. 3A), and predicted to induce a conformational change to protein structure (Fig. 3B-D), and CARS were found to be expressed in the cytoplasm of neurons within the human temporal cortex (Fig. 3E). Moreover, single-cell transcriptome analysis showed that the standard deviation (SD) of Cars expression fold change is significantly low compared with that of dosage-sensitive genes (Fig. 3F), indicating Cars expression is strictly required in normal brain/spinal cord and its expression alteration is intolerant. More importantly, we observed a decreased aminoacylation activity of mutant CARS, as well as a known mutant CARS (c.2061) that has been reported to reduce the aminoacylation activity [41] via spectrophotometric assay in vitro (Fig. 3G-H). The fitted curve of the initial rate estimated a 20% decrease in the activity of mutant CARS compared with that of CARS WT (Table 3). These results indicated that CARS mutation altered the molecular dynamics of WT CARS and caused a decreased activity to charge tRNACys with L-cysteine in protein synthesis. Taken together with bioinformatic prediction, statistical evidence, and molecular evidence in vitro, we suggested that CARS mutation is the genetic cause of disease.

The impact of CARS mutation. A Evolutionary conservation. The red triangle indicates that the mutated amino acid at codon-795 of CARS orthologs is highly conserved. B–D Prediction of 3D protein structure. The human CARS protein structure is displayed as a ball-and-stick model B. Compared with the wild type C, the mutated variant D destroyed one of three backbone hydrogen bonds and changed the α-helix to a loop. E CARS expression in human brain cortex. Paraffin-embedded temporal-cortex sections (Netherlands Brain Bank) from a 92-year-old control male were stained with the CARS antibody (Novus Biological, NBP1-86624). F The strict requirement of CARS expression in mice brain and spinal cord. The standard deviation (SD) of fold change of gene expression is used to evaluate the spectrum of expression variation. A low SD value indicates a gene requires strict expression. Dosage-sensitive genes are known to be intolerant of expression alteration and present lower SD values. Loss-of-function tolerant genes are known to be tolerant of expression alteration and present higher SD values. The significance of Mann-Whitney U-tests exhibits that CARS (redpoint) expression is strictly required in mice brain/spinal cord, indicating CARS expression is intolerant to be altered. G–H The aminoacylation activity of CARS in vitro. Curves in a green rectangle of G were detailed in H. The curves of the initial rate (pmol/min) at different concentrations of L-cysteine are fitted by the Michaelis-Menten formula. Compared with WT, a 20% decrease in the activity of mutant CARS was estimated by the fitted curves. The differences in curves between WT and a mutant CARS (c.2061) known to reduce activity demonstrate our experiment results are reliable.
Discussion
In the present study, we discovered a novel late-onset parkinsonism/SCA complex in a 90-member family, which presented core characteristics in all nine affected members. The affected members in this family displayed a variety of other clinical features consistent with those of rare neurodegenerative diseases, such as vertical gaze palsies, facial grimacing, cognitive impairment, peripheral neuropathy, autonomic dysfunction, blood-vessel dysfunction, and bladder dysfunction, as well as rare symptoms such as erectile dysfunction, stridor, and cold hands. Interestingly, posterity's families exhibited specific non-core clinical manifestations. The brain MRI findings in all four available members suggested that loci of pathological changes were not limited to the cerebellum and brainstem but were also present in the basal ganglia, mesencephalic red nucleus, and the SN. The pathological characteristics not only consisted of atrophy and a “hot cross bun” sign in the pons but also included abnormal iron deposition and a reduction of rCBF. These findings suggest that this disorder is a novel rare neurodegenerative disease, namely a parkinsonism/SCA complex. However, these clinical manifestations may exist in different SCA subtypes. SCAs—particularly SCA2, SCA3, SCA6, SCA8, and SCA17—can exhibit parkinsonism [42]. The “hot cross bun” sign in the pons of axial T2-weighted images may occur in certain SCA subtypes (SCA1, SCA2, SCA3, SCA6, SCA7, SCA17, SCA8, and SCA34) despite being more a common feature MSA [43,44,45]. Additionally, most of these SCAs present a shorter expansion of CAG repeats. However, none of these SCA subtypes can solely represent the diverse clinical manifestations of affected members in the pedigree that we analyzed in the present study. In addition, impaired horizontal eye movement has been reported in many SCA subtypes, whereas abnormal vertical eye movement is a typical sign of progressive supranuclear palsy, which is a rare symptom and generally presents as a relatively pure cerebellar phenotype in SCA37 [46]. In our present study, the affected member (III-7) with vertical gaze palsies and other neurodegenerative signs cannot be considered as having a subtype of SCA37. Although gait ataxia was the first symptom of all affected family members in the present study, a 22-year-old asymptomatic carrier (V-23) showed reduced rCBF only in the left putamen and caudate nuclei, but not in the dentate nuclei, which has not been reported for any SCA subtype [47]. Furthermore, an affected member (IV-10) had iron accumulation in the basal ganglia, mesencephalic red nucleus, SN, and cerebellar dentate nucleus, which in combination with other neurodegenerative signs, should be considered a rare neurodegenerative disease rather than SCA17 [48]. Additionally, dynamic DNA-repeat expansions in 12 SCA loci were identified to be within normal ranges and none of the pathogenic point mutations found in the pedigree of the present study were related to SCAs, PD, or MSA; hence, these results did not provide any genetic evidence for known SCA subtypes. Moreover, the clinical manifestations found in the present pedigree were different from those of MSA. MSA generally progresses rapidly. Early and severe autonomic nerve failure is a major feature of most MSA patients, with 78.3% having orthostatic hypotension, 86.9% having urinary incontinence, and 94.3% having erectile dysfunction. To the best of our knowledge, no previous study has reported such extensive, rich, and specific clinical and neuropathological manifestations of affected members within such a large pedigree [46, 49,50,51,52,53,54], which cannot be explained by other known disorders or by the functions of genes implicated in these disorders [55,56,57].
In the genetic analysis, we identified a CARS missense mutation as the pathogenic variant in this large domain pedigree. CARS, namely CARS1, is responsible for charging tRNAs with cysteine during protein synthesis. CARS is a member of ARSs, in which each ARS is responsible for ligating a specific amino acid to its cognate tRNA during protein synthesis. Recent studies have shown that mutations in ARSs cause a variety of neurological disorders [58]. Many mutants result in loss of tRNA aminoacylation function [59].Aminoacylation experiment revealed that mutant AARS [60], AARS2 [61], MARS [62], QARS [63], VARS [64], NARS1 [65], KARS [66], HARS1 [67], SARS [68], and CARS [39] severely reduces enzymic activity. Kuo ME’s study showed that CARS bi-allele caused a multi-system, recessive disease that includes microcephaly, developmental delay, and brittle hair and nails [39]. The pathogenic variants R341H and S359L reduced 40% and 70% of CARS activity, respectively. In contrast, our study showed that the CARS E795V variant causes a 20% decreasing activity of aminoacylation in protein synthesis. CARS genes are dosage-sensitive in mice brains and spinal cords. The SD value of CARS gene expression is lower than the median of ARSs genes that presented significantly low SDs compared with dosage-sensitive genes (P < 8.5×10-12) or loss-of-function tolerant genes (P < 0.05). Loss-of-function tolerant genes are tolerant of expression alteration and present low SD values. Oppose to loss-of-function tolerant genes, dosage-sensitive genes are intolerant of expression alteration and present high SD values. These results suggested that the 20% decreasing activity of aminoacylation caused a loss of function effect on the CARS gene function. Although loss of function is the mechanism of diseases caused by CARS pathogenic variants, the reduction of CARS activity is differed by E795V(20%), R341H(40%), and S359L(70%) mutant. The R341H and S359L affect the catalytic domain that activates and transfers cysteine to tRNA. E795V was localized in the C-terminal extension, which is different from that of loss of function caused by R341H, S359L, and other ARS pathogenic variants. On the other hand, the loss of function of DARS [69], RARS [70], KARS [66], HARS1 [67], SARS [68], and NARS2/PARS2 [71] were reported to cause neurological diseases that share ataxic feature, indicating an important role of ARS function in the pathogenesis of ataxia. HARS1 pathogenic variant caused a multisystem ataxic syndrome. DARS, RARS, KARS, and AARS2 pathogenic variants caused ataxia with or without leukoencephalopathy. SARS pathogenic variant caused complex spastic paraplegia with microcephaly, intellectual disability, and ataxia. NARS2 and PARS2 pathogenic variants caused Alpers–Huttenlocher syndrome with ataxia. Our study of the CARS pathogenic variant expands the genetic spectrum of ARS-related ataxia.
In summary, we reported that a CARS pathogenic variant (E795V) is associated with a domain parkinsonism/SCA presentation. These results may broaden the spectrum of human diseases and provide insight into the genetic architecture of ARS-related neurological disorders.
Availability of Data and Materials
The necessary code was released on GitHub (https://github.com/liuhankui/WGS-linkage). Analyses were performed using public software packages: quality control of genome sequence (SOAPnuke, https://github.com/BGI-flexlab/SOAPnuke); alignment (BWA, https://github.com/lh3/bwa); variant calling of SNV and INDEL (GATK, https://github.com/broadinstitute/gatk), repeat-expansion (ExpansionHunter, https://github.com/Illumina/ExpansionHunter), CNV (CNVnator, https://github.com/abyzovlab/CNVnator), and SV (LUMPY, https://github.com/arq5x/lumpy-sv; SVtyper, https://github.com/hall-lab/svtyper); variant annotation (VEP, https://github.com/Ensembl/ensembl-vep); linkage analysis (MERLIN, http://csg.sph.umich.edu/abecasis/merlin/index.html); co-segregation analysis (https://github.com/magnusdv/segregatr); protein structure prediction (I-TASSER, https://zhanglab.ccmb.med.umich.edu/I-TASSER); data visualization (R, https://www.r-project.org).
References
Dueñas AM, Goold R, Giunti P. Molecular pathogenesis of spinocerebellar ataxias. Brain 2006, 129: 1357–1370.
Schöls L, Peters S, Szymanski S, Krüger R, Lange S, Hardt C. Extrapyramidal motor signs in degenerative ataxias. Arch Neurol 2000, 57: 1495–1500.
van Gaalen J, Giunti P, van de Warrenburg BP. Movement disorders in spinocerebellar ataxias. Mov Disord 2011, 26: 792–800.
Ashizawa T, Öz G, Paulson HL. Spinocerebellar ataxias: Prospects and challenges for therapy development. Nat Rev Neurol 2018, 14: 590–605.
Durr A. Autosomal dominant cerebellar ataxias: Polyglutamine expansions and beyond. Lancet Neurol 2010, 9: 885–894.
Schöls L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: Clinical features, genetics, and pathogenesis. Lancet Neurol 2004, 3: 291–304.
Chen Y, Chen Y, Shi C, Huang Z, Zhang Y, Li S, et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7: gix120.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25: 1754–1760.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10: giab008.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010, 20: 1297–1303.
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 2010, 26: 2069–2070.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature 2020, 581: 434–443.
Cao Y, Li L, Xu M, Feng Z, Sun X, Lu J, et al. The ChinaMAP analytics of deep whole genome sequences in 10, 588 individuals. Cell Res 2020, 30: 717–731.
Gao Y, Zhang C, Yuan L, Ling Y, Wang X, Liu C, et al. PGG.Han: The Han Chinese genome database and analysis platform. Nucleic Acids Res 2020, 48: D971–D976.
Dolzhenko E, Deshpande V, Schlesinger F, Krusche P, Petrovski R, Chen S, et al. ExpansionHunter: A sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 2019, 35: 4754–4756.
Shakkottai VG, Fogel BL. Clinical neurogenetics: Autosomal dominant spinocerebellar ataxia. Neurol Clin 2013, 31: 987–1007.
Tankard RM, Bennett MF, Degorski P, Delatycki MB, Lockhart PJ, Bahlo M. Detecting expansions of tandem repeats in cohorts sequenced with short-read sequencing data. Am J Hum Genet 2018, 103: 858–873.
Rafehi H, Read J, Szmulewicz DJ, Davies KC, Snell P, Fearnley LG, et al. An intronic GAA repeat expansion in FGF14 causes the autosomal-dominant adult-onset ataxia SCA27B/ATX-FGF14. Am J Hum Genet 2023, 110: 1018.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 2011, 21: 974–984.
Quinlan AR, Hall IM. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26: 841–842.
Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: A probabilistic framework for structural variant discovery. Genome Biol 2014, 15: R84.
Chiang C, Layer RM, Faust GG, Lindberg MR, Rose DB, Garrison EP, et al. SpeedSeq: Ultra-fast personal genome analysis and interpretation. Nat Methods 2015, 12: 966–968.
Abel HJ, Larson DE, Regier AA, Chiang C, Das I, Kanchi KL, et al. Mapping and characterization of structural variation in 17, 795 human genomes. Nature 2020, 583: 83–89.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002, 30: 97–101.
Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson’s disease. Lancet Neurol 2020, 19: 170–178.
Liu Y, Niu L, Liu X, Cheng C, Le W. Recent progress in non-motor features of Parkinson’s disease with a focus on circadian rhythm dysregulation. Neurosci Bull 2021, 37: 1010–1024.
Tseng FS, Foo JQX, Mai AS, Tan EK. The genetic basis of multiple system atrophy. J Transl Med 2023, 21: 104.
Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, et al. ClinVar: Improvements to accessing data. Nucleic Acids Res 2020, 48: D835–D844.
Jarvik GP, Browning BL. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am J Hum Genet 2016, 98: 1077–1081.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009, 4: 1073–1081.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010, 7: 248–249.
Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet 2016, 48: 1581–1586.
Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010, 7: 575–576.
Roy A, Kucukural A, Zhang Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat Protoc 2010, 5: 725–738.
Liu H, Guan L, Deng M, Bolund L, Kristiansen K, Zhang J, et al. Integrative genetic and single cell RNA sequencing analysis provides new clues to the amyotrophic lateral sclerosis neurodegeneration. Front Neurosci 2023, 17: 1116087.
Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 2018, 360: 176–182.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60, 706 humans. Nature 2016, 536: 285–291.
Dang VT, Kassahn KS, Marcos AE, Ragan MA. Identification of human haploinsufficient genes and their genomic proximity to segmental duplications. Eur J Hum Genet 2008, 16: 1350–1357.
Kuo ME, Theil AF, Kievit A, Malicdan MC, Introne WJ, Christian T, et al. Cysteinyl-tRNA synthetase mutations cause a multi-system, recessive disease that includes microcephaly, developmental delay, and brittle hair and nails. Am J Hum Genet 2019, 104: 520–529.
Cestari I, Stuart K. A spectrophotometric assay for quantitative measurement of aminoacyl-tRNA synthetase activity. J Biomol Screen 2013, 18: 490–497.
Goel P, Parvez S, Sharma A. Genomic analyses of aminoacyl tRNA synthetases from human-infecting helminths. BMC Genomics 2019, 20: 333.
Park H, Kim HJ, Jeon BS. Parkinsonism in spinocerebellar ataxia. Biomed Res Int 2015, 2015: 125273.
Ozaki K, Doi H, Mitsui J, Sato N, Iikuni Y, Majima T, et al. A novel mutation in ELOVL4 leading to spinocerebellar Ataxia (SCA) with the hot cross bun sign but lacking erythrokeratodermia: A broadened spectrum of SCA34. JAMA Neurol 2015, 72: 797–805.
Heidelberg D, Ronsin S, Bonneville F, Hannoun S, Tilikete C, Cotton F. Main inherited neurodegenerative cerebellar ataxias, how to recognize them using magnetic resonance imaging? J Neuroradiol 2018, 45: 265–275.
Way C, Pettersson D, Hiller A. The ‘hot cross bun’ sign is not always multiple system atrophy: Etiologies of 11 cases. J Mov Disord 2019, 12: 27–30.
Serrano-Munuera C, Corral-Juan M, Stevanin G, San Nicolás H, Roig C, Corral J, et al. New subtype of spinocerebellar ataxia with altered vertical eye movements mapping to chromosome 1p32. JAMA Neurol 2013, 70: 764–771.
Xing W, Wang XY, Liao XX, Liao WH, Shen L. Spin labeling artery method perfusion MRI study of SPG4 and SCA3/MJD. Magn Reson Imaging 2014, 32: 1330–1334.
Claassen J, Gerding WM, Kastrup O, Uslar E, Goericke S, Timmann D. Excessive brain iron accumulation in spinocerebellar ataxia type 17. Neurology 2015, 84: 212–213.
Shimazaki H, Vazifehmand R, Heidari MH, Khorram-Khorshid HR, Saber S, Hejazi S, et al. A large family with spinocerebellar ataxia type 6 in Iran: A clinical and genetic study. Arch Iran Med 2008, 11: 459–462.
Rosa AL, Molina I, Kowaljow V, Conde CB. Brisk deep-tendon reflexes as a distinctive phenotype in an Argentinean spinocerebellar ataxia type 2 pedigree. Mov Disord 2006, 21: 66–68.
Wang JL, Yang X, Xia K, Hu ZM, Weng L, Jin X, et al. TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain 2010, 133: 3510–3518.
Sun H, Satake W, Zhang C, Nagai Y, Tian Y, Fu S, et al. Genetic and clinical analysis in a Chinese parkinsonism-predominant spinocerebellar ataxia type 2 family. J Hum Genet 2011, 56: 330–334.
Cadieux-Dion M, Turcotte-Gauthier M, Noreau A, Martin C, Meloche C, Gravel M, et al. Expanding the clinical phenotype associated with ELOVL4 mutation: Study of a large French-Canadian family with autosomal dominant spinocerebellar ataxia and erythrokeratodermia. JAMA Neurol 2014, 71: 470–475.
Wang C, Xu Y, Feng X, Ma J, Xie S, Zhang Y, et al. Linkage analysis and whole-exome sequencing exclude extra mutations responsible for the parkinsonian phenotype of spinocerebellar ataxia-2. Neurobiol Aging 2015, 36(545): e1-545.e7.
Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet 1983, 1: 1151–1155.
Seidel K, Siswanto S, Brunt ERP, den Dunnen W, Korf HW, Rüb U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol 2012, 124: 1–21.
Rossi M, Perez-Lloret S, Doldan L, Cerquetti D, Balej J, Millar Vernetti P, et al. Autosomal dominant cerebellar ataxias: A systematic review of clinical features. Eur J Neurol 2014, 21: 607–615.
Boczonadi V, Jennings MJ, Horvath R. The role of tRNA synthetases in neurological and neuromuscular disorders. FEBS Lett 2018, 592: 703–717.
Antonellis A, Green ED. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu Rev Genomics Hum Genet 2008, 9: 87–107.
Simons C, Griffin LB, Helman G, Golas G, Pizzino A, Bloom M, et al. Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am J Hum Genet 2015, 96: 675–681.
Kuo ME, Antonellis A, Shakkottai VG. Alanyl-tRNA synthetase 2 (AARS2)-related Ataxia without leukoencephalopathy. Cerebellum 2020, 19: 154–160.
van Meel E, Wegner DJ, Cliften P, Willing MC, White FV, Kornfeld S, et al. Rare recessive loss-of-function methionyl-tRNA synthetase mutations presenting as a multi-organ phenotype. BMC Med Genet 2013, 14: 106.
Zhang X, Ling J, Barcia G, Jing L, Wu J, Barry BJ, et al. Mutations in QARS, encoding glutaminyl-tRNA synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures. Am J Hum Genet 2014, 94: 547–558.
Stephen J, Nampoothiri S, Banerjee A, Tolman NJ, Penninger JM, Elling U, et al. Loss of function mutations in VARS encoding cytoplasmic valyl-tRNA synthetase cause microcephaly, seizures, and progressive cerebral atrophy. Hum Genet 2018, 137: 293–303.
Wang L, Li Z, Sievert D, Smith DEC, Mendes MI, Chen DY, et al. Loss of NARS1 impairs progenitor proliferation in cortical brain organoids and leads to microcephaly. Nat Commun 2020, 11: 4038.
Sun C, Song J, Jiang Y, Zhao C, Lu J, Li Y, et al. Loss-of-function mutations in Lysyl-tRNA synthetase cause various leukoencephalopathy phenotypes. Neurol Genet 2019, 5: e565.
Galatolo D, Kuo ME, Mullen P, Meyer-Schuman R, Doccini S, Battini R, et al. Bi-allelic mutations in HARS1 severely impair histidyl-tRNA synthetase expression and enzymatic activity causing a novel multisystem ataxic syndrome. Hum Mutat 2020, 41: 1232–1237.
Verdura E, Senger B, Raspall-Chaure M, Schlüter A, Launay N, Ruiz M, et al. Loss of seryl-tRNA synthetase (SARS1) causes complex spastic paraplegia and cellular senescence. J Med Genet 2022, 59: 1227–1233.
Wolf NI, Toro C, Kister I, Latif KA, Leventer R, Pizzino A, et al. DARS-associated leukoencephalopathy can mimic a steroid-responsive neuroinflammatory disorder. Neurology 2015, 84: 226–230.
Nafisinia M, Sobreira N, Riley L, Gold W, Uhlenberg B, Weiß C, et al. Mutations in RARS cause a hypomyelination disorder akin to Pelizaeus-Merzbacher disease. Eur J Hum Genet 2017, 25: 1134–1141.
Mizuguchi T, Nakashima M, Kato M, Yamada K, Okanishi T, Ekhilevitch N, et al. PARS2 and NARS2 mutations in infantile-onset neurodegenerative disorder. J Hum Genet 2017, 62: 525–529.
Acknowledgments
We thank the participants and their families for their involvement in this study. This work was supported by the NIH-NIA Research Project (R21AG036454), the National Key R&D Program of China (2016YFC0901500), CAMS Innovation Fund for Medical Sciences (2016-I2M-1-004), the National Key R&D Program of China (2016YFC1305900), the National Natural Science Foundation of China (3203004), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB02030001), and Shenzhen Municipal of Government of China (JCYJ20170412153248372 and JCYJ20180507183615145).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethics Approval and Consent to Participate
The written informed consent was approved by PUMCH and was obtained from each of the thirty available participants. The pedigree study was approved by the Institutional Review Board of PUMCH. The genetic data sharing was approved (2020BAT0031) by the Human Genetic Resources Administration of China.
Additional information
Han-Kui Liu, Hong-Lin Hao and Hui You contributed equally to this work.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, HK., Hao, HL., You, H. et al. A Cysteinyl-tRNA Synthetase Mutation Causes Novel Autosomal-Dominant Inheritance of a Parkinsonism/Spinocerebellar-Ataxia Complex. Neurosci. Bull. (2024). https://doi.org/10.1007/s12264-024-01231-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12264-024-01231-0