
Abstract
Background and Aims
The liver plays a key role in the storage, metabolism and homeostasis of fat-soluble vitamins. We investigated the relation of Vitamin(Vit)A/D/E serum levels with severity of liver disease and portal hypertension (PHT).
Methods
VitA/D/E serum levels were assessed in 234 patients with advanced chronic liver disease (ACLD, i.e. hepatic venous pressure gradient [HVPG] ≥ 6 mmHg). Patients with hepatocellular carcinoma, pre-/post-hepatic PHT, TIPS or liver transplantation were excluded.
Results
Most patients were male (n = 153; 65%) with a median age of 57.6 (49.7–64.5) years. Thirty-two (14%) patients had HVPG 6–9 mmHg, 66 (28%) 10-15 mmHg, and 136 (58%) ≥ 16 mmHg, respectively. VitD deficiency (25-OH-vitamin-D <50 nmol/L) was found in 133 (57%) with higher prevalence in Child-Turcotte-Pugh (CTP)-C: 85% vs. B: 66% vs. A: 47% (p < 0.001). VitD levels displayed significant but weak correlations with hepatic dysfunction and PHT. VitE levels were normal in 227 (97%) patients and displayed no relevant association with hepatic dysfunction or PHT. Only 63 (27%) patients had normal (>1.05 µmol/L) VitA levels, while 58 (25%) had mild (0.70–1.04 µmol/L), 71 (30%) moderate (0.35–0.69 µmol/L), and 42(18%) severe(<0.35 µmol/L) VitA deficiency. VitA correlated with HVPG (Rho = −0.409), CTP score (Rho = −0.646), and serum bile acid levels (Rho = −0.531; all p < 0.001). The prevalence of decompensated ACLD (dACLD) continuously increased with severity of VitA deficiency (no: 40% vs. mild: 51% vs. moderate: 67% vs. severe: 91% had dACLD; p < 0.001). CTP score (per point; OR 2.46; 95%CI 1.80–3.37; p <0.001), age (per year; OR 0.95; 95%CI 0.92–0.98; p = 0.001) and elevated bile acid levels(>10 µmol/L; OR 3.62; 95%CI 1.61–8.14; p = 0.002) were independently associated with VitA deficiency.
Conclusion
VitA and VitD but not VitE deficiencies are highly prevalent in ACLD. VitA deficiency strongly correlates with hepatic dysfunction, PHT and bile acid levels and is associated with decompensated ACLD.
Trial registration number
NCT03267615.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fat-soluble vitamins A (retinol), D (cholecalciferol), E (tocopherols and tocotrienols), and K (phylloquinone and menaquinones) are lipophilic molecules with distinct physiological properties. While dietary intake is an important (or even exclusive) source for all vitamins, the lipophilicity of fat-soluble vitamins requires mediation of bile acids (BA) for intestinal uptake [1].
Dietary uptake is the exclusive source of vitamin A (VitA) in humans [2]. Notably, 60–95% of VitA is stored in the liver of healthy individuals, while only minor fractions are located in extrahepatic tissues [3]. Importantly, activated hepatic stellate cells (HSCs) are the main driver of fibrogenesis upon liver injury by producing extracellular matrix proteins. During their activation process, HSCs lose their lipid droplets containing retinyl esters [4, 5]. In patients with non-alcoholic fatty liver disease (NAFLD) undergoing bariatric surgery, reduced levels of serum and hepatic retinol, as well as retinoic acid (RA) have been observed, being inversely correlated with grade of hepatic steatosis and severity of non-alcoholic steatohepatitis (NASH) [6]. In other etiologies of (advanced) chronic liver disease, VitA deficiency was reported in a considerable percentage of patients and associated with higher fibrosis stages or cirrhosis [7,8,9].
Vitamin D (VitD) is mainly derived from the endogenous synthesis in the skin followed by two critical steps of hydroxylation in the liver and the kidneys [10]. Importantly, a previous study on patients with cirrhosis displayed a link between severe vitamin D deficiency (VitDDef) and increased levels of inflammatory biomarkers as well as the risk of hepatic decompensation [11]. Similarly, disease severity and mortality were associated with VitDDef [12, 13].
Vitamin E (VitE) functions as an antioxidant by scavenging peroxyl radicals and regulating the oxidation process of polyunsaturated fatty acids, and is widely reported as a modulator and promoter of immunity [14]. However, while the direct effect of VitE on immunity-/oxidative stress-related pathways may be overestimated, prior studies have demonstrated an association between disease severity and oxidative stress in NAFLD patients [15].
This study aimed to investigate the prevalence of vitamin A, D, and E deficiencies in prospectively recruited patients with advanced chronic liver disease (ACLD) and to determine their association with severity of liver disease and portal hypertension (PHT). Furthermore, we assessed the previously unreported relation to serum levels of bile acids and fibrosis markers to address pathophysiological concepts of vitamin A homeostasis.
Patients and methods
Study design
234 patients with ACLD [defined by hepatic venous pressure gradient (HVPG) ≥ 6 mmHg] undergoing hepatic vein catheterization at the Vienna Hepatic Hemodynamic Lab of the Medical University of Vienna were consecutively included in the prospective VICIS study (NCT03267615) between 01/2017 and 03/2020. Patients with non-cirrhotic PHT, pre- or post-hepatic PHT, hepatocellular carcinoma, history of transjugular intrahepatic portosystemic shunt (TIPS) implantation or liver transplantation were excluded (Supplementary Fig. S1). Furthermore, patients under treatment with non-selective betablockers (NSBB) were excluded. More specifically, patients either had never received NSBB (including carvedilol) or paused NSBB intake 5 days before HVPG measurement. Patients’ medical records were reviewed to obtain relevant clinical information, laboratory parameters, and prescription vitamin supplements. Compensated ACLD (cACLD) was defined as the absence of hepatic decompensation events prior to HVPG measurement, i.e. ascites, hepatic encephalopathy, and variceal bleeding [16].
Analysis of laboratory parameters
All reported laboratory parameters were assessed from blood samples obtained via the catheter introducer sheath placed in the internal jugular vein for HVPG measurement. Detailed information towards laboratory analyses is depicted in the supplementary material (“Supplementary Methods”).
HVPG measurements and transient elastography
HVPG measurements were performed by trained physicians of the Vienna Hepatic Hemodynamic Lab following a defined standard operating procedure in fasting condition [17]. Detailed steps of the procedure are delineated in the supplementary material (“Supplementary Methods”).
Statistics
Statistical analyses were performed using IBM SPSS Statistics 26 (IBM, Armonk, New York, USA) and GraphPad Prism 8 (GraphPad Software, La Jolla, California, USA). Continuous variables are reported as mean ± standard error of the mean (SEM) or median and interquartile range (IQR), and categorical variables are presented as numbers (n) and proportions (%) of patients. Comparisons of continuous variables were performed using Student’s t test or Mann–Whitney U test, as applicable. Post-hoc analysis was performed using Dunn’s multiple comparison test. Categorical variables were compared with Chi squared or Fisher‘s exact test, as applicable. Correlation between parameters were assessed by calculation of either Spearman or Pearson correlation coefficients dependent on the distribution of the respective parameters. Risk factors for moderate or severe vitamin A deficiency (VitADef), or VitDDef, were calculated by uni- and multivariate logistic regression analysis, respectively. Parameters achieving a p value ≤ 0.10 in univariate analysis were subsequently included in multivariate analysis. In all analyses, a two-sided p value ≤ 0.05 was defined to denote statistical significance.
Compliance with ethical standards
This study was conducted in accordance with the 1964 Helsinki declaration and its later amendments and approved by the local ethics committee of the Medical University of Vienna (EK1262/2017). All patients gave written informed consent to liver vein catheterizations and provided written consent to be enrolled in the VICIS study (NCT03267615). All authors had access to the study data and reviewed and approved the final manuscript.
Results
Patient characteristics
Most of the patients included in this study were male (n = 153/234, 65.4%), while the median age was 57.6 (49.7–64.5) years. Alcohol-related liver disease (ALD; n = 96, 41%) and viral hepatitis (n = 47, 20%) represented the predominant ACLD etiologies. Median HVPG was 18 (12–20) mmHg, and 202 (86%) patients had clinically significant portal hypertension (CSPH, i.e. an HVPG ≥ 10 mmHg). More specifically, HVPG was 6–9 mmHg in 32 patients (14%), 10–15 mmHg in 66 (28%) and ≥ 16 mmHg in 136 patients (58%). The majority of our study cohort was classified as Child-Turcotte-Pugh (CTP) stage A (n = 131, 56%), 83 (35%) CTP stage B, and 20 (9%) CTP stage C (Supplementary Table S1). Only 1 (0.4%) patient received prescription VitA supplements, whereas no prescription for VitE was recorded (of note, VitE is available as an “over-the-counter” supplement). Furthermore, 59 (25%) patients reported prescription VitD supplements. Twenty-one (9%) of these patients had VitD levels within the normal range, whereas 22 (9%) and 16 (7%) patients still had VitD insufficiency and deficiency, respectively. In only 6 (2.6%) patients, potential vitamin prescriptions were insufficiently recorded.
Prevalence of vitamin A, D, and E deficiency
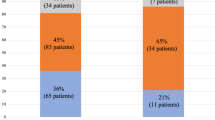
The minority of patients (n = 63, 27%) in our study cohort had vitamin A serum levels within the normal range, whereas 53 (23%) had mild VitADef, 76 (32%) had moderate VitADef, and 42 (18%) had severe VitADef. When stratifying patients by severity of PHT (i.e., 6–9, 10–15, ≥ 16 mmHg), VitADef prevalence incremented with rising HVPG (p < 0.001; Fig. 1). More importantly, stratification by CTP stage revealed a pronounced stepwise increase of VitADef severity across groups: 2 (1.5%) patients with CTP A, 22 (27%) with CTP B, and 18 (90%) with CTP C had severe VitADef (p < 0.001; Fig. 1a; Supplementary Table S2). Furthermore, the proportion of patients with decompensated ACLD (dACLD) continuously rose with VitADef severity grade as compared to compensated patients: 40% without VitADef, 51% with mild, 67% with moderate, and 91% with severe VitADef had dACLD (p < 0.001; Fig. 2a).

Vitamin A, D, and E serum levels in patients stratified by (a, b, c) Child-Turcotte-Pugh (CTP) stage and (d, e, f) hepatic venous pressure gradient (HVPG). Different colours in the background indicate ranges between cut-offs for Vitamin A, D, and E deficiencies as specified in the methods section. VitA vitamin A, VitD vitamin D, VitE vitamin E, HVPG hepatic venous pressure gradient, CTP Child-Turcotte-Pugh, ns not significant, (*) p < 0.05, (***) p < 0.001

Prevalence of compensated and decompensated advanced chronic liver disease in patients stratified by the presence and severity of vitamin A, D, and E deficiency. VitA vitamin A, VitD vitamin D, VitE vitamin E, cACLD compensated advanced chronic liver disease, dACLD decompensated advanced chronic liver disease
Similarly, only 41 (17%) patients had VitD serum levels within the normal range, whereas 60 (26%) and 133 (57%) had VitD insufficiency and deficiency, respectively. VitD levels also gradually decreased across HVPG and CTP strata, however, the discrimination between groups was less pronounced as compared to VitA (Fig. 1e, p = 0.009; Fig. 2b, p = 0.008). Nevertheless, VitDDef was observed among 62 (47%) patients with CTP A, 54 (66%) with CTP B, and 17 (85%) with CTP C (p < 0.001; Fig. 1b). Patients reporting VitD prescription supplementation had higher VitD levels across individual CTP and HVPG strata (Supplementary Fig. S2).
In contrast, vitamin E deficiency (VitEDef) was observed in only 7 (3%) patients. Severity of portal hypertension or disease stage was not associated with serum levels of VitE (Figs. 1c, f, 2c).
Correlation between vitamin A, D, E levels and parameters of hepatic (dys-)function
VitA serum levels showed a significant negative correlation with HVPG (Rho = −0.409, 95%CI: −0.51 to [−0.29]; p < 0.001), and even stronger association with MELD (Rho = −0.552, 95%CI: −0.64 to [−0.45]; p < 0.001; Fig. 3a) and CTP score (Rho = −0.646, 95%CI: −0.72 to [−0.56]; p < 0.001) (Table 1). Similarly, serum BA levels were significantly associated with VitA (Rho = −0.531, 95%CI −0.62 to [−0.43]; p < 0.001; Fig. 2b). Furthermore, surrogate parameters for liver fibrosis displayed inverse correlation with VitA: Rho = −0.393 (95%CI −0.52 to [−0.26]; p < 0.001; available in n = 174) for vibration-controlled transient elastography (VCTE) and Rho = -0.571 (95%CI −0.66 to [−0.47]; p < 0.001; Fig. 2c) for enhanced liver fibrosis score (ELF).

Correlation between vitamin A serum levels and Model for End-stage Liver Disease (MELD) score, bile acid serum levels, and enhanced liver fibrosis (ELF) score. VitA vitamin A, MELD Model for end-stage liver disease, BA bile acids, ELF enhanced liver fibrosis score
Furthermore, VitA levels strongly correlated with any single parameter of CTP and MELD scores reflecting hepatic synthesis (Table 1, Supplementary Fig. S3). As for the good correlation of VitA with Vitamin K-dependent coagulation parameters, i.e., INR (Rho = −0.699, 95%CI −0.76 to [−0.62], p < 0.001) and Protein C activity (Rho = 0.777, 95%CI 0.72–0.83, p < 0.001), we also observed strong association with vitamin K-independent antithrombin III (ATIII; Rho = 0.739, 95%CI 0.67–0.79; p < 0.001) (Supplementary Fig. S3).
VitD levels displayed significant but weak correlations with PHT and hepatic dysfunction. Similarly, weak but statistically significant associations were found for parameters reflecting hepatic synthesis capacity (Table 1).
Lastly, we observed no meaningful associations between VitE and PHT, disease severity, or hepatic synthesis. Furthermore, neither BA levels nor ELF score showed a significant correlation with VitE (Table 1).
Differences in hepatic (dys-)function between high and low vitamin A quintiles
Due to the strong association between hepatic dysfunction/disease severity and VitA, we aimed to assess differences between patients stratified by high and low quintiles of VitA serum levels (Supplementary Table S3). Consequently, quintile 1 (Q1) represented patients with the lowest 20% of VitA levels, Q2-Q4 represented percentiles 20–80, and Q5 represented patients with the highest 20% VitA levels.
Interestingly, both age and sex displayed significant differences between low and high quintiles. Patients in Q5 (i.e. high VitA levels) were older than patients in lower quintiles (63.7 vs. 54.0 years; p = 0.001). In contrast, male patients were overrepresented in Q5, indicating that women had significantly lower levels of vitamin A (male sex 79.6% vs. 55.1%; p = 0.034).
Concordantly, HVPG (median 19 mmHg in Q1 vs. 12 mmHg in Q5), CTP (8 points in Q1 vs. 5 points in Q5) and MELD (16 points in Q1 vs. 9 points in Q5) score, as well as the prevalence of dACLD (84% in Q1 vs. 41% in Q5) significantly differed between quintiles (all p < 0.001).
Similarly, VitK-dependent and -independent coagulation parameters displayed strong differences between VitA quintiles (Fig. 4): median INR yielded 1.7 in Q1 vs. 1.2 in Q5, AT-III activity was almost half with 43% in Q1 vs. 83% in Q5, similar to protein C activity with 33% in Q1 and 84% in Q5 (all p < 0.001).

Comparison of coagulation parameters in patients stratified by vitamin A quintiles. Green colour in the background indicates normal ranges for AT-III and Protein C activity, respectively. Q1–Q5 quintile 1–5; INR international normalized ratio; AT-III antithrombin-III, (***) p < 0.001
Conversely, body-mass index (BMI) as a basic parameter for cachexia/obesity/nutritional status was similar between vitamin A strata (p = 0.121).
Independent risk factors for vitamin A, D, and E deficiency
Furthermore, independent risk factors for vitamin A/D/E deficiencies were assessed using binary logistic regression analysis (Table 2). Age (per year; OR 0.97, 95% CI 0.94–0.99, p = 0.003), HVPG (per mmHg; OR 1.16, 95%CI 1.10–1.22, p < 0.001), CTP score (per point; OR 2.44, 95% CI 1.89–3.15, p < 0.001), and elevated serum BA (OR 7.06, 95% CI 3.73–13.4, p < 0.001) were associated with VitADef on univariate analysis (Table 2). On multivariate analysis, CTP score (per point; OR 2.25, 95% CI 1.67–3.04, p < 0.001) and elevated serum BA levels (OR 3.53, 95% CI 1.61–7.76, p = 0.002) emerged as independent risk factors for VitADef, whereas age (per year; OR 0.95, 95% CI 0.92–0.98, p = 0.001) was associated with the absence of moderate or severe VitADef.
Similarly, HVPG (per mmHg; OR 1.06, 95%CI 1.02–1.11, p = 0.008) and CTP score (per point; OR 1.40, 95% CI 1.17–1.67, p < 0.001) but not age or serum BA levels–were significantly associated with VitDDef on univariate analysis (Table 2). Only CTP score remained an independent risk factor for VitDDef (per point; OR 1.31, 95% CI 1.07–1.60, p = 0.008).
Importantly, when performing these analyses using either HVPG or CTP score only, both of these parameters emerged as independent risk factors for VitA and VitD deficiency, respectively, which might be attributed to collinearity between HVPG and CTP score in reflecting the severity of liver disease (Supplementary Tables S4, S5).
Conversely, we were not able to identify any risk factor for VitE deficiency (Supplementary Table S6).
Independent risk factors for decompensated liver disease
Since VitA and VitD deficiency was more prevalent in patients with dACLD, we assessed whether VitA and VitD levels were independently associated with dACLD (Supplementary Table S7). We chose to use MELD for this analysis of dACLD patients only, as the decompensation events ascites and encephalopathy are included in the CTP score. On univariate analysis, HVPG and MELD, as well as VitA and VitD serum levels emerged as risk factors for dACLD. However, only MELD (per point; OR 1.22, 95%CI 1.09–1.36; p < 0.001) and HVPG (per mmHg; OR 1.19, 95%CI 1.11–1.27; p < 0.001) remained independently associated with dACLD.
Discussion
This study is the first to simultaneously describe VitA/D/E status in a large prospective cohort of 234 ACLD patients with characterized severity of PHT. We found a VitADef in 73% of our patients and its prevalence increased with CTP stages and was associated with dACLD. Similarly, 57% had VitDDef, which also displayed a distinct association with disease severity. Conversely, we found that only 3% had low VitE serum levels and did not observe an association of VitE levels with liver dysfunction or PHT severity. Of note, our study provides novel data by investigating the relationship of these fat-soluble vitamins with serum BA levels as well as ELF score (both available in about 95% of patients), which is a well-established and an increasingly used non-invasive surrogate parameter for fibrosis.
The liver represents the key regulator of VitA homeostasis, its storage in HSCs, intestinal absorption by BA production and its systemic distribution by the synthesis of retinol-binding protein 4 (RBP4) and transthyretin [18]. As mentioned above, prior studies on patients with NAFLD reported reduced VitA levels both in the serum and the liver [6], being inversely correlated with the grade of hepatic steatosis and severity of NASH [19]. It is reasonable to hypothesize that during the progression of fibrosis, HSCs lose their essential ability to store VitA, subsequently leading to progressive VitADef as fibrosis accumulates in the liver. Ultimately, this process might be reflected by reduced VitA serum levels. This assumption is supported by data from the literature [7,8,9], as well as the moderate/strong correlation of VitA levels with markers of hepatic dysfunction and, importantly, ELF score, in our study. However, it has to be acknowledged that VitA serum levels may not mirror hepatic VitA content [20].
Furthermore, VitADef may also be due to inadequate intake or intestinal malabsorption. In this regard, it seems very interesting that elevated bile acids were independently associated with VitADef. It might be argued that serum levels might not reflect the availability of bile acids in the intestines and that impaired bile acid homeostasis and biliary excretion may rather reflect impaired liver function in cirrhosis [21]. Intriguingly, retinoic acid is required for activation of retinoid X receptor-alpha (RXR-α) as a heterodimeric partner of the farnesoid X receptor (FXR) and other key nuclear receptors required for transactivation of genes maintaining proper bile acid homeostasis and excretion [22]. Importantly, elevated serum BA levels may also be a reflection of portosystemic shunting, and thereby, the degree of PHT [23, 24].
Dietary intake of VitA was not systematically assessed in our patients, which represents an important limitation of this study. Interestingly, a previous study on patients with cirrhosis reported that adequate VitA intake did not prevent VitA deficiency [25]. Similarly, another study on patients with (A)CLD applied the relative-dose-response method to assess hepatic VitA storages after oral VitA challenge and reported that neither duration nor dosage of VitA supplementation was independently associated with an effective test response. In contrast, low RBP was the only independent parameter associated with failed test response [26]. Since RBP is synthesized in the liver, it might be hypothesized that VitA serum levels are closely associated with its synthetic capacity and that low serum VitA levels are simply a consequence of impaired hepatic function. Several findings of our study support this hypothesis: First, CTP score – reflecting important aspects of hepatic dysfunction was independently associated with VitADef. Second, we observed a moderate/strong correlation of VitA with all laboratory parameters reflecting hepatic synthesis capacity, including vitamin K-dependent and independent coagulation parameters. For example, AT-III and Protein C activity were reduced by nearly 50% in the lowest vs. highest VitA quintile. Third, the independent association of CTP score with VitD (25-OH-vitamin D) deficiency further underlines the role of hepatic dysfunction towards decreased vitamin serum levels as this metabolic step in the course of VitD synthesis is localized in the liver [10].
Finally, we observed that patients with low VitA levels were significantly younger than patients with high VitA levels, which was also confirmed on multivariate regression analysis. At this point, we can only speculate that patients undergoing HVPG measurement (i.e., had developed ACLD) at a younger age had a more progressive course of the disease, which might be related to pronounced VitA depletion.
Correction of VitA levels in ACLD, however, is challenging: First, simple supplementation might not lead to effective amelioration of VitA serum levels, depending on the severity of liver disease [25, 26]. Second, it remains unclear whether VitADef (as assessed by serum VitA levels) really impacts on downstream signaling relevant for modulation of liver disease. Lastly, overdosing of VitA may cause hepatotoxicity which may be of particular concern in patients with ACLD [27]. However, the strong link between VitADef and fibrosis may indicate that treatment strategies promoting fibrosis regression will also ameliorate VitA homeostasis and vice versa.
VitDDef was highly common in our study cohort, being present in 57% of patients and additional 26% with VitD insufficiency, according to commonly used cut-offs for 25-OH-vitamin D levels [10]. Of note, comparisons to other studies regarding the prevalence of VitDDef are difficult to draw, as there are considerable differences in cut-offs and patient characteristics, however, other studies have demonstrated similar results [11, 12]. As indicated previously, a recent meta-analysis confirmed the association between VitD deficiency and mortality in patients with cirrhosis [13].
In our study, VitD levels were clearly associated with disease stage and severity of PHT. As mentioned above, only severity of hepatic dysfunction (i.e. CTP score) remained an independent risk factor for VitDDef, which seems particularly interesting in regard to the observation that elevated BA levels were independently associated with VitADef but not with VitDDef. While VitA supply is exclusively dependent on dietary uptake (mediated by BA [1]), VitD is mainly provided by endogenous synthesis/metabolization [10]. Therefore, the identification of hepatic (dys-)function – but not elevated BA levels – as the main determinant for 25-OH-vitamin D deficiency seems consistent with physiological concepts of VitD metabolism.
Importantly, a previous RCT by Pilz et al. demonstrated that VitD supplementation effectively increases VitD serum levels in patients with cirrhosis [28], while another study reported rapid declines of VitD levels after cessation of supplementation [29]. Furthermore, the efficacy of VitD supplementation towards osteopenia/osteoporosis remains unclear, as parathyroid hormone levels were not affected by VitD supplementation in the study by Pilz et al [28] and since several studies displayed no benefits of VitD supplementation on bone density/osteoporosis in patients with liver disease [30,31,32]. Similarly, a systematic review on VitD supplementation in chronic liver disease suggested limited efficacy of VitD supplementation on mortality, but importantly no effects on morbidity and health-related quality of life [33]. Nevertheless, in the absence of significant toxicity related to VitD supplementation in patients with ACLD, VitD supplementation seems safe, and thus, it may be argued that ACLD patients with VitD deficiency should still receive VitD supplements in case of low VitD levels.
Interestingly, our study clearly refutes that VitEDef is a regular or relevant condition in ACLD. First of all, only 7 (3%) patients fulfilled laboratory criteria for VitEDef. This observation is particularly relevant in regard to previous studies reporting lower levels of VitE in patients with NASH as compared to healthy subjects [34]. Of note, the study by Erhardt et al. [34] reported mean VitE levels of 22.4 µmol/L and 26.8 µmol/L in NASH patients and controls, respectively, which was quite similar to our study (median 23.6 µmol/L). Therefore, we can only speculate whether “significantly” lower VitE serum levels – however, still within normal range between healthy controls and patients have an impact on disease or may even be classified as VitEDef based on their potential implications on ACLD. Nevertheless, RCT displayed a significantly higher rate of histological improvement upon VitE treatment as compared to placebo in NASH patients [35], which was also suggested by a meta-analysis on VitE supplementation in NASH [36]. Similarly, a recent publication reported a remarkable reduction of risk for death or liver transplantation in compensated NASH patients receiving VitE supplements (adjusted HR 0.30) [37]. However, next to the retrospective and non-randomized study design, adherence to VitE intake in the treatment group was not assessed [37]. In any case, we did not observe any differences in VitE levels across CTP or HVPG strata or any meaningful correlation with parameters of hepatic (dys-)function. Of note, our study only included 25 (11%) NASH patients that exclusively displayed VitE levels above the cut-off for VitEDef.
Our study has several limitations: First, we could not provide data on hepatic VitA content, as well as a profile of different VitA metabolites in serum. Second, specific binding proteins for these vitamins were not measured, which prevents us from reporting their relation to vitamin serum levels. Third, we did not perform a systematic assessment of dietary vitamin intake, which would include both regular food intake as well as over-the-counter vitamin supplements, as well as current adherence towards and prior duration of prescription vitamin supplement intake. Fourth, the limited duration of potential follow-up in our study cohort prevents us from reporting follow-up events such as mortality and (further) hepatic decompensation in these patients at this point, which would have allowed to assess the prognostic value of VitA/D/E deficiencies. While these limitations must be acknowledged, we provide novel and important data on the strong association between vitamin deficiencies and ACLD severity as well as the severity of PHT in a large series of prospectively recruited patients. Furthermore, this study displays a close link between VitA levels and ELF score, as well as an independent association between elevated BA levels and VitADef.
In summary, our study demonstrates a considerable prevalence of VitA and VitD deficiency in patients with ACLD, while VitE deficiency was uncommon. VitA and VitD deficiencies are closely linked to hepatic dysfunction, which may be explained by advanced fibrosis and/or synthetic dysfunction impacting on the uptake, storage, and metabolism of these vitamins. The prognostic value of VitA as well as its role in liver disease progression should be assessed in future studies.
Abbreviations
- ACLD:
-
Advanced chronic liver disease
- ATIII:
-
Antithrombin III
- BA:
-
Bile acids
- BMI:
-
Body-mass index
- cACLD/dACLD:
-
Compensated/decompensated advanced chronic liver disease
- CSPH:
-
Clinically significant portal hypertension
- CTP:
-
Child-Turcotte-Pugh
- ELF:
-
Enhanced liver fibrosis score
- FXR:
-
Farnesoid X receptor
- HSC:
-
Hepatic stellate cells
- HVPG:
-
Hepatic venous pressure gradient
- IQR:
-
Interquartile range
- MELD:
-
Model for end-stage liver disease
- NAFLD:
-
Non-alcoholic fatty liver disease
- NASH:
-
Non-alcoholic steatohepatitis
- NSBB:
-
Non-selective betablockers
- PHT:
-
Portal hypertension
- RA:
-
Retinoic acid
- RXR-α:
-
Retinoid X receptor-alpha
- SEM:
-
Standard error of the mean
- TIPS:
-
Transjugular intrahepatic portosystemic shunt
- VCTE:
-
Vibration-controlled transient elastography
- VitA:
-
Vitamin A
- VitADef :
-
Vitamin A deficiency
- VitD:
-
Vitamin D
- VitDDef :
-
Vitamin D deficiency
- VitE:
-
Vitamin E
- VitEDef :
-
Vitamin E deficiency
References
Di Ciaula A, et al. Bile acid physiology. Ann Hepatol 2017;16(Suppl. 1):s4–s14
Grune T, et al. β-Carotene is an important vitamin A source for humans. J Nutr 2010;140(12):2268S-2285S
Haaker MW, Vaandrager AB, Helms JB. Retinoids in health and disease: a role for hepatic stellate cells in affecting retinoid levels. Biochim Biophys Acta 2020;1865:158674
Blaner WS, et al. Hepatic stellate cell lipid droplets: a specialized lipid droplet for retinoid storage. Biochim Biophys Acta 2009;1791(6):467–473
Testerink N, et al. Replacement of retinyl esters by polyunsaturated triacylglycerol species in lipid droplets of hepatic stellate cells during activation. PLoS One 2012;7(4):e34945
Chaves GV, et al. Association between liver vitamin A reserves and severity of nonalcoholic fatty liver disease in the class III obese following bariatric surgery. Obes Surg 2014;24(2):219–224
Peres WAF, et al. Vitamin A deficiency in patients with hepatitis C virus-related chronic liver disease. Br J Nutr 2011;106(11):1724–1731
Kataria Y, et al. Retinoid and carotenoid status in serum and liver among patients at high-risk for liver cancer. BMC Gastroenterol 2016;16:30
Chaves GV, et al. Vitamin A and retinol-binding protein deficiency among chronic liver disease patients. Nutrition 2015;31(5):664–668
Holick MF. Vitamin D deficiency. N Engl J Med 2007;357(3):266–281
Kubesch A, et al. Vitamin D deficiency is associated with hepatic decompensation and inflammation in patients with liver cirrhosis: A prospective cohort study. PLoS One 2018;13(11):e0207162
Triantos C, et al. Vitamin D-related immunomodulation in patients with liver cirrhosis. Eur J Gastroenterol Hepatol 2019;32:867–876
Yang F, et al. The value of severe vitamin D deficiency in predicting the mortality risk of patients with liver cirrhosis: a meta-analysis. Clin Res Hepatol Gastroenterol. 2019;43(6):722–729
Lee GY, Han SN. The role of vitamin E in immunity. Nutrients 2018;10(11):1614
Hardwick RN, et al. Diversity in antioxidant response enzymes in progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos 2010;38(12):2293–2301
EASL. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J Hepatol 2018;69(2):406–460
Reiberger T, et al. Measurement of the Hepatic Venous Pressure Gradient and Transjugular Liver Biopsy. J Vis Exp 2020. https://doi.org/10.3791/58819
Saeed A, et al. Disturbed Vitamin A Metabolism in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2017;10(1):29
Liu Y, et al. Association of serum retinoic acid with hepatic steatosis and liver injury in nonalcoholic fatty liver disease. Am J Clin Nutr 2015;102(1):130–137
Ukleja A, et al. Nutritional assessment of serum and hepatic vitamin A levels in patients with cirrhosis. J Parent Enter Nutr 2002;26(3):184–188
Kakiyama G, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol 2013;58(5):949–955
Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol 2014;11(1):55–67
Poupon RE, et al. Effect of portacaval shunt on serum bile acid concentration in patients with cirrhosis. Digestion 1977;16(1–2):138–145
Ohkubo H, et al. Role of portal and splenic vein shunts and impaired hepatic extraction in the elevated serum bile acids in liver cirrhosis. Gastroenterology 1984;86(3):514–520
de Paula TP, Ramalho A, Braulio VB. The effectiveness of relative dose response to retinol intake as an evaluation of vitamin A status of cirrhotic patients. J Hum Nutr Diet 2010;23(6):583–589
Arantes FPW, et al. Assessment of the relative dose-response test as indicators of hepatic vitamin A stores in various stages of chronic liver disease. Nutr Clin Pract 2013;28(1):95–100
Institute of Diabetes and Digestive and Kidney Diseases. Vitamin A, in LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda: National Institute of Diabetes and Digestive and Kidney Diseases; 2012
Pilz S, et al. Effects of vitamin D supplementation on serum 25-hydroxyvitamin D concentrations in cirrhotic patients: a randomized controlled trial. Nutrients 2016;8(5):278
Rode A, Fourlanos S, Nicoll A. Oral vitamin D replacement is effective in chronic liver disease. Gastroenterol Clin Biol 2010;34(11):618–620
Yurci A, et al. Efficacy of different therapeutic regimens on hepatic osteodystrophy in chronic viral liver disease. Eur J Gastroenterol Hepatol 2011;23(12):1206–1212
Herlong HF, Recker RR, Maddrey WC. Bone disease in primary biliary cirrhosis: histologic features and response to 25-hydroxyvitamin D. Gastroenterology 1982;83(1 Pt 1):103–108
Matloff DS, et al. Osteoporosis in primary biliary cirrhosis: effects of 25-hydroxyvitamin D3 treatment. Gastroenterology 1982;83(1 Pt 1):97–102
Bjelakovic G, et al. Vitamin D supplementation for chronic liver diseases in adults. Cochrane Database Syst Rev 2017;11:Cd011564
Erhardt A, et al. Plasma levels of vitamin E and carotenoids are decreased in patients with Nonalcoholic Steatohepatitis (NASH). Eur J Med Res 2011;16(2):76–78
Sanyal AJ, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 2010;362(18):1675–1685
Xu R, et al. Association between vitamin E and non-alcoholic steatohepatitis: a meta-analysis. Int J Clin Exp Med 2015;8(3):3924–3934
Vilar-Gomez E, et al. Vitamin E improves transplant-free survival and hepatic decompensation among patients with nonalcoholic steatohepatitis and advanced fibrosis. Hepatology 2020;71(2):495–509
Funding
Open access funding provided by Medical University of Vienna. BeSi was supported by a Gilead International Research Scholarship awarded to TR and MT.
Author information
Authors and Affiliations
Contributions
Study design (BeSi, GS, MM, TR). Extraction of data (BeSi, AS, EE). Statistical analysis (BeSi, MM, TR). Drafting of the manuscript (BeSi, GS, TR). Critical revision for important intellectual content (all authors).
Corresponding author
Ethics declarations
Conflict of interest
BeSi received travel support from AbbVie and Gilead. BeSc received travel support from Abbvie and Gilead. PS received speaking honoraria from Bristol-Myers Squibb and Boehringer-Ingelheim, consulting fees from PharmaIN, and travel support from Falk and Phenex Pharmaceuticals. DB received travel support from AbbVie and Gilead. TB received speaker honoraria from BMS, travel support from Abbvie, BMS, and Gilead; travel grant, financial award/grant from Medis. MP is an investigator for Bayer, BMS, Lilly, and Roche; he received speaker honoraria from Bayer, BMS, Eisai, and MSD; he is a consultant for Bayer, BMS, Ipsen, Eisai, Lilly, MSD, and Roche; he received travel support from Bayer and BMS. MT received speaker fees from BMS, Falk Foundation, Gilead, Intercept and MSD; advisory board fees from Albireo, BiomX, Boehringer Ingelheim, Falk Pharma GmbH, Genfit, Gilead, Intercept, MSD, Novartis, Phenex and Regulus. He further received travel grants from Abbvie, Falk, Gilead and Intercept and unrestricted research grants from Albireo, Cymabay, Falk, Gilead, Intercept, MSD and Takeda. MM has served as a speaker and/or consultant and/or advisory board member for AbbVie, Bristol-Myers Squibb, Gilead, W.L. Gore & Associates and Janssen. TR received grant support from Abbvie, Boehringer-Ingelheim, Gilead, MSD, Philips Healthcare, Gore; speaking honoraria from Abbvie, Gilead, Gore, Intercept, Roche, MSD; consulting/advisory board fee from Abbvie, Bayer, Boehringer-Ingelheim, Gilead, MSD, Siemens; and travel support from Boehringer-Ingelheim, Gilead and Roche. GS, AFS, AS, EE, and RP declare no conflict of interest.
Ethical approval
This study was conducted in accordance with the 1964 Helsinki declaration and its later amendments and approved by the local ethics committee of the Medical University of Vienna (EK1262/2017). All patients gave written informed consent to liver vein catheterizations and provided written consent to be enrolled in the VICIS study (NCT03267615).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Simbrunner, B., Semmler, G., Stadlmann, A. et al. Vitamin A levels reflect disease severity and portal hypertension in patients with cirrhosis. Hepatol Int 14, 1093–1103 (2020). https://doi.org/10.1007/s12072-020-10112-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12072-020-10112-3