
Abstract
Purpose
The subclassification of adrenal cancers according to the WHO classification in ordinary, myxoid, oncocytic, and sarcomatoid as well as pediatric types is well established, but the criteria for each subtype are not sufficiently determined and the relative frequency of the different types of adrenal cancers has not been studied in large cohorts. Therefore, our large collection of surgically removed adrenal cancers should be reviewed o establish the criteria for the subtypes and to find out the frequency of the various types.
Methods
In our series of 521 adrenal cancers the scoring systems of Weiss et al., Hough et al., van Slooten et al. and the new Helsinki score system were used for the ordinary type of cancer (97% of our series) and the myxoid type (0.8%). For oncocytic carcinomas (2%), the scoring system of Bisceglia et al. was applied.
Results
Discrepancies between benign and malignant diagnoses from the first thee classical scoring systems are not rare (22% in our series) and could be resolved by the Helsinki score especially by Ki-67 index (more than 8% unequivocally malignant). Since all our cancer cases are positive in the Helsinki score, this system can replace the three elder systems. For identification of sarcomatoid cancer as rarest type in our series (0.2%), the scoring systems are not practical but additional immunostainings used for soft tissue tumors and in special cases molecular pathology are necessary to differentiate these cancers from adrenal sarcomas. According to the relative frequencies of the different subtypes of adrenal cancers the main type is the far most frequent (97%) followed by the oncocytic type (2%), the myxoid type (0.8%) and the very rare sarcomatoid type (0.2%).
Conclusions
The Helsinki score is the best for differentiating adrenal carcinomas of the main, the oncocytic, and the myxoid type in routine work. Additional scoring systems for these carcinomas are generally not any longer necessary. Signs of proliferation (mitoses and Ki-67 index) and necroses are the most important criteria for diagnosis of malignancy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
According to the WHO classification 2017 of adrenocortical tumors [12], adrenal carcinomas are subdivided into main type, myxoid type, oncocytic type, and sarcomatoid type and a pediatric type. This subtyping is generally very important for the histological diagnosis of adrenal cancer, since the identification of malignancy is different for each subtype (Table 1). The malignancy of the main type and its myxoid subtype should be determined using the scoring system of Weiss et al. [39] supported by the scoring systems of van Slooten et al. [38] and of Hough et al. [15] or using the more simplified Helsinki score [29]. For oncocytic carcinomas, a special scoring system by Bisceglia et al. [4] has been established. The sarcomatoid adrenal cancers do not need a scoring system, since their malignancy is out of doubt. For pediatric cancers, the system of Wienecke et al. [40] should be used. The scoring systems are necessary for differentiation carcinomas from adenomas, but should be used only for specimens of adrenal resections and not for biopsies or metastases.
Detailed characteristics for identification of the subtypes are not found in the literature. Data of their frequency are sparse. In a series of 67 adrenal cancers, nine tumors were classified as oncocytic carcinomas [27]. Therefore, we reviewed our large collection of adrenal cancers for structural details in the subtypes and their relative frequency.
Aims of our studies are answers to the following questions:
-
1.
Which scoring system is the best for the main type of adrenal cancers?
-
2.
Are more than one scoring system necessary for identifying malignancy of the main type?
-
3.
Which are the most important criteria for diagnoses of the different subtypes?
-
4.
What are the relative frequencies of the different subtypes?
Materials and methods
A collection of adrenal cancers from 521 patients treated between 1993 and 2005 were available for review. Pediatric carcinomas were not included. Paraffin-embedded specimens were sent together with some clinical data and macroscopic findings for second consultant diagnosis (WS).
Paraffin sections were stained with hematoxylin–eosin, PAS, and elastica-van Gieson, and additional immunostainings were performed: keratin Kl-1 or AE1/AE3, synaptophysin, chromogranin A, melan A, inhibin, SF-1, p53, and Ki-67.
The first step of our studies in each case was the assessment of possible cancer subtype. In the second step for assessing the malignancy, we used for the main subtype and the myxoid subtype the scoring system of Weiss et al. [39] (malignancy > 3) supported by the scoring systems of van Slooten et al. [38] (malignancy > 8) and of Hough et al. [15] (malignancy > 2.0). The Helsinki score [29] with the criteria necroses, mitoses, and Ki-67 index was additionally used for the final decision of malignancy if the three other scoring systems come to indeterminate levels between benign and malignant data. In oncocytic tumors, the system of Bisceglia et al. [4] was applied. Since pediatric tumors were not in our collection the scoring of Wieneke et al. [40] could not be used.
Results
The following histological findings of the different subtypes base on our experiences from our large collection of adrenal cancers. Most of these descriptions are also found in textbooks of adrenocortical tumors [12, 21, 32].
Main type
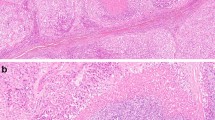
The main type of adrenal cancer is characterized by diffuse growth pattern (Table 2) (Fig. 1). More solid type or nested, trabecular, or fascicular formations are rare. The intratumorous vessels are mostly of sinusoidal type with small endothelial layer and sparse or lacking muscular cell elements within thin walls. Circumscribed but present in more than two diameters of high power fields (HPF) or more extensive invasions of these vessels are often accompanied by thromboses that are followed by small or extensive, in necroses. The venous vessels in the periphery near the capsule can show circumscribed or multiple invasions. Partial of complete infiltrations of the fibrous capsules has to be noted. Fibroses may be present as an irregular network, but are more often found in larger fields. Some myxoid areas may be present, but should not exceed 40–50% of tumor volume. The tumor cells are slightly or strongly pleomorphic with increased chromatin. Nucleoli may be regular and small or irregular and increased. The number of mitoses is very different and has to be counted per high power fields. The cytoplasm shows mostly very few small lipid vacuoles or is free of visible lipid. Strong lipid accumulations or more differentiated spongiocytes as in normal adrenal cortex are rare.

Main type with diffuse pattern, strong atypia, and thrombosis of sinusoidal vessel. Hematoxylin–eosin staining, 360×
In 29 (5.8%) of main type cancers, the index of Weiss et al., (malignancy > 3) was lower than four, but in 16 of these, the index of Hough et al., (malignancy > 2.0) was higher than 2.01. Of the remaining 13 cases, eight tumors showed a van Slooten et al., index (malignancy > 8) higher than eight. The 476 cancers with Weiss et al., score more than three (malignant) showed not malignant levels in the two other systems in 70 cases. In these, Hough et al., score was less than two (not malignant) in 62 cases and van Slooten et al., index lower than eight (not malignant) in 24 tumors. In five cancers with non-malignant data in all three scoring systems, the diagnosis of adrenal carcinoma is based on the Helsinki score with Ki-67 indexes of more than 8.5.
Myxoid subtype
In our collection, we diagnosed a myxoid adrenal carcinoma if more than half of tumor volume was composed of myxoid matrix and the included tumor cells. When less than 50% we designate that as a main type cancer with myxoid parts. Otherwise, the myxoid subtype shows nearly all characteristics of the main type. The large areas of myxoid cell-free formations are strongly stained for alcian-blue (Fig. 2). The growth pattern is diffuse, solid, or trabecular (Table 2).

Myxoid type with strongly increased, Astra-blue myxoid stroma. Astra-blue staining, 220×
Oedematous areas are sometimes very similar and have to be differentiated from myxoid areas by negative staining for alcian-blue.
Oncocytic subtype
The oncocytic subtype is always diffuse in architecture (Table 2) (Fig. 3). The cells can be very pleomorphic harboring nuclei with strongly increased chromatin und irregular nucleoli. The cytoplasm has to be strongly eosinophilic with diffuse dense staining. Lipid vacuoles are lacking. All other criteria are similar to the main type. The substantial criteria for differentiation from oncocytic adenomas are an increased mitotic index (> 5/50 HPF), atypical mitoses, and invasion of veins. One main criterion is enough or sufficient for diagnosis of malignancy. Minor important criteria are size (more than 100 mm in diameter) and weight of tumor (> 200 g), invasion of sinusoidal vessels and capsule, and necroses.

Oncocytic type with diffuse growth pattern, strong atypia, dense acidophilic cytoplasm, and no lipid vacuoles. Hematoxylin–eosin staining, 440×
Sarcomatoid subtype
The sarcomatoid subtype is characterized by a mostly very irregular, often partly spindle cell-like epithelial component and by sarcomatoid mesenchymal formations with spindle cell or fascicular formations (Table 2) (Fig. 4). For the differentiation of the sarcomatoid part, immunostainings are essential. By these, rhabdomyoblastic, leiomyoblastic, fibroblastic, lipoblastic, chondroid, or osteoid areas can be identified. In some cases, additional methods of molecular pathology are necessary.

Sarcomatoid type with outlined fascicular pattern, spindly pleomorphic cells with atypical nuclei. Hematoxylin–eosin staining, 360×
Comparison of scoring systems
For evaluation of special importance of different criteria in the four scoring systems (Table 3), we gathered the criteria into the groups growth pattern, cell structures, proliferation parameters, proportion of spongiocytes, invasion of vessels or capsule, and regressive changes. The diffuse growth pattern is very important only in the system of Hough et al. [15]. The atypical cell structures (nuclear atypia, nuclear hyperchromasia, and enlarged nucleoli) are most important in the system of van Slooten et al. [38]. If all of these criteria for atypical cell structures are present, the threshold value is exceeded. In two other systems, these structures are much lower important reaching a value of up to 19% for reaching the lowest value for diagnosing malignancy. The signs of proliferation (mitoses) are differently important: in the system of van Slooten et al. [38], the value for malignancy can be fulfilled by one criterion of increased proliferation (mitoses in > 2/10 HPF). The Helsinki score is the only one taking the Ki-67 index into account (value three or more than 5/50 mitoses per HPF plus the number of Ki-67 index). Regressive changes are very important in the systems of Van Slooten et al. score [38] (value 70%) and Hough et al. score [15] (value 33%) and most important in the Helsinki score. Since all cancers of our collection showed a number of more than the threshold value in the Helsinki score [29], this system appeared to be the most reliable procedure—the gold standard—in differentiating adrenal carcinomas from cortical adenomas.
Relative frequencies of subtypes
Our data of subtyping show that the main type comprehends 97% of all adrenocortical cancers, the oncocytic type 2%, the myxoid type 0.8%, and the rarest sarcomatoid type 0.2% (Table 2).
Discussion
Main type
From our studies, the most characteristic features of the main type are the signs of increased proliferation (mitoses > 5/50 HPF, Ki-67 index > 5%) and the amount of necroses (diameter > 2 HPF). Less important criteria are the growth pattern, the infiltration of capsule and vessels, and the amount of lipid vacuoles in the cytoplasm.
The histomorphology, immunostaining, and differential diagnosis of the adrenocortical main type cancer have been published in the WHO classification of 2004 [23] and 2017 [12] but also in other textbooks [8, 21] and are reflected by the scoring systems of Weiss et al. [39], van Slooten et al. [38], and of Hough et al. [15] and by the Helsinki score [29]. All these characteristics correspond to our findings in our large collection.
Discrepancies between benign and malignant diagnoses from data of the classical three scoring systems lead to the diagnosis “indeterminate malignancy”. This could be canceled by the Helsinki score [29] by the criteria necroses, mitoses, and (most important) Ki-67 index more than eight. Therefore, it should be discussed whether or not the Helsinki score can replace the classical three scoring systems and be used alone.
Myxoid subtype
Our criterion for the myxoid subtype was the enormous amount of myxoid fields with alcian bue-positivity going beyond half of tumor volume. Cancers with smaller myxoid areas were assigned to the main type as all other criteria are identical with the main type.
All reported myxoid adrenal tumors are endocrine active. About half of them are benign [16, 31]. The areas of myxoid alterations comprehend between 5 and 90% of tumor volume [28].
Differences of myxoid cancers in their prognosis compared with the main type carcinomas could not be found [28].
Oncocytic subtype
In contrast to the main type, the oncocytic cancer type is more complicated and controversially discussed in the literature. Only about 20% of adrenocortical oncocytic tumors are malignant [19, 26]. To assess malignancy, Bisceglia et al. [4] created a special scoring system with mayor criteria (Mitotic rate of more than 5 per HPF, atypical mitotic figures, or venous invasion). Only one of these main criteria should be sufficient for diagnosis of malignancy, whereas other criteria (nuclear pleomorphism, nucleoli) of other scoring systems are completely irrelevant. In our opinion, oncocytic tumors are nearly entirely composed of oncocytes, whereas others [4] define them by oncocytic predominance. We agree with Bisceglia et al. [4], Mearini et al. [26], and Summer et al. [36] that oncocytic adrenocortical tumors are hormonally inactive probably due to the strongly increased but malfunctioning mitochondria. Others have reported that between 10 and 20% of oncocytic adrenocortical tumors produce hormones [42] and one case report was published in which Cushing’s syndrome was induced by this tumor type [2]. We believe that these are not oncocytic active tumors but adrenocortical tumors especially of lipid-poor compact cell type that are more active than lipid-rich cells. The differentiation from oncocytic tumors may be possible when a minority of tumor cells show lipid vacuoles in the cytoplasm which are lacking in oncocytic tumors. In animal experiments, it could be shown that increased stimulation of adrenocortical cells induces a reduction of spongiocytes and an increase of compact cells [33].
Sarcomatoid subtype
If an adrenocortical malignant tumor shows areas of epithelial differentiation and greater areas of sarcomatous differentiation, the term “carcinosarcoma” [11, 20, 22, 37] should be used. If a more homogenous tumorous proliferation possesses some characteristics of sarcomas and some of carcinomas, the term “sarcomatoid carcinoma” [6, 10, 14, 18, 18, 24, 35] is appropriate. The sarcomatous features including their immunostainings have similarities with leiomyosarcoma [5], rhabdomyosarcoma [7, 11, 37], spindle cell sarcoma [5, 11, 22, 35], or osteosarcoma [3].
One tumor showed remnants of adrenocortical features—a precursor lesion?—in an otherwise dedifferentiated sarcoma [34]. If no adrenocortical features could be found, an adrenal sarcoma is likely [13, 17, 25]. Immunostainings are generally necessary for differentiation and, in single cases, molecular pathology [34].
Comparison of scoring systems
The four scoring systems (Table 3) have different priorities. The Weiss score [39] stresses the invasion of capsule and vessels (3 points), the van Sooten score [38], the atypical cell structures, and the mitotic index, the Hough score [15], the atypical cell structures, the regressive changes, and the diffuse growth pattern.
The Helsinki score [29] based on studies of 167 consecutive adrenal cancers that were identified with the Weiss score and clinical follow-up checks. By integrating the Ki-67 index in the differentiation of carcinomas from adenomas, the authors developed their system which showed a sensitivity of 100% and a specificity of 99.6%.
In our studies, all adrenal cancers showed a scoring number of more than the threshold value of 8.5 in the Helsinki score. Therefore, we can confirm the data of Pennanen et al. [29] and emphasize that the Helsinki score is the most reliable procedure in differentiating adrenal carcinomas from cortical adenomas.
Molecular pathology
Molecular pathology became very important for choice of treatment and prognosis of malignant tumors, but for adrenal cancers, tumor programmed death-ligand 1 (PD-L1) expression and MSI-H/MMR-D status were not associated with objective response [30] Adrenocortical carcinoma is a Lynch syndrome-associated cancer. Three of four patients carried a pathogenic germ-line mutation in a mismatch repair gene [1]. Whether or not these molecular alterations correlate to subtypes of adrenal cancer is not known and should be studied.
Prognosis
The median overall prognosis amount to 3–4 years [9]. 5 year survival comes to 60–80% if the tumor is confined to adrenal space, to 35–50% if the cancer is locally advanced, and to 0–25% if metastases exist [9]. Further important prognostic markers are Ki-67 index, Weiss score, mitotic index, R-status [9], and chromosomal aberrations [41]. An adjuvant mitotane therapy is necessary if Ki-67 index exceeds 10%, and an advanced stage or an R1 status exists [9].
Statistic
The statistical data from our large collection concerning the relative frequency of oncocytic adrenal cancers (2%) are in strong contrast to data (22%) found by others [27]. The main type is in the huge majority of our collection, and the subtypes comprehend 3% of all adrenal cancers.
Conclusion
Answering the questions in introduction, we confirm
-
1.
that the Helsinki score is the best for differentiating adrenal carcinomas of the main and myxoid type in routine work,
-
2.
that additional scoring systems for these carcinomas are generally not any longer necessary,
-
3.
that signs of proliferation (mitoses and Ki-67 index) and necroses are the most important criteria for diagnoses of the different subtypes,
-
4.
that, according to the relative frequencies of the different subtypes of adrenal cancers, the main type is the far most frequent (97%) followed by the oncocytic type (2%), the myxoid type (0.8%), and the very rare sarcomatoid type (0.2%).
References
Albers CA, Newbury-Ecob R, Ouwehand WH, Ghevaert C. New insights into the genetic basis of TAR (thrombocytopenia absent radii) syndrome. Curr Opin Genet Dev. 2013;23:316–23.
Alexander A, Paulose RP. Oncocytic variant of adrenal carcinoma presenting as Cushing’s syndrome. J Assoc Physicians India. 1996;46:235–7.
Barksdale SK, Marincola FM, Jaffe G. Carcinosarcoma of the adrenal cortex presenting with mineralocorticoid excess. Am J Surg Pathol. 1993;17:941–5.
Bisceglia M, Ludovico O, Di Mattia A, Ben-Dor D, Sandbank J, Pasquinelli G, Lau SK, Weiss LM. Adrenocortical oncocytic tumors: report of 10 cases and review of the literature. Int J Surg Pathol. 2004;12:231–43.
Coli A, Di Giorgio A, Castri F, Destito C, Marin AW, Bigotti G. Sarcomatoid carcinoma of the adrenal gland: a case report and review of literature. Pathol Res Pract. 2010;206:59–65.
Collina G, Maldarizzi F, Betts CM, Eusebi V. Primary sarcomatoid carcinoma of the adrenal gland; first case report. Virchows Arch A Pathol Anat Histopath. 1989;415:161–7.
Decorato JW, Gruber M, Petti M, Levowitz BS. Adrenal carcinosarcoma. J Surg Oncol. 1990;45:134–6.
Duan K, Giordano TJ, Mete O. Adrenal cortex. In: Mete O, Asa SL, editors. Endocrine pathology. Cambridge: Cambridge University Press; 2016. p. 588–627.
Fassnacht M, Dekkers OM, Else T, Gaudin E, Berruti A, de Krijger RR, Haak HR, Mihail R, Assie G, Terzolo M. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrin. 2018;179:G1–46.
Feng YC, Yang ZG, Chen TW, Su XY, Deng W, Wang QL. Adrenal sarcomatoid carcinoma: a rare case depicted on multi-detector row computed tomography. Indian J Med Sci. 2010;64:37–40.
Fischler DF, Nunez C, Levin HS, McMahon JT, Sheeler LR, Adelstein DJ. Adrenal carcinosarcoma presenting in a woman with clinical signs of virilization—a case-report with immunohistochemical and ultrastructural findings. Amer J Surg Pathol. 1992;16:626–31.
Giordano TJ, Chrousos GP, de Krijger RR, Gill A, Kawashima A, Koch CA, Medeiros LJ, Merino M, Papathomas TG, Papotti M, Sasano HR, Weiss LM. Tumours of the adrenal cortex. In: Lloyd RV, Osamura RY, Klöppel G, Rosai J, editors. WHO classification of tumours of endocrine organs. Heidelberg-Berlin: Springer; 2017. p. 161–78.
Hart J, Mandavilli S. Epithelioid angiosarcoma a brief diagnostic review and differential diagnosis. Arch Pathol Lab Med. 2011;135:268–72.
Hayashi T, Gucer H, Mete O. A mimic of sarcomatoid adrenal cortical carcinoma: epithelioid angiosarcoma occurring in adrenal cortical adenoma. Endocr Pathol. 2014;25:404–9.
Hough AJ, Hollifield JW, Page DL, Hartmann WH. Prognostic factors in adrenal cortical tumours. Am J Clin Pathol. 1979;72:390–9.
Hsieh MS, Chen JH, Lin LW. Myxoid adrenal cortical carcinoma presenting as primary hyperaldosteronism: case report and review of the literature. Int J Surg Pathol. 2011;19:803–7.
Hung YP, Hornick JL. Immunohistochemical biomarkers of mesenchymal neoplasms in endocrine organs: diagnostic pitfalls and recent discoveries. Endocr Pathol. 2018;29:189–98.
Ishikawa N, Nagase M, Takami S, Araki A, Ishikawa N, Koike C, Shiina H, Maruyama R. A case report of bilateral sarcomatoid carcinoma of adrenal glands with adrenal insufficiency. Int J Surg Pathol. 2016;24:743–8.
Kakimoto S, Yushita Y, Sanefuji T, Kondo A, Fujishima N, Kishikawa M, Matsumoto K. Nonhormonal adrenocortical adenoma with oncocytoma-like appearances. Hinyokika Kiyo. 1986;32:757–63.
Kao CS, Grignon DJ, Ulbright TM, Idrees MT. A case report of adrenocortical carcinosarcoma with oncocytic and primitive neuroectodermal-like features. Hum Pathol. 2013;44:1947–55.
Lack EE. Adrenal cortical carcinoma. In:Lack EE (eds) Tumors of the adrenal glands and extraadrenal paraganglia, Ed 1, pp131–160. Armed Forces Institute of Pathology, Washington. 2007.
Lee MS, Park IA, Chi JG, Ham EK, Lee KC, Lee CW. Adrenal carcinosarcoma a case report. J Korean Med Sci. 1997;12:374–7.
Lloyd RV, Tischler AS, Kimura N, McNicol AM, Young WF Jr, Weiss LM, Bertagna X, Chrousos GP, Kawashima A, Kleihues P, Giordano TJ, Medeiros LJ, Merino MJ, Ordonez NG, Sasano H, Koch CA, Thompson LDR, Komminoth P, Chetty R, Capella C, Albores-Saavedra J, Lam KY, Cheng L, Ulbright TM. Tumours of the adrenal gland. In: DeLellis RA, Lloyd RV, Heitz PU (eds) Pathology and genetics. Tumours of endocrine tumours, Ed 1, pp 135–173. International Agency for Research and Cancer (IARC), Lyon. 2004.
Mark D, Boyd C, Eatock F. Adrenal sarcomatoid carcinoma: a case report and review of the literature. Ulster Med J. 2014;83:89–92.
McLaughlin SA, Schmitt TM, Huguet KL, Menke DM, Nguyen JH. Myofibrosarcoma of the adrenal gland. Am Surg. 2005;71:191–3.
Mearini L, Del Sordo R, Costantini E, Nunzi E, Porena M. Adrenal oncocytic neoplasm: a systematic review. Urol Int. 2013;91:125–33.
Mills JK, Khalil M, Pasieka J, Kong SY, Xu Y, Harvey A. Oncocytic subtypes of adrenal cortical carcinoma: aggressive in appearance yet more indolent in behavior? Surgery. 2019;166:524–33.
Papotti M, Volante M, Duregon E, Delsedime L, Terzolo M, Berruti A, Rosai J. Adrenocortical tumors with myxoid features: a distinct morphologic and phenotypical variant exhibiting malignant behavior. Amer J Surg Pathol. 2010;34:973–83.
Pennanen M, Heiskanen I, Sane T, Remes S, Mustonen H, Haglund C, Arola J. Helsinki score-a novel model for prediction of metastases in adrenocortical carcinomas. Hum Pathol. 2015;46:404–10.
Raj N, Zheng YY, Kelly V, Katz SS, Chou J, Do RKG, Capanu M, Zamarin D, Saltz LB, Ariyan CE, Untch BR, O’Reilly EM, Gopalan A, Berger MF, Olino K, Segal NH, Reidy-Lagunes DL. PD-1 blockade in advanced adrenocortical carcinoma. J Clin Oncol. 2020;38:71.
Raparia K, Ayala AG, Sienko A, Zhai QJ, Ro JY. Myxoid adrenal cortical neoplasms. Ann Diagn Pathol. 2008;12:344–8.
Saeger W. Nebennierenrinde. In: Amann K, Kain R, Klöppel G (eds) Urogenitale und endokrine Organe, Gelenke und Skelett, Ed 3. Aufl: pp 735–772. Springer, Heidelberg. 2015.
Saeger W, Mitschke H. Licht-und elektronenmikroskopische untersuchungen an der zona glomerulosa der rattennebenniere nach carbenoxolon. Virchows Arch A Path Anat. 1973;358:45–59.
Saeger W, Mohren W, Behrend M, Iglauer P, Wilczak W. Sarcomatoid adrenal carcinoma: case report with contribution to pathogenesis. Endocr Pathol. 2017;28:139–45.
Sturm N, Moulai N, Laverriere M, Chabre O, Descotes J, Brambilla E. Primary adrenocortical sarcomatoid carcinoma: case report and review of literature. Virchows Arch. 2008;452:215–9.
Sumner E, Acar BC, Acker MR. Oncocytic adrenocortical carcinoma: a rare adrenal tumor subtype. Can J Urol. 2017;24:8865–7.
Thway K, Ohms D, Shah C, Flora R, Shipley J, Fisher C. Oncocytic adrenal cortical carcinosarcoma with pleomorphic rhabdomyosarcomatous metastases. Am J Surg Pathol. 2012;36:470–7.
van Slooten H, Schaberg A, Smeenk D, Moolenaar AJ. Morphologic characteristics of benign and malignant adrenocortical tumors. Cancer. 1985;55:766–73.
Weiss LM, Medeiros LJ, Vickery AL. Pathologic features of prognostic significance in adrenal cortical carcinoma. Am J Surg Pathol. 1989;13:202–6.
Wieneke JA, Thompson LDR, Heffess CS. Adrenal cortical neoplasms in the pediatric population—a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol. 2003;27:867–81.
Wittekind C. TNM Klassifikation maligner Tumoren (Union for International Cancer Control), Ed. 8, pp 1–313. Willey-VCH, Weinheim. 2017.
Wong DD, Spagnolo DV, Bisceglia M, Havlat M, McCallum D, Platten MA. Oncocytic adrenocorticalneoplasms—a clinicopathologic study of 13 new cases emphasizing the importance of their recognition. Hum Pathol. 2011;42:489–99.
Zhu C, Zheng A, Mao X, Shi B, Li X. Primary adrenal sarcomatoid carcinoma metastatic to the lung: case report and review of the literature. Oncol Lett. 2016;11:3117–22.
Acknowledgements
The authors (WS, SM) would like to thank all clinicians and pathologists for sending paraffin blocks of adrenocortical tumors for a second opinion or consultant diagnosis which were the base of these studies.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Informed consent
For this type of study, no informed consent is required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Minner, S., Schreiner, J. & Saeger, W. Adrenal cancer: relevance of different grading systems and subtypes. Clin Transl Oncol 23, 1350–1357 (2021). https://doi.org/10.1007/s12094-020-02524-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-020-02524-2