
Abstract
Perfluorinated alkyl substances (PFAS) are anthropogenic substances and can only come from polluted sources. There are many classes of PFAS which can be transformed to perfluoalkyl carboxylic acids and perfluoroalkyl sulfonic acids in the environment. Once they are in this form, they are environmentally mobile and extremely stable with half-lives of decades. These compounds are not innocuous and are implicated in causing many different diseases. In this publication, a method for the extraction and quantitation of perflurocarboxylic acids and perfluorosulfonic acids, using high-resolution liquid chromatography mass spectrometry (HRMS), is evaluated. A QuEChERS extraction method was performed on tomatoes, strawberries and milk purchased from different supermarkets on different days. Recoveries from milk and tomatoes were between 80 and 120%. Matrix effects were significant for strawberries and tomatoes and stable isotope internal standards were required to compensate. PFBA was found in all 3 food types. The total PFAS was 0.68 ng g−1 in milk, 0.20 ng g−1 in strawberries and 0.36 ng g−1 in tomatoes. This study demonstrates the importance of using a robust analytical method to investigate the PFAS content of complex food matrices and in particular the contribution of short-chain PFAS to overall dietary consumption.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Perfluorinated alkyl substances (PFAS) are manmade fluorinated organic compounds which are ubiquitous in the environment. They were first manufactured in the 1940s and have many industrial and domestic uses from fire-fighting foams to grease repellents in fast food boxes (Hamid et al. 2018; Glüge et al. 2020). They have exceptional chemical stability which means that conventional waste treatment processes such as biodegradation are ineffective (Wei et al. 2019). Human exposure to PFAS can be through multiple routes including drinking water, food and indoor air and dust (Eschauzier et al. 2013; Graber et al. 2019; Johnson 2022), and they are also known to bioaccumulate (Ghisi et al. 2019; Lesmeister et al. 2020). A major route into the human food chain is via leaching from contaminated soil into grass which is consumed by grazing animals, and other plants which may be directly consumed by humans (Melo et al. 2022). It is estimated that most humans have PFAS in their bloodstream (Göckener et al. 2020; Graber et al. 2019; US EPA 2021) and they have also been detected in breastmilk (Zheng et al. 2021). Exposure to PFAS has been associated with health problems such as kidney, testicular and pancreatic cancer as well as reproductive and developmental effects (USEPA 2021; EEA 2019; Melo et al. 2022; Schrenk et al. 2020; White et al. 2011).
According to the Organisation for Economic Co-operation and Development (OECD) in 2018, there were over 4700 PFAS worldwide from 42 different chemical families (OECD 2018). Perfluorooctanoic acid (PFOA) and perfulorooctane sulfonic acid (PFOS) are two most commonly found PFAS in groundwater and drinking water. Campo et al. (2014) analysed influent, effluent and sludge samples from 16 wastewater treatment plants from different locations in Spain for 21 PFAS. Over 2 years, only 2 of the samples were free from PFAS and it was estimated this contributed between 16 and 67 g day−1 of PFAS to the receiving rivers. In an extreme case, Eschauzier et al. (2013) reported a combined total PFAS of 4400 ng L−1 in landfill leachate and Busch et al. (2010) reported 8060 ng L−1 and 12,819 ng L−1 in treated leachate and untreated leachate, respectively.
Increasing concern about PFAS led US EPA to work with chemical companies to reduce the production of PFOA and PFOS and in 2016, set a combined PFOA and PFOS drinking water advisory level of 70 ng L−1 (US EPA 2016a, b). In Europe, PFOA and PFOS were added to the Stockholm convention on Persistent Organic Pollutants in 2017 (UN Environment 2017). However, the industry response was to manufacture structurally similar PFAS by varying the carbon chain length (Buck et al. 2011). This is legitimate business practice but did nothing to reduce the amount of PFAS in the environment and only exacerbated the problem by increasing the number of different PFAS making them more difficult to analyse and quantify. Precursor compounds can potentially breakdown to more persistent PFAS which due to their extreme stability, will recirculate in the environment for decades to come. The perfluoroalkyl carboxylic acids (PFCAs) and perfluoroalkyl sulfonic acids (PFSAs) are often the ultimate degradation product for these precursor contaminants and their presence can be used as markers of PFAS contamination.
Although dietary intake is considered a major source of human exposure to PFAS, there are no universally recognised standards or regulations to routinely measure PFAS in food. In September 2020, the European Food Safety Authority (EFSA) recommended a maximum intake of 4.4 ng of PFAS per kg body weight per week (European Food Safety Authority 2020). Similarly, there are current regulations for PFOA and PFOS which apply to drinking water and if the concentration of either exceeds 0.3 µg L−1 monitoring is put in place (UKDRI 2021).
Although a maximum intake has been specified, there are no regulations to routinely measure PFAS in food or drinking water. However, there is growing evidence of PFAS in food, probably from bio-accumulation of contaminants present in fertilisers or groundwater, but this is still unclear (Stahl et al. 2009). There is also evidence to suggest that this contamination could be from sewage sludge treated agricultural soil (Johnson 2022; Pepper et al. 2021). Our publication establishes the need for more routine testing of everyday foods.
The PFCA and PFSA transformation products with a carbon chain length of 6 or less are regarded as short-chain PFAS, C7 and greater are long-chain PFAS. A study by Higgins and Luthy (2006) demonstrated the longer chain PFAS were more likely to adsorb to the soil and therefore less likely to be taken up by the plant. However, the C8 chain lengths were the lowest C chain length in the study.
The short-chain PFAS are more easily taken up by plants from the soil but have a shorter metabolic half-life in humans ranging from a few days to a month and are excreted unchanged, whereas the longer chain versions can persist in the human body for years (European Food safety Authority 2020). Food matrices are very varied, and PFAS content is also expected to vary depending on the fat or water content of the product as well as growing conditions. PFAS have been previously detected in cows’ milk and dairy products (Hill et al. 2022; Young et al. 2012). Processing and packaging are possible sources of PFAS contamination as well as animal feed or grass fertilised using sewage sludge. Milk could be a significant contributor of dietary PFAS especially to young children. Food with a high water content, e.g. strawberries and tomatoes, is likely to accumulate PFAS from groundwater irrigation as well as fertilisers. The aim of this study is to quantify various endpoint PFCAs and PFSAs in tomato, strawberry and milk. Little is known about the PFAS content of foods such as tomato and strawberry which are not only grown as commercial crops but often are grown domestically in gardens or allotments where groundwater contamination may be significant (Scher et al. 2018). Development of a robust method of detecting PFAS in these representative food matrices is important in assessing the potential contribution to overall PFAS exposure and associated risk to health.
Liquid chromatography mass spectrometry (LCMS) has become the method of choice for determination of trace pollutants in complex matrices. It is highly specific as detection is based on the chemical composition of the analyte of interest. Therefore, for quantitative methods the analyte can be determined by retention time, precursor ion and product ion reducing the risk of false positives. This makes it capable of quantifying multiple analytes in one sample providing more comprehensive information about the occurrence of contaminants in an individual sample. The analysis is further enhanced by the development of low cost and efficient extraction techniques, e.g. QuEChERS (quick, easy, cheap, effective, rugged and safe) method, to enrich the analyte in the sample and remove extraneous material which might affect the analysis. The QuEChERS method was originally developed for determination of pesticide residues in food. It uses organic solvent, water and NaCl which often does not require additional clean-up steps such as solid-phase extraction (SPE). This makes it low-cost and easy to use. However, even when the sample is extracted, interferences can still remain. This is called matrix effect and can be a problem especially when using an electrospray ion-source (Nasiri et al. 2021). Although a specific analyte ion is used for the analysis there are still background ions which can adversely affect the analyte signal. Matrix effect can cause an artificial decrease or increase in the signal known as ion suppression or ion enhancement respectively, and there is a need to demonstrate the effect the matrix has on the different analytes. Although PFAS described in this paper are from two homologous series and may be considered to behave in a similar manner in the mass spectrometer, they each elute at a different retention time and have the potential to co-elute with different contaminants thereby causing different matrix effects. The matrix effect, along with the recovery efficiency of the target analytes using a QuEChERS method, will be determined.
PFAS are implicated in causing many diseases and their presence and recirculation in food and the environment is a public health issue. The analysis of these pollutants is difficult due to their ubiquitous nature and number of different chemical derivatives.
This publication describes a specific, accurate and robust method to determine PFAS in different food matrices which is essential to understanding the overall dietary exposure of PFAS.
Experimental
Materials
Perfluorobutanoic acid (PFBA) ≥ 95.5%, perfluoropentanoic acid (PFPeA) ≥ 96.5%, perfluorohexanoic acid (PFHxA), perfluoroheptanoic acid (PFHpA) ≥ 97.0%, PFOA ≥ 95.5%, perfluorononanoic acid (PFNA) ≥ 96.5%, perfluorodecanoic acid (PFDA) ≥ 97.5%, perfluoroundecanoic acid (PFUdA) ≥ 94.5%, perfluorododecanoic acid (PFDoA), perfluorobutane sulfonic acid (PFBS) ≥ 96.5%, perfluorohexane sulfonic acid (PFHxS), sodium chloride and magnesium sulphate were purchased from Sigma-Aldrich. PFOS and the internal standards 13C PFBA, 13C PFHxA, 13C PFOA, and 13C PFOS were purchased from LGC. Optima acetonitrile and Optima methanol were from Fisher Scientific. To minimise the use of the pollutants, approximately 2 mg of each analyte was weighed on an analytical balance and used to prepare a stock solution in methanol at 2 mg mL−1. The stock solutions were made on 4 different occasions and used to prepare calibration lines and spiking solutions for recovery and matrix effect.
The tomatoes, strawberries and milk were purchased from two different supermarkets and one independent shop in separate locations on different days. The tomatoes were wrapped in plastic, the milk was in a plastic carton and the strawberries had no wrapping.
Instrumentation and LCMS Conditions
All analysis was carried out at Glasgow Caledonian University, Department of Applied Science, Glasgow, UK. The mass spectrometer used was a Thermo Scientific Q-exactive Orbitrap instrument, connected to a Dionex Ultimate 3000 RS Pump, Dionex Ultimate 3000 RS Autosampler (temperature controlled at 10 °C) and Dionex Ultimate 3000 RS Column Compartment (temperature controlled at 30 °C). The LC column was a Waters Xselect HSS T3 2.5 μm 2.1 × 150 mm Column XP fitted with a C18 guard cartridge. The flow rate was set to 0.2 mL min−1 and the injection volume was 10 μL. Gradient elution was chosen: solution (A) was methanol. Solution (B) was 10 mM ammonium formate and formic acid at pH 3.5 in 18 MΩ water. Ninety-five percent of (B) was run for 1 min, then up to 95% (A) over 7 min, and held for 5 min. It was then returned to initial conditions in 1 min and equilibrated with 95% of (B) for 7 min. A Raptor PFAS Delay column (50 × 2.1 mm) was fitted after the LC pumps and before the injection system in order to remove PFAS from the solvent system. Stainless steel tubing was used throughout.
The mass spectrometer was operated in negative ion mode. The electrospray conditions were set with a sheath gas of 45 arbitrary units, auxiliary gas 10 arbitrary units, capillary and auxiliary gas temperature 300 °C. The spray voltage was − 4.5 kV and the mass resolution 17,500.
Calibration Line
The 10-point calibration line was prepared in acetonitrile/water (20/80) over the range 0.1–50 ng mL−1. The internal standards, 13C PFBA, 13C PFHxA, 13C PFOA and 13C PFOS, were added to the calibration standards and the samples at a concentration of 10 ng mL−1. The peak area for the internal standard should be the same throughout the analysis unless there are interferences. The response is calculated as a ratio of analyte peak area/internal standard peak area and is called the response ratio. The calibration line is created using the response ratio at the different concentrations and the concentration of the unknown determined from it.
Recovery Experiment
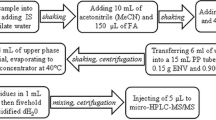
A simple QuEChERS method was developed to extract PFAS from the food matrices. The recoveries were performed on tomato and milk. Tomato was emulsified using a stick blender until homogeneous and the milk was taken straight from the carton. In a centrifuge tube, the matrix (5 g) was spiked with the PFAS mixture to a concentration of 10 ng mL−1 for all the analytes except for PFOS which was spiked at 1 ng mL−1, followed by 5 mL of acetonitrile + 1 g of NaCl. The mixture was vortexed for 5 min then centrifuged for 20 min at 400 rpm. The organic (upper) layer (4 mL) was collected, dried at 40 °C then reconstituted in 0.8 mL of acetonitrile/water 20/80 with sonication for 15 min. The samples were filtered through a Whatman 0.45 μm nylon filter before analysis.
Matrix Effect Experiment and Quantitation
The QuEChERS method was scaled up to 200 g of matrix for tomato, milk and strawberry. Different volumes of acetonitrile were collected due to scale up issues with the different matrices. The volume of acetonitrile collected was measured, the batch dried down as described above and reconstituted in the appropriate amount of acetonitrile/water 20/80. The batch of extracted matrix was filtered through a Whatman 0.45 μm nylon filter. The blanks had no further treatment. Aliquots were spiked to a concentration of 5, 10 and 25 ng mL−1 except for PFOS which was spiked at 0.5, 1 and 2.5 ng mL−1.
The matrix effect was calculated using the following equation.
where matrix is the concentration determined in the spiked matrix and solvent is the same concentration spiked into acetonitrile/water 20/80.
Results
Specificity
The risk of false positives using HRMS is greatly reduced as each analyte is quantified based on retention time, precursor and product ion. Table 1 describes the retention time and transitions used for each of the PFAS analysed.
Instrument Performance
Before implementation of the delay column PFBA was determined in most samples and blanks. This was not a consistent amount and varied greatly between samples with no discernible pattern. Once the delay column was in place, the results were consistent and blanks, such as solvent blanks, were free from PFBA. The overall chromatography was improved when ammonium formate was included in the mobile phase as it removed the peak tailing and improved the peak shape (Fig. 1). Figure 1 A–K show the extracted ion chromatogram for each of the PFAS which is an interference free signal. This is highly specific, similar to a fingerprint, as the selection is based on the charged molecular structure of the analyte (precursor ion), how it breaks up when an energy is applied (product ion) and the chromatographic retention time. In high resolution mass spectrometry the mass of an ion is accurate to 4 decimal places and is so accurate it can be used to determine the elemental composition of an unknown. Figure 1 L–N show the total ion chromatogram in full-scan mode for each of the different matrices and therefore illustrates all the ionisable components and consequently potential interferences. The strawberry and tomato contain earlier eluting compounds consistent with more polar compounds such as flavonoids and carotenoids. Whereas the milk has more later eluting compounds consistent with less polar lipids. The matrices would be expected to be different as they each contain different mixtures. This is why the matrix effect experiment needs to be done for each different food for all the analytes. The chromatograms are normalized to the highest peak and, although strawberry looks noisier, it is because most of the peaks are at a similar level whereas in tomato the peak at 10.27 min has a higher response and dominates the chromatogram.

A–K extracted ion chromatograms for the PFAS and L–N total ion chromatogram of the matrices over the scan range m/z = 120–1800 amu
Calibration Data
Table 2 shows the inter-day calibration data for the PFAS for a 10-point calibration. The data was processed using a 1/X2 regression (excluding the origin) and a % relative error of ± 30 for the points as acceptance criteria for better accuracy at low concentration. The limit of quantitation was set at the lowest calibration standard concentration which fitted this criterion. The average inter-day relative error was from 7 calibration sets run over 4 days.
Limit of Quantitation
The limit of quantitation (LOQ) was calculated by examining the calibration data over several datasets. Therefore, to achieve an LOQ value, the most reliable and lowest (Evard et al. 2016; Miller and Miller 2010; Analytical Methods Committee 2001; Eurachem 2014) concentration in the calibration line was used. This is based on the % relative error of the data point and how reproducible this value is over several experiments. Based on these criteria, the LOQ for PFBA, PFBS, PFHpA, PFNA, PFDA, and PFUdA is 0.25 ng mL−1 and 0.1 ng mL−1 for the others. Samples designated < LOQ had a value between zero and the LOQ. If no peak was determined, this was not detected (ND). Due to the sample preparation concentration factor, the data was quantified based on the LOQ from the calibration line and divided by 5.
The data is summarized for each analyte in Table 1. The calibration range is also reported where the LOQ is the lowest acceptable calibration concentration.
Recovery
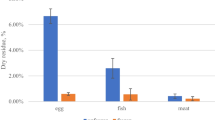
The recovery data from milk is over 2 days with 9 extractions each day and for tomato is over 3 days and 9 extractions each day. Most of the recoveries are between 80 and 120%, only PFOS was slightly lower (Fig. 2). Due to PFBA in the blank samples, the recovery was calculated by subtracting the amount of PFBA in the sample from the spiked recovery sample and comparing to the solvent. For milk, the recovery for PFBA was 112.6% and for tomato the average recovery was 114.8%.

Recovery of PFAS from tomato and milk extraction
Results Matrix Effect
A matrix effect between 80 and 120% is regarded as no effect. If the matrix is below 80%, there is ion suppression and above 120% ion enhancement (Table 3).
PFAS in Food
The amount of PFAS determined in each of the foods is summarized in Table 4 where the data has been corrected for the concentration factor. Most analytes were not detected (ND) in the samples. In some cases, analyte was detected but was below the limit of quantitation (< LOQ) which is specified in Table 1.
Discussion
Recovery
The recovery data for tomato and milk was generally good and between 80 and 120%. This was with the exception of PFOS in tomato which was low at 69.3%. For the other analytes, the recovery from milk is consistently lower which may be due to protein binding (Forsthuber et al. 2020) of the PFAS. Although it is lower, the recoveries are still acceptable.
Matrix Effect
In milk, signal enhancement is observed for most of the analytes without the internal standard. This is well compensated when the internal standard is included even when it is not the matched stable isotope. In tomato and strawberry, matrix suppression is observed. In tomato, inclusion of the internal standard compensates for the ion suppression effects, including some of those which are not matched. However, in strawberry, only the data with the matched internal standards compensate well. The analytes elute between 8 and 12 min (Fig. 1), and this is the noisiest part of the matrix for tomato and strawberry. Strawberry shows most contamination across the analyte peak area despite having the QuEChERS extraction. Although the extraction method removed much of the extraneous matter and gave good recoveries for tomato and strawberry, further clean-up is required, for example using solid-phase extraction. The lower recoveries for milk are possibly due to protein binding of the PFAS. The full-scan chromatogram for milk shows less contamination across the same retention time. This might explain why there is no suppression but does not explain the signal enhancement. There is less interference in the tomato matrix for PFBA and PFBS which is reflected in the matrix effect results (Zhou et al. 2017).
PFAS in Food
The current EFSA recommended maximum intake for PFAS is 4.4 ng kg−1 body weight per day (Schrenk et al. 2020). The average weight for a UK man and woman is 83.6 kg and 70.2 kg, respectively (Office for National Statistics 2010) and therefore a maximum ∑PFAS intake of 368 ng and 309 ng for each. Based on the concentration determined in food stuffs described in Table 4, consuming in excess of 542 g milk, 1840 g strawberry or 1023 g tomato would exceed the intake for a man and 453 g milk, 1540 g strawberry or 856 g tomato for a woman. Although this might seem a lot to consume, only 3 foods were tested and if other foods have similar concentrations of PFAS, there will be an accumulative effect throughout the day. The daily maximum intake for an average 3-year-old child weighing 14 kg is 61.6 ng. As little as 100 g of milk would contribute 68 ng of PFAS alone and a helping of 308 g strawberry or 171 g tomato would also pass the maximum daily intake. The half-life of PFBA in humans is less than longer chain PFAS. However, they are still excreted unchanged and will continue to circulate in the environment for decades with the potential to be re-consumed.
Conclusion
This study describes a cheap and easy method to extract PFAS from food matrices and a robust analysis. The method has demonstrated the requirement for a delay column to remove unwanted PFAS contamination which greatly affects the quantitation. Although good inter-day calibration data and recoveries were achieved the data demonstrates the variation of signal by the different matrices. In some cases a surrogate stable isotope internal standard can suffice which was successful for milk and partially successful for tomato. However, it was only the analytes with the matched internal standards in strawberry which compensated for matrix effect. This demonstrates that although the described QuEChERS method is useful to enrich the analyte, it does not adequately remove interferences in some for example in strawberry and requires further sample treatment for the long-chain PFAS.
PFBA had a matched internal standard, good recovery and minimal matrix effect and was found in all the test substances. This demonstrates even with a ban on the use of C8 PFAS the problem of these substances in food is still an issue and should be monitored on a more regular basis, especially for the shorter chain length PFAS which are becoming more common.
Only a small selection of the 1000 s of known PFAS were quantified. The ∑PFAS for those analysed was 0.68 ng g−1, 0.20 ng g−1 and 0.36 ng g−1 for milk, tomato and strawberry, respectively. These are just a few foods consumed on a daily basis and only a small selection of PFAS were quantified and an average 3 year old could easily exceed the daily limit.
This publication describes a robust method for the analysis of PFAS and provides information of dietary exposure of PFAS in everyday foods. Future work would expand the food items tested to include all meals in a normal days food.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Analytical Methods Committee (2001). What should be done with results below the detection limit? Mentioning the unmentionable. AMC Technical Briefs, 5, 1–2. Retrieved from http://scholar.google.com/scholar?hl=en&btnG=Search&q=intitle:amc+technical+briefs#0
Buck RC et al (2011) Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins. Integr Environ Assess Manag 7(4):513–541. https://doi.org/10.1002/ieam.258
Busch J et al (2010) Polyfluoroalkyl compounds in landfill leachates. Environ Pollut 158(5):1467–1471
Campo J et al (2014) Distribution and fate of perfluoroalkyl substances in Mediterranean Spanish sewage treatment plants. Sci Total Environ 472:912–922
EEA (2019) “Emerging Chemical Risks in Europe — ‘PFAS.’” Europe/Emerging-Chemical-Risks-in-Europe, 1–16. Retrieved from https://www.eea.europa.eu/publications/emerging-chemical-risks-in-europe/emerging-chemical-risks-in-europe
EFSA (2020) PFAS in food: EFSA assesses risks and sets tolerable intake. EFSA Available at: https://www.efsa.europa.eu/en/news/pfas-food-efsa-assesses-risks-and-sets-tolerable-intake
Eschauzier C et al (2013) Perfluorinated alkylated acids in groundwater and drinking water: identification, origin and mobility. Sci Total Environ 458–460:477–485
Eurachem (2014) The fitness for purpose of analytical methods. A laboratory guide to method validation and related topics. https://eurachem.org/images/stories/Guides/pdf/MV_guide_2nd_ed_EN.pdf. Accessed 11.05.13
Evard H et al (2016) Tutorial on estimating the limit of detection using LC-MS analysis, part II: practical aspects. Anal Chim Acta 942:40–49
Forsthuber M et al (2020) Albumin is the major carrier protein for PFOS, PFOA, PFHxS, PFNA and PFDA in human plasma. Environ Int 137 https://doi.org/10.1016/j.envint.2019.105324
Ghisi R et al (2019) Accumulation of perfluorinated alkyl substances (PFAS) in agricultural plants: a review. Environ Res 169:326–341
Glüge J et al (2020) An overview of the uses of per- and polyfluoroalkyl substances (PFAS). Environ Sci: Process Impacts 22(12):2345–2373. https://doi.org/10.1039/D0EM00291G
Göckener B et al (2020) Human biomonitoring of per- and polyfluoroalkyl substances in German blood plasma samples from 1982 to 2019. Environ Int 145. https://doi.org/10.1016/j.envint.2020.106123
Graber JM et al (2019) Per and Polyfluoroalkyl Substances (PFAS) Blood levels after contamination of a community water supply and comparison with 2013–2014 NHANES. J Eposure Sci Environ Epidemiol 29(2):172–182. https://doi.org/10.1038/s41370-018-0096-z
Hamid H et al (2018) Review of the fate and transformation of per- and polyfluoroalkyl substances in landfills. Environ Pollut 235:74–84. https://doi.org/10.1016/j.envpol.2017.12.030
Higgins CP, Luthy RG (2006) Sorption of perfluorinated surfactants on sediments. Environ Sci Technol 40(23):7251–7256. https://doi.org/10.1021/es061000n
Hill NI et al (2022) A sensitive method for the detection of legacy and emerging per- and polyfluorinated alkyl substances (PFAS) in dairy milk. Anal Bioanal Chem 414(3):1235–1243. https://doi.org/10.1007/s00216-021-03575-2
Johnson GR (2022) PFAS in soil and groundwater following historical land application of biosolids. Water Res 211:118035. https://doi.org/10.1016/j.watres.2021.118035
Lesmeister L et al. (2021). Extending the knowledge about PFAS bioaccumulation factors for agricultural plants – A review. Sci Total Environ 766. https://doi.org/10.1016/j.scitotenv.2020.142640
Melo TM et al (2022) Ecotoxicological effects of per- and polyfluoroalkyl substances (PFAS) and of a new PFAS adsorbing organoclay to immobilize PFAS in soils on earthworms and plants. J Hazard Mater 433:128771. https://doi.org/10.1016/j.jhazmat.2022.128771
Miller and Miller (2010) Statistics and Chemometrics for Analytical Chemistry. Prentice Hall
Nasiri A et al (2021) Overview, consequences, and strategies for overcoming matrix effects in LC-MS analysis: a critical review. Analyst 146(20):6049–6063. https://doi.org/10.1039/D1AN01047F
OECD (2018) Comprehensive global database of PFASs. Access through Portal on Per and Poly Fluorinated Chemicals. https://www.oecd.org/chemicalsafety/portal-perfluorinated-chemicals/
Office for National Statistics (2010) ‘Average’ Briton highlighted on UN World Statistics Day. https://www.ons.gov.uk/ons/about-ons/get-involved/events/events/un-world-statictics-day/-average--briton-highlighted-on-un-world-statistics-day.pdf. Accessed 20 Feb 2018
Pepper IL et al (2021) Incidence of PFAS in Soil following long-term application of class B biosolids. Sci Total Environ 793:148449. https://doi.org/10.1016/j.scitotenv.2021.148449
Scher DP et al (2018) Occurrence of perfluoroalkyl substances (PFAS) in garden produce at homes with a history of PFAS-contaminated drinking water. Chemosphere 196:548–555. https://doi.org/10.1016/j.chemosphere.2017.12.179
Schrenk D et al (2020) Risk to human health related to the presence of perfluoroalkyl substances in food. EFSA J 18(9). https://doi.org/10.2903/j.efsa.2020.6223
Stahl T et al (2009) Carryover of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonate (PFOS) from soil to plants. Arch Environ Contam Toxicol 57(2):289–298. https://doi.org/10.1007/s00244-008-9272-9
UKDRI (2021) UK drinking water inspectorate. Guidance on the water supply (Water Quality) regulations 2016 1 specific to PFOS (perfluorooctane sulphonate) and PFOA (perfluorooctanoic acid) concentrations in drinking water. https://www.dwi.gov.uk/private-water-supplies/pws-installations/guidance-on-the-water-supply-water-quality-regulations-2016-specific-to-pfos-perfluorooctane-sulphonate-and-pfoa-perfluorooctanoic-acid-concentrations-in-drinking-water/#1-introduction
UN Environment (2017) The 16 New POPs, p 25. http://www.pops.int/TheConvention/ThePOPs/TheNewPOPs/tabid/2511/Default.aspx
US EPA (2016a) “FACT SHEET PFOA & PFOS Drinking Water Health Advisories.” EPA 800-F-16-003. https://www.epa.gov/sites/default/files/2016a-06/documents/drinkingwaterhealthadvisories_pfoa_pfos_updated_5.31.16.pdf
US EPA (2016b) “Biomonitoring Perfluorochemicals (PFCs).” https://www.epa.gov/americaschildrenenvironment/biomonitoring-perfluorochemicals-pfcs. Accessed 17 Jan 2023
U.S. EPA (2021) IRIS toxicological review of perfluorobutanoic acid (PFBA) and related salts (Public Comment and External Review Draft, 2021). U.S. Environmental Protection Agency, Washington, DC, EPA/635/R-20/424
Wei Z et al (2019) Treatment of per- and polyfluoroalkyl substnaces in landfill leachate: status, chemistry and prospects. Environ. Sci Water ResTechnol 5:1814–1835. https://doi.org/10.1039/C9EW00645A
White SS et al (2011) Endocrine disrupting properties of perfluorooctanoic acid. J Steroid Biochem Mol Biol 127(1–2):16–26. https://doi.org/10.1016/j.jsbmb.2011.03.011
Young WM et al (2012) Determination of perfluorochemicals in cow’s milk using liquid chromatography-tandem mass spectrometry. J Agric Food Chem 60(7):1652–1658. https://doi.org/10.1021/jf204565x
Zheng G et al (2021) Per and polyfluoroalkyl substances (PFAS) in breast milk: concerning trends for current-use PFAS. Environ Sci Technol 55(11):7510–7520. https://doi.org/10.1021/acs.est.0c06978
Zhou W, Yang S, Wang PG (2017) Matrix effects and application of matrix effect factor. Bioanalysis 9(23):1839–1844. https://doi.org/10.4155/bio-2017-0214
Funding
The LCMS/MS system used in this study was funded by the noPILLS project, as part of EU funding from the Transnational Territorial Cooperation programme INTERREG IVB NWE. The studentship was funded by the British Mass Spectrometry Society and the Chromatographic Society. The standards were purchased from RSC funding.
Author information
Authors and Affiliations
Contributions
Joanne Roberts was responsible for the conceptualization and funding acquisition. Joanne Roberts and Moyra McNaughtan wrote the main text. Hector de las Heras Prieto contributed to most of the practical work with some participation from Joanne Roberts. Hector de las Heras Prieto and Joanne Roberts were responsible for the data analysis. Tables 1 and 2 and Fig. 1 were prepared by Hector de las Heras Prieto. Tables 3 and 4 and Fig. 2 were prepared by Joanne Roberts. All three authors reviewed the manuscript before submission.
Corresponding author
Ethics declarations
Ethics Approval
This study does not contain experiments involving animals or humans.
Informed Consent
Not applicable.
Conflict of Interest
Joanne Roberts declares that she has no conflict of interest. Moyra McNaughtan declares that she has no conflict of interest. Hector de las Heras Prieto declares that he has no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Roberts, J., McNaughtan, M. & de las Heras Prieto, H. Unwanted Ingredients—Highly Specific and Sensitive Method for the Extraction and Quantification of PFAS in Everyday Foods. Food Anal. Methods 16, 857–866 (2023). https://doi.org/10.1007/s12161-023-02451-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-023-02451-2