
Abstract
Major depressive disorder (MDD) is one of the leading causes of disability worldwide, and its incidence is expected to increase. Despite tremendous efforts to understand its underlying biological mechanisms, MDD pathophysiology remains elusive and pharmacotherapy outcomes are still far from ideal. Low-grade chronic inflammation seems to play a key role in mediating the interface between psychological stress, depressive symptomatology, altered intestinal microbiology, and MDD onset. We review the available pre-clinical and clinical evidence of an involvement of pro-inflammatory pathways in the pathogenesis, treatment, and remission of MDD. We focus on caspase 1, inducible nitric oxide synthase, and interferon gamma, three inflammatory systems dysregulated in MDD. Treatment strategies aiming at targeting such pathways alone or in combination with classical therapies could prove valuable in MDD. Further studies are needed to assess the safety and efficacy of immune modulation in MDD and other psychiatric disorders with neuroinflammatory components.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Major depressive disorder (MDD) is a psychiatric disorder with significant morbidity, mortality, disability, and economic burden worldwide [1, 2]. In addition to the psychosocial and psychophysical dysfunctions associated with MDD, several conditions are often comorbid, including but not limited to obesity, type-2 diabetes, heart conditions, autoimmune diseases, neurodegenerative disorders, cancer, and intestinal conditions [3,4,5,6,7]. Multiple hypotheses have been formulated attempting to describe the elusive pathophysiology of MDD, including the monoamine hypothesis, the neurotrophic hypothesis, the glutamate hypothesis, the cytokine (or macrophage) hypothesis, and the microbiota-inflammasome hypothesis [8,9,10,11,12,13]. However, no single hypothesis seems to fully explain the onset, course, and remission of the disease. To complicate matters further, antidepressant drugs present numerous side effects and are effective only in a subset of patients [14,15,16]. Newer therapeutic strategies involve drugs acting on neuroplasticity-related pathways, gut microbiome modulation, and deep brain stimulation surgery [17,18,19]. Nevertheless, the quest for a better understanding of the molecular underpinnings of this disease represents an essential step in the identification of efficacious therapeutic strategies that could target the causal biological mechanisms of MDD.
Emerging evidence suggests that dysregulated neuro-immune pathways underlie depressive symptomatology in at least a subset of MDD patients [2, 20,21,22,23,24,25]. Three crucial inter-linked networks seem to influence the bidirectional communication between the brain, the immune system, and the intestinal microbiome, namely (a) increased oxidative stress, driven by nitric oxide (NO) overproduction, (b) chronic inflammation, driven by caspase 1 (CASP1), and Nod-like receptors family pyrin domain containing 3 (NLRP3) inflammasome over activation, and (c) central nervous system (CNS) T cell-helper 1 (Th1) lymphocyte infiltration, driven by interferon-gamma (IFNG). These three networks are strictly interlinked and present several levels of reciprocal regulation. For example, NO is a critical negative modulator of the NLRP3 inflammasome, while being necessary for IFNG-mediated suppression of interleukin-1 beta (IL1B) processing [26, 27]. Moreover, CASP1 regulates IFNG production via producing IL18, while IFNG modulates the CASP1 system [28]. Similarly, transcription of inducible nitric oxide synthase (NOS2) can be activated by IFNG [29]. Lastly, CASP1 is involved in the epigenetic regulation of NOS2 [30]. These multidirectional interactions suggest the importance of observing and therapeutically approaching these pathways as a whole rather than as insular entities. The possible involvement of these three systems in MDD is briefly summarized here and will be described in detail throughout this review.
Reactive oxygen species (ROS) are produced during cell metabolism, and are largely quenched by the endogenous antioxidant machinery [31]. However, excess of oxidative products can elicit oxidative stress and cause protein, lipid, and/or DNA damage [32]. Preclinical and clinical studies suggest that chronic stress exposure is associated with increased ROS production [33,34,35,36,37,38,39,40]. One of the free radicals produced during psychological stress is NO, mainly by NOS2 [41]. Inflammatory factors play key roles in tissue repair and in defense against pathogens [42, 43]. However, pathological activation of inflammatory cascades caused by stress and other insults can alter brain function and increase the likelihood of developing MDD and comorbid conditions [44,45,46]. CASP1, a protease that in the NLRP3 inflammasome renders the mature forms of IL1B and IL18, is also activated by stress [47, 48]. It has been shown that reactive T cells infiltrate the brain where they produce pro-inflammatory cytokines in response to CNS antigens [49]. Lastly, IFNG is a powerful inducer of indoleamine 2,3-dioxygenase 1 (IDO1), which degrades tryptophan increasing kyneurine and quinolinic acid, leading to hyposerotonergia and hyperglutamatergia, involved in MDD [9, 50, 51].
Recently, the role of the gut microbiome in mental health and illness has come to the forefront in psychiatry [52, 53]. Increasing evidence suggests the existence of a gut-brain-axis, a communication network that integrates brain and gut function, which plays a fundamental role in health and disease [54]. Such communication occurs via the endocrine and immune systems, the vagus nerve, and the bacterial metabolome [55,56,57]. It is becoming clear that the gut-brain-axis is an entity directly involved in modulating stress systems like the hypothalamic-pituitary-adrenal (HPA) axis, via its effects on the immune and endocrine systems, which affect behavior and mood and that can lead to MDD [53, 58, 59]. Given its central role in modulating immune processes and brain function, and given that MDD is characterized by altered gut microbiome composition, consensus is growing that manipulating the gut microbiota could represent a therapeutic tool in the treatment of MDD [19, 60]. In this review, we will summarize the pre-clinical and clinical evidence supporting the involvement of CASP1, NOS2, and IFNG in the pathophysiological processes underlying depressive symptomatology.
Communication Between the Brain, the Immune System, and the Gut Microbiome
Although the CNS is considered to have its “own” immune system, independent from the peripheral immune system, it is accepted that the two constantly communicate and cooperate, that the CNS is involved in regulating immunity, and that immune responses in the periphery lead to behavioral changes [66, 67].
Stress-mediated upregulation of pro-inflammatory cytokines [such as IL1, IL6, tumor necrosis factor (TNF), and IFNG] leads to endocrine and neurochemical responses, such as sympathetic nervous system (SNS), hypothalamic-pituitary-adrenal (HPA) axis, and microglial activation. SNS stimulation triggers epinephrine and norepinephrine release in the locus coeruleus and adrenal medulla, which result in an upregulation of pro-inflammatory signaling. SNS activation in response to stress pushes the CNS to “steer” immunity towards pro-inflammatory and antiviral responses [23]. At the same time, norepinephrine modulates pro-inflammatory cytokines transcription via beta-adrenergic receptor stimulation [68].
This leads to HPA axis activation by hypothalamus-secreted corticotropin releasing hormone (CRH) and arginine vasopressin (AVP). CRH stimulates adrenocorticotropic hormone (ACTH) release from the pituitary gland, which stimulates glucocorticoids release by the adrenal gland. Glucocorticoids interact with the glucocorticoid receptor (NR3C1) and the mineralocorticoid receptors (NR3C2), activating anti-inflammatory cascades and inhibiting Th1-driven pathways. This upregulates anti-inflammatory gene expression to avoid side effects [69,70,71,72,73]. The gut microbiome modulates HPA axis processes. In fact, germ-free rodents have greater plasma ACTH and corticosterone spikes compared to wild-type in response to stressors, while displaying altered anxiety-like behavior [74]. This exaggerated response can be reversed by early stage (but not later stage) recolonization with Bifidobacterium infantis [74]. Interestingly, the brain regions presenting the highest concentrations of pro-inflammatory cytokines are the prefrontal cortex, the hypothalamus, and the hippocampus, areas involved in cognition, mood, and antidepressant response [75, 76].
Increased concentrations of brain cytokines trigger the activation of microglia, immune cells inhabiting the brain parenchyma, representing chief innate immune cells in the brain [67, 77]. Depending on the temporal and qualitative cytokine profile, stress-induced microglial activation can either stimulate neuroprotection or neurodegeneration [78]. Not surprisingly, the gut microbiome modulates microglia homeostasis and maturation, while reduced gut microbiome complexity impairs microglia function [79]. Altogether, these stress-induced inflammatory events alter neurotransmitter systems, such as serotonin (5HT) and dopamine (DA), exacerbating depressive symptoms [80, 81]. Interestingly, the gut microbiome is also involved in neurotransmitter modulation, either via producing neurotransmitters, consuming them, or responding to them [82]. This raises the intriguing possibility that by altering gut microbiota composition, it might become possible to modulate neurotransmitter systems in pathological states, including MDD (Reviewed by [82]).
Glucocorticoids have the effect of restoring homeostasis [83]. However, in MDD, the HPA axis can become hyperactive. This phenomenon is underlined by increased cortisol, blunted ACTH response to CRH, glucocorticoid resistance, impairment in gluco- and mineral-corticoid signaling, and enlargement of the pituitary and adrenal glands [84,85,86,87,88]. Antidepressant drugs normalize the HPA axis and enhance the expression and function of corticosteroids [89, 90]. Peripheral cytokines can cross the blood-brain barrier (BBB) via (a) CNS lymphatic vessels, (b) active transport and a leaky or compromised BBB, (c) crossing at circumventricular organs, and (d) binding to receptors in the blood vessels that course through the brain [91,92,93,94]. Moreover, cytokines can affect brain function indirectly, through vagal nerve activation or by binding to cell-surface proteins found in brain endothelial cells [91, 93, 95, 96].
Cytokines can be produced in the gut in response to bacterial virulence factors (such as LPS), and in response to bacterial translocation to physiologically sterile enteric compartments (“leaky gut”) [97]. It was proposed that the leaky gut phenomenon contributes to MDD [98]. In fact, stress is known to compromise gut epithelial barrier integrity, allowing gut bacteria to access the enteric nervous system and immune cells [99]. Intestinal inflammation is a major contributor to changes in gut microbiome composition and function that are associated with disease (Reviewed in [100]). IFNG triggers the production of hydrogen peroxide and the epithelial expression of NOS2, which elevates the concentration of NO, in turn favoring the expansion of facultative anaerobic clades and hindering enterocyte proliferation [100, 101]. The resulting inflamed intestine perpetuates the production of pro-inflammatory cytokines and inflammogenic microbial metabolites, which affect brain processes and precipitate MDD onset while increasing the likelihood of comorbid conditions [99, 102]. Lastly, cytokines are produced de novo in the brain in response to stress [103,104,105].
Psychoneuroimmune Interactions and the Cytokine Hypothesis of Depression
Psychoneuroimmunology studies the reciprocal interactions between behavioral traits and the immune system, mediated by the nervous and endocrine systems [106]. In MDD, increasing evidence suggests that the communication networks existing between the microbiota and the nervous, immune, and endocrine systems lie at the crossroads of psychosocial stress, onset of depressive symptomatology and antidepressant response [107]. Studies suggest anti-inflammatory, endocrine,- and entero-regulatory effects of antidepressants, antidepressant effects of anti-inflammatory medications, and differential responses to antidepressants driven by polymorphisms in inflammation-related genes [108,109,110,111,112]. With regard to the immune players of such communication, cytokines have gained increasing attention over the past 20 years. Cytokines are pleiotropic signaling molecules with immunomodulatory function expressed constitutively and on-demand in the periphery and in the CNS and have been associated in at least a subset of patients with onset, course, and severity of neuropsychiatric disorders, as well as with the response to therapeutic drugs [113,114,115,116,117,118,119,120,121,122].
Exposure to psychological stressors primes the immune system towards the creation of a pro-inflammatory environment in the brain, a phenomena called sterile inflammation, which prepares the CNS and the body to trigger a potential full-blown immune response [123, 124]. While this program is essential for coping with the stressor and restoring homeostasis, it requires high amounts of energy and has collateral damage potential. In fact, repeated or chronic stress exposure results in a sustained inflammatory milieu in the brain which can lead to the development of MDD and comorbid illnesses [23, 125].
These lines of evidence led to the “cytokine hypothesis” (or “macrophage hypothesis”) of depression, which proposes that cytokines and an out-of-balance brain-immune communication are key MDD milestones [126,127,128,129,130]. This hypothesis is supported by mounting evidence: (a) illnesses characterized by chronic inflammatory responses (e.g., type-1 diabetes and systemic lupus erythematous) are associated with increased depression rates [4, 6], (b) administration of pro-inflammatory cytokines as a therapeutic strategy (e.g., IFNA administration in cancer and hepatitis-C) induces a dose-response depressive symptomatology and molecular features of MDD [131,132,133,134,135], and (c) pro-inflammatory cytokines administration in vivo induces sickness or depressive-like behavior [22, 136]. Lastly, polymorphisms in inflammation-related genes associate with increased MDD susceptibility and differential antidepressant response [25]. These layers of evidence suggest that neuroinflammation is involved in MDD, providing fertile ground to investigate diagnostic and therapeutic opportunities in neuro-immuno-psychiatry.
Major Depression and Dysregulated Inflammatory Pathways
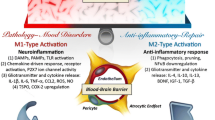
Psychoneuroimmunology research has highlighted that at least a subgroup of MDD patients present with a systemic low-grade chronic inflammatory profile underlined by increased T cell, monocytic, microglial, and astrocytic activation [23, 24, 137, 138]. This is characterized by increased Th1 cytokines such as IL1, IL2, IL6, TNF, and IFNG, decreased Th2 cytokines such as IL4 and IL10, and decreased regulatory T cells [128, 139,140,141,142,143,144]. The resulting skewed inflammatory balance triggers multi-level dysfunctions, such as metabolism, neurotransmission, gut microbiome, and neurogenesis alterations [137, 145, 146]. Accordingly, the neurotrophic hypothesis of depression suggests that MDD patients have inflammation-driven decreased neurogenesis, which leads to atrophy of brain areas such as the hippocampus and the prefrontal cortex [147,148,149,150]. Not surprisingly, pro-inflammatory cytokines and increased glucocorticoids production downregulate neurotrophins (such as brain derived- and nerve-growth factor) and neurogenesis during and following stress, while antidepressants reverse such decreases [151, 152]. The gut microbiome is also involved in regulating neuroplasticity and neurogenesis; germ-free mice display altered neurogenesis and BDNF expression in the dentate gyrus, while antibiotic treatment impairs neurogenesis [74, 153, 154] (Fig. 1).

Major depression and dysregulated inflammatory pathways
Cytokine Signaling and Nitrosative Stress
Oxidative stress is involved in MDD pathophysiology [155]. Stress exposure leads to ROS upregulation via cytokine-induced NOS2 induction, an event that heightens the overall oxidative stress, activating a feedback loop (co-activation state) that produces more cytokines [138]. Oxidative stress is characterized by the generation of ROS, which contributes to protein and DNA damage, and can result in irreversible brain function changes, leading to neurodegeneration and cognitive impairments [156]. Oxidative processes are gaining attention in psychiatry, since an expanding body of research suggests the involvement of these pathways in MDD [24, 40, 138, 157,158,159].
The involvement of oxidative and nitrosative stress in MDD is confirmed by the increased oxidative (such as NO, arachidonic acid, malondialdehyde, and 8-hydroxy-2-deoxyguanosine) and nitrosative (such as immunoglobulin (M IgM)- antibodies directed against phosphatidylitol and nitro-bovine serum albumin) stress markers in MDD patients, together with decreased levels of antioxidants (such as vitamins C and E) [160,161,162,163,164]. Interestingly, the concentration of oxidative stress markers correlates with depression severity and chronicity, as well as with antidepressant response [40, 138, 161, 165]. Accordingly, some antioxidant compounds have antidepressant properties, and antidepressants (such as paroxetine) partially reverse oxidative damage by enhancing the protective antioxidant status following stress [158, 166,167,168].
Of crucial importance for this work, the NO system is being investigated in MDD, because NO levels are increased in MDD and in animal models of stress, while NO inhibition has antidepressant effects (discussed in detail below) [37, 164, 169,170,171]. Increased levels of oxidative and nitrosative molecules can easily damage neurons, since they are particularly vulnerable to free radicals [172]. Moreover, the brain presents lower concentrations of antioxidants compared to other organs, making it more susceptible to free radicals [160]. Unsurprisingly, some areas (i.e., the subfields Cornu Ammonis (CA)1) and CA4) of the hippocampus (a brain region involved in mood regulation and adult neurogenesis) are the most sensitive to oxidative damage [24].
The Role of Caspase 1 in MDD
As mentioned above, stress triggers “sterile inflammation,” initiated by endogenous danger signal recognition, termed damage-associated molecular patterns (DAMPs), by glial cells, macrophages, and oligodendrocytes [124, 181, 182]. DAMPS are nuclear, cytosolic, mitochondrial, or extracellular molecules normally hidden from the immune system that upon activation are exposed and released in the extracellular space, where they stimulate an immune activation [124, 183]. In line with this understanding, increased levels of DAMPs have been found in rodent blood and hippocampus following stress exposure [103, 184].
Once released in the extracellular space, DAMPs function as alarm signals, alerting immune cells through pattern recognition receptors, to get ready for a potential full-blown immune response [182, 185, 186]. It has been hypothesized that such processes could represent an adaptive characteristic of the acute stress response; for example, if an animal were running away from a predator and were wounded during the chase, it might have better chances of surviving if its immune system were primed and ready to respond [187]. Another theory, one that places this mechanism in a modern context, suggests that such stress responses are activated when an individual is exposed to social evaluation, rejection, isolation, exclusion or conflict, possibly due to the potentially physically harmful significance of such social situations throughout history [188].
Together, DAMPs activation and release induce the transcriptional upregulation of a number of immune genes, such as IL1B, IL6, and TNF. This results in the creation of a pro-inflammatory milieu in the brain and periphery, and in the activation of the afferent nerves, which in turn leads to de novo production of pro-inflammatory cytokines in the brain and culminates with the onset of depressive-like behavior [22, 136, 189].
Further, DAMP activation results in the assembly of inflammasomes [186, 190] A peculiar role is played by the NLRP3 inflammasome, that consists of the NLRP3 protein, the adaptor apoptosis-associated speck-like protein containing a CARD (ASC), and the cysteine-protease CASP1 [47]. Upon inflammasome assembly, the inactive procaspase 1 zymogen is proteolitically cleaved into the enzymatically active heterodimer [191, 192]. In turn, activated CASP1 cleaves pro-IL1B and pro-IL18 into their mature, releasable, bioactive isoforms [47, 193]. Increased circulating levels of IL1B activate the HPA axis, which increases glucocorticoids production. [72]
CASP1 and NLRP3 transcripts and their protein products are increased in peripheral blood mononuclear cells (PBMC) from MDD patients compared to controls, while antidepressants decrease such hyperactivity [61]. Similarly, IL1B and IL18 are increased in MDD, and their levels correlate with the severity of depression [61] (Table 1). Correspondingly, antidepressants decrease IL1B levels [109].
Casp1−/− mice display decreased depressive- and anxiety-like behaviors, while being protected by the exacerbation of depressive-like behavior following chronic stress [19, 173]. Similarly, minocycline-treated mice display resilience in developing depressive-like behavior following stress, and this effect is accompanied by the expansion of bacterial clades with anti-inflammatory properties, which could help explain minocycline’s antidepressant effects [19] (Table 2).
CASP1−/− mice have the same behavioral and inflammatory responses to systemic lipopolysaccharide (LPS) administration as wild-type (wt) mice, but are resistant to the development of depressive-like behavior and to pro-inflammatory cytokines increase following intracerebroventricular LPS administration [194]. Moreover, CASP1−/− mice are resistant to lethal LPS doses and have decreased levels of inflammation-induced brain and systemic transcription [195,196,197]. Significantly for this review, CASP1 and the NLRP3 inflammasome are involved in the development of depressive-like behavior in stress models and are increased in MDD [61, 173]. At the same time, pathological shifts in gut microbiota composition and leaky gut trigger an increase in pro-inflammatory signaling, which increases the risk of developing depressive symptomatology and comorbid illnesses [198]. Such evidence has led to the formulation of the microbiota-inflammasome hypothesis of major depression and comorbid systemic illnesses [58]. This hypothesis suggests that pathological gut microbiome shifts upregulate pro-inflammatory pathways exacerbating depressive symptomatology and increasing the likelihood of developing comorbid conditions [58].
Interleukin-1B (IL1B)
IL1B binds to the interleukin-1 receptor (IL1R1), which results in the activation of many acute-phase inflammation genes, such as NOS2, IL6, and cyclooxygenase type 2 [192, 199]. Recently, it was suggested that NLRP3 inflammasome activation mediates IL1B orchestrated inflammation (that results in depressive-like behavior) in the prefrontal cortex following stress, and that fluoxetine reverses such changes [173, 175]. Accordingly, mice lacking the IL1 receptor are resistant to developing depressive-like behavior following chronic stress while being protected against the decrease in neurogenesis observed in wt mice following stress [176, 177].
Interleukin-1A (IL1A)
IL1A shares features with IL1B and is an equally potent pro-inflammatory cytokine [207]. However, IL1A also presents differences to IL1B. For example, unlike the IL1B precursor which is not active, both the pro-IL1A and the cleaved IL1A are active ligands of the IL1R1 [208]. Moreover, while IL1B is released, IL1A can be secreted or membrane-bound, although the factors that control such translocation have not been fully elucidated yet [207, 209]. Finally, while IL1B is produced on-demand in immune cells, IL1A is constitutively expressed in a variety of cell types but can be produced by immune cells in response to insults [210]. Interestingly, IL1A-mediated activation of p38-MAPK inhibits NR3C1 function, suggesting that the mechanism conferring glucocorticoid resistance in MDD could be associated with IL1A [211]. To the best of our knowledge, no studies have investigated anxiety- and depressive-like phenotypes in IL1A−/− mice.
Interleukin-18 (IL18)
IL18 is a prototypical Th1 cytokine for its ability to stimulate IFNG activity, and it is expressed in macrophages and dendritic cells [212]. Circulating IL18 increases during stress and in response to HPA axis activation [213]. IL18 binds to the IL18 receptor (IL18R) activating p38-MAPK, c-Jun N-terminal kinase, and NFKB1 cascades, which potentiate antimicrobial and antiviral immunity [214, 215]. Although IL18 is known for its ability to promote both Th1- and Th2-related inflammatory responses, its predominant role in enhancing Th1 activity makes this cytokine a candidate therapeutic target in Th1-related inflammatory and autoimmune diseases, including MDD [212].
IL18 is increased in MDD and in panic disorder [62, 63]. IL18 gene promoter variants (rs187238 and rs1946518) associate with higher IL18 transcription and increased MDD susceptibility in patients exposed to stressful events. IL18−/− mice have decreased IFNG production and impaired natural killer cell activity and abnormal Th1 responses [216]. Moreover, IL18−/− mice display decreased depressive- and anxiety-like behavior, as well as gene expression changes across various brain regions [178, 217]. In addition, immobilization stress in mice induces pro-IL18 via ACTH and a superoxide-activated CASP1 pathway [218]. Given that IL6 is not induced in response to stress in IL18−/− mice, it seems that IL18 mediates stress-induced IL6 upregulation [218]. Lastly, IL18 is involved in stress-induced microglial activation in rodents while contributing to dopaminergic degeneration [179, 180].
Interleukin-33 (IL33)
IL33 has alarmin and transcription factor roles and triggers predominantly Th2 responses (such as the induction of IL4, IL5, IL13, and anti-inflammatory gene expression) [221]. Like other members of the IL1 family, IL33 can be beneficial or detrimental, depending on its spatio-temporal expression. IL33 is constitutively expressed and localized in the cytoplasm. However, if a barrier is breached and IL33 is released from destroyed cells, it acts as an alarmin upon binding the IL33 receptor (ST2) [222]. The signaling cascade in response to ST2 activation modulates hundreds of genes with a pattern that resembles that of IL1R1 activation [223].
Two single nucleotide polymorphisms in the IL33 gene (rs11792633 and rs7044343) moderate the correlation between history of childhood abuse and recurrent depression in women [65]. Moreover, patients with a history of recurrent depression have greater peripheral levels of IL33 and IL1B [65]. Finally, IL33 is expressed in the paraventricular nucleus of the hypothalamus and in the prefrontal cortex of rats exposed to acute stress, suggesting that stress induces IL33 expression in those brain regions [65].
The Role of Inducible Nitric Oxide Synthase in MDD
NO is a small intercellular and intracellular signaling molecule with a very short half-life (3–6 s) that freely diffuses across cell membranes. NO plays important roles in the brain modulating pathways such as neurogenesis, neurotransmission, synaptic plasticity, learning, and pain [224]. NO also regulates emotional and cognitive processes, suggesting that it could be involved in the etiology of MDD and anxiety disorders [225]. Three isoforms of the NOS enzyme produce NO: NOS2, neuronal (NOS1), and endothelial (NOS3), all of which have specific spatio-temporal patterns of regulation. In this review, we will focus on the inducible isoform since it is considered the most relevant to MDD.
Over the past two decades, several lines of evidence have brought NO and specifically the NOS2 isoform to the forefront in psychiatry: (a) the levels of NO and its metabolites are increased in MDD patients and suicide attempters compared to controls [171, 200, 201], (b) NOS2 transcription is increased in the peripheral blood of patients with recurrent depressive disorder [202], (c) a polymorphism (-1026C/A) in the NOS2 promoter associates with recurrent depressive disorder risk [203], (d) IgM against NO adducts are elevated in MDD patients, suggesting that the protein damage created by NO results in the formation of immunogenic peptides, that in turn activate an autoimmune-like response [204, 205], (e) the selective serotonin reuptake inhibitor paroxetine is a NOS2 inhibitor [206, 226], (f) adjuvant NOS2 inhibition enhances the efficacy of serotonergic antidepressants [169], and (g) NOS2 is increased in the hippocampus and cerebral cortex in mice following stress, and NOS2 inhibition results in antidepressant-like effects in rodents [38, 219, 220] (Tables 3-4).
The architecture of the NOS2 promoter region suggests that this gene has a tight and complex pattern of transcriptional control since it is rich in positive and negative regulatory regions, and it is responsive to many transcription factors, cytokines, and bacterial by-products [29]. NOS2 is synthesized on-demand in macrophages and microglia [227]. In fact, whereas there is no detectable physiological NOS2 expression in the brain, a profound transcriptional upregulation of the NOS2 gene can be observed in response to traumatic events such as ischemia and systemic inflammation, most likely through activation of the NOS2 promoter by inflammation-related molecules [29, 39, 196, 228, 229]. Following induction, NOS2 produces NO continuously until the proteasome degradation pathway inactivates the enzyme [230].
Several studies have targeted the NO system in pre-clinical MDD research, yielding promising results. For example, NO decreases norepinephrine production, decreases nitrate and nitrite levels in the hippocampus and cerebral cortex, and decreases serotonin turnover in the frontal cortex [231,232,233]. Moreover, NO inhibits the dopamine transporter, indirectly increasing the availability of inter-synaptic dopamine [234]. Finally, several molecules such as bupropion (a norepinephrine-dopamine reuptake inhibitor), venlafaxine (a serotonin-norepinephrine reuptake inhibitor), mementine (an NMDA receptor antagonist), and berberine (a plant alkaloid), all of which produce antidepressant-like effects, modulate this signaling pathway [235].
It is accepted that anaerobic bacteria in the gut prevent the expansion of facultative anaerobic bacteria, at least partially by limiting the host-mediated production of oxygen and nitrate [236]. Antibiotic-mediated disruption of the gut microbiota increases the production of host nitrate in the gut [237]. This allows an expansion of the facultative anaerobic Enterobacteriaceae, which includes potentially pathogenic gram-negative bacteria, such as Escherichia coli (this effect is likely not to be limited to E. coli, although the latter has been the focus of investigation to date). These bacteria produce the virulence molecule LPS, which triggers depressive-like behavior and increases serotonin degradation in the brain [237, 238]. This alteration is mediated by NOS2; therefore, its inhibition prevents E. coli overgrowth [237]. Therefore, rectifying aberrant NO signaling could have a therapeutic role in altered gut microbiology-induced depressive symptoms [239]. Accordingly, stimulation of colonic epithelial cancer cells by IFNG induces NOS2-mediated NO production, while butyrate (one of the main anti-inflammatory short chain fatty acids (SCFAs)) blunts NO production [237]. This result suggests that a diet rich in substrates for SCFAs production could have antidepressant-like effects via its repercussions on gut microbiome composition and inflammatory processes. Together, these findings suggest that modulation of the NO system could represent a useful approach in treating MDD and in keeping of a healthy gut microbiome.
The Role of Interferon-Gamma in MDD
IFNG is a pleiotropic soluble cytokine which orchestrates cellular programs via transcriptional and translational gene control. IFNG is produced by immune cells such as lymphocytes, cytotoxic lymphocytes, B cells, and antigen-presenting cells [240, 241]. The IFNG receptor (IFNGR) is expressed on almost all cell types, and its activation triggers the janus kinase 1 and 2 (JAK1/2) signal transducer and activator of transcription 1 (STAT1) pathway, as well as additional pathways, such as the extracellular-signal-regulated-kinase 1/2 (ERK1/2) [242, 243]. Activation of the IFNGR results in the transcription of genes with IFNG-stimulated response elements (ISREs) within their promoter region until STAT1 dissociates following complete dephosphorylation within 1–2 h [244, 245]. The genes transcribed in response to IFNGR activation are at least 200, together with many micro RNAs and long non-coding RNAs [246] (for a database see [247]). At the same time, after IFNGR stimulation, the secondary transcription factors IRF1, IRF2, and interferon consensus sequence binding protein are upregulated. This in turn results in the transcriptional induction of a subset of inflammatory-related genes such as NOS2 (stimulated by IRF1) and guanylate-binding protein. Finally, IFNG can activate and be activated by CASP [248,249,250,251].
Ex vivo PBMC from MDD patients display increased IFNG and neopterin production upon stimulation, as well as decreased tryptophan bioavailability [252]. Nevertheless, IFNG transcriptional levels (together with those of TNF) in patients with multiple sclerosis correlate with the severity of the depressive symptomatology during flare-ups [253]. At the same time, most categories of antidepressants suppress the IFNG/IL10 ratio through suppressing IFNG and stimulating IL10 [254, 255]. These findings (Table 5) suggest that MDD patients have increased systemic IFNG and neopterin production by activated T cells and macrophages. This could be responsible for an upregulation of the enzyme IDO1 (since the latter presents 2 ISREs at the promoter region that lead to maximum promoter activity) and consequent tryptophan depletion through upregulation of the kyneurine/tryptophan pathway, events that decrease serotonin availability and increase the toxic metabolite kyneurine [252, 258,259,260]. Accordingly, a polymorphism (CA repeat, rs3138557) in the IFNG gene correlates with lower serum tryptophan and 5-hydroxindolacetic acid (the main metabolite of serotonin) and higher levels of kyneurine, suggesting that carriers of the CA allele might be more susceptible to developing MDD [256]. Similarly, the presence of the high producer T allele +874(T/A) polymorphism (rs2430561) associates with increased IDO1 activity [257]. Interestingly, IFNG signaling drives Th1 development [261, 262]; therefore, early increased signaling of IFNG by traumatic events could be involved in the Th1/Th2 shift towards Th1 in MDD [141].
IFNG−/− mice do not show developmental defects but present compromised immune responses and increased susceptibility to infections [263]. With regard to their behavior, IFNG−/− mice display decreased anxiety- and depressive-like behaviors as well as heightened emotionality in several paradigms [264,265,266]. These behaviors are underlined by (a) increased serotonergic and noradrenergic activity (i.e., greater metabolite accumulation) in the central amygdaloid nucleus, together with (b) increased baseline plasma corticosterone, (c) decreased neurogenesis in the hippocampus, and (d) decreased levels of nerve-growth factor in the prefrontal cortex, suggesting that IFNG modulates anxiety and depressive states and is involved in CNS plasticity [264, 265]. On the other hand, while IFNG deficiency does not confer resistance to a chronic stress regimen in mice, it attenuates monoamine, corticoid, and cytokine alterations in response to stressors [264] (Table 6).
IFNG signaling promotes leaky gut and bacterial translocation. In fact, in vitro experiments have highlighted that low-dose IFNG dramatically increases the translocation of opportunistic pathogens, and high-doses disrupt tight junctions [267]. Lastly, IFNG levels affect the representation of specific bacterial species while being up- or downregulated by specific commensals [97]. For example, the degradation of tryptophan to the metabolite tryptophol inhibits IFNG production, while IFNG levels dictate the presence and expansion of specific bacterial taxa [97]. Given this evidence for an involvement of IFNG in pathways relevant to depressive symptoms and gut dysbiosis, targeting IFNG and/or its receptor could hold potential in the quest for novel MDD therapies.
Conclusions and Future Directions
Convergent pre-clinical and clinical evidence points towards an involvement of central and peripheral inflammatory pathways and the gut microbiome in the response to psychological stressors and in the onset, treatment, and remission of MDD. Future randomized controlled trials should investigate the safety and efficacy of decreasing CASP1-, NOS2,- and IFNG-mediated pathways in MDD patients. Reduced activity of those pro-inflammatory mediators could be achieved via pharmacological inhibition or gut microbiome manipulation. The latter approach can involve diet, probiotics supplementation, and fecal microbiota transplantation. This could lead to the development of novel antidepressant strategies acting upon the dysregulated inflammatory milieu observed in MDD. Because inhibiting such pathways might hinder physiological immune processes, particular care should be taken when developing immunomodulatory and gut microbiota-directed therapies.
References
Murray CJ, Lopez AD (1997) Alternative projections of mortality and disability by cause 1990-2020: global burden of disease study. Lancet 349(9064):1498–1504. https://doi.org/10.1016/S0140-6736(96)07492-2
Maes M, Leonard B, Fernandez A, Kubera M, Nowak G, Veerhuis R, Gardner A, Ruckoanich P et al (2011) (Neuro)inflammation and neuroprogression as new pathways and drug targets in depression: From antioxidants to kinase inhibitors. Prog Neuro-Psychopharmacol Biol Psychiatry 35(3):659–663. https://doi.org/10.1016/j.pnpbp.2011.02.019
Levitan RD, Davis C, Kaplan AS, Arenovich T, Phillips DI, Ravindran AV (2012) Obesity comorbidity in unipolar major depressive disorder: refining the core phenotype. J Clin Psychiatry 73(8):1119–1124. https://doi.org/10.4088/JCP.11m07394
Katon WJ (2008) The comorbidity of diabetes mellitus and depression. Am J Med 121(11 Suppl 2):S8–S15. https://doi.org/10.1016/j.amjmed.2008.09.008
Halaris A (2009) Comorbidity between depression and cardiovascular disease. Int Angiol 28(2):92–99
Kayser MS, Dalmau J (2011) The emerging link between autoimmune disorders and neuropsychiatric disease. J Neuropsychiatry Clin Neurosci 23(1):90–97. https://doi.org/10.1176/appi.neuropsych.23.1.90
Brintzenhofe-Szoc KM, Levin TT, Li Y, Kissane DW, Zabora JR (2009) Mixed anxiety/depression symptoms in a large cancer cohort: prevalence by cancer type. Psychosomatics 50(4):383–391. https://doi.org/10.1176/appi.psy.50.4.383
Myint AM, Kim YK (2003) Cytokine-serotonin interaction through IDO: a neurodegeneration hypothesis of depression. Med Hypotheses 61(5–6):519–525
Muller N, Schwarz MJ (2007) The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry 12(11):988–1000. https://doi.org/10.1038/sj.mp.4002006
Lesch KP, Beckmann H (1990) The serotonin hypothesis of depression. Fortschr Neurol Psychiatr 58(11):427–438. https://doi.org/10.1055/s-2007-1001206
Sanacora G, Treccani G, Popoli M (2012) Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 62(1):63–77. https://doi.org/10.1016/j.neuropharm.2011.07.036
Duman RS, Monteggia LM (2006) A neurotrophic model for stress-related mood disorders. Biol Psychiatry 59(12):1116–1127. https://doi.org/10.1016/j.biopsych.2006.02.013
Inserra A, Rogers GB, Licinio J, Wong ML (2018) The microbiota-Inflammasome hypothesis of major depression. Bioessays 40(9):e1800027. https://doi.org/10.1002/bies.201800027
Rush AJ, Trivedi MH, Wisniewski SR, Stewart JW, Nierenberg AA, Thase ME, Ritz L, Biggs MM et al (2006) Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med 354(12):1231–1242. https://doi.org/10.1056/NEJMoa052963
Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, Niederehe G, Thase ME et al (2006) Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 163(11):1905–1917. https://doi.org/10.1176/ajp.2006.163.11.1905
Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, Norquist G, Howland RH et al (2006) Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 163(1):28–40. https://doi.org/10.1176/appi.ajp.163.1.28
Dandekar MP, Fenoy AJ, Carvalho AF, Soares JC, Quevedo J (2018) Deep brain stimulation for treatment-resistant depression: an integrative review of preclinical and clinical findings and translational implications. Mol Psychiatry 23(5):1094–1112. https://doi.org/10.1038/mp.2018.2
Huang YJ, Lane HY, Lin CH (2017) New treatment strategies of depression: based on mechanisms related to neuroplasticity. Neural Plast 2017:4605971. https://doi.org/10.1155/2017/4605971
Rogers GB, Keating DJ, Young RL, Wong ML, Licinio J, Wesselingh S (2016) From gut dysbiosis to altered brain function and mental illness: mechanisms and pathways. Mol Psychiatry 21(6):738–748. https://doi.org/10.1038/mp.2016.50
Vogelzangs N, Duivis HE, Beekman AT, Kluft C, Neuteboom J, Hoogendijk W, Smit JH, de Jonge P et al (2012) Association of depressive disorders, depression characteristics and antidepressant medication with inflammation. Transl Psychiatry 2:e79. https://doi.org/10.1038/tp.2012.8
Maes M (2008) The cytokine hypothesis of depression: inflammation, oxidative & nitrosative stress (IO&NS) and leaky gut as new targets for adjunctive treatments in depression. Neuro Endocrinol Lett 29(3):287–291
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9(1):46–56. https://doi.org/10.1038/nrn2297
Miller AH, Maletic V, Raison CL (2009) Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65(9):732–741. https://doi.org/10.1016/j.biopsych.2008.11.029
Leonard B, Maes M (2012) Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depression. Neurosci Biobehav Rev 36(2):764–785. https://doi.org/10.1016/j.neubiorev.2011.12.005
Wong ML, Dong C, Maestre-Mesa J, Licinio J (2008) Polymorphisms in inflammation-related genes are associated with susceptibility to major depression and antidepressant response. Mol Psychiatry 13(8):800–812. https://doi.org/10.1038/mp.2008.59
Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, Sassetti CM (2013) Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat Immunol 14(1):52–60. https://doi.org/10.1038/ni.2474
Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, He Z, Zeng Y et al (2013) Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res 23(2):201–212. https://doi.org/10.1038/cr.2013.6
Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, Quintal L, Sekut L et al (1997) Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 386(6625):619–623. https://doi.org/10.1038/386619a0
Xie QW, Whisnant R, Nathan C (1993) Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J Exp Med 177(6):1779–1784
Buzzo CL, Medina T, Branco LM, Lage SL, Ferreira LC, Amarante-Mendes GP, Hottiger MO, De Carvalho DD et al (2017) Epigenetic regulation of nitric oxide synthase 2, inducible (Nos2) by NLRC4 inflammasomes involves PARP1 cleavage. Sci Rep 7:41686. https://doi.org/10.1038/srep41686
Karihtala P, Soini Y (2007) Reactive oxygen species and antioxidant mechanisms in human tissues and their relation to malignancies. APMIS 115(2):81–103. https://doi.org/10.1111/j.1600-0463.2007.apm_514.x
Liu J, Wang X, Shigenaga MK, Yeo HC, Mori A, Ames BN (1996) Immobilization stress causes oxidative damage to lipid, protein, and DNA in the brain of rats. FASEB J 10(13):1532–1538
Patki G, Solanki N, Atrooz F, Allam F, Salim S (2013) Depression, anxiety-like behavior and memory impairment are associated with increased oxidative stress and inflammation in a rat model of social stress. Brain Res 1539:73–86. https://doi.org/10.1016/j.brainres.2013.09.033
Miyashita T, Yamaguchi T, Motoyama K, Unno K, Nakano Y, Shimoi K (2006) Social stress increases biopyrrins, oxidative metabolites of bilirubin, in mouse urine. Biochem Biophys Res Commun 349(2):775–780. https://doi.org/10.1016/j.bbrc.2006.08.098
Shao Y, Yan G, Xuan Y, Peng H, Huang QJ, Wu R, Xu H (2015) Chronic social isolation decreases glutamate and glutamine levels and induces oxidative stress in the rat hippocampus. Behav Brain Res 282:201–208. https://doi.org/10.1016/j.bbr.2015.01.005
Noh SR, Cheong HK, Ha M, Eom SY, Kim H, Choi YH, Paek D (2015) Oxidative stress biomarkers in long-term participants in clean-up work after the Hebei Spirit oil spill. Sci Total Environ 515-516:207–214. https://doi.org/10.1016/j.scitotenv.2015.02.039
Olivenza R, Moro MA, Lizasoain I, Lorenzo P, Fernandez AP, Rodrigo J, Bosca L, Leza JC (2000) Chronic stress induces the expression of inducible nitric oxide synthase in rat brain cortex. J Neurochem 74(2):785–791
Madrigal JL, Moro MA, Lizasoain I, Lorenzo P, Castrillo A, Bosca L, Leza JC (2001) Inducible nitric oxide synthase expression in brain cortex after acute restraint stress is regulated by nuclear factor kappaB-mediated mechanisms. J Neurochem 76(2):532–538
Yoshida T, Waeber C, Huang Z, Moskowitz MA (1995) Induction of nitric oxide synthase activity in rodent brain following middle cerebral artery occlusion. Neurosci Lett 194(3):214–218
Chung CP, Schmidt D, Stein CM, Morrow JD, Salomon RM (2013) Increased oxidative stress in patients with depression and its relationship to treatment. Psychiatry Res 206(2–3):213–216. https://doi.org/10.1016/j.psychres.2012.10.018
Peng YL, Liu YN, Liu L, Wang X, Jiang CL, Wang YX (2012) Inducible nitric oxide synthase is involved in the modulation of depressive behaviors induced by unpredictable chronic mild stress. J Neuroinflammation 9:75. https://doi.org/10.1186/1742-2094-9-75
Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22(2):240–273, table of contents. https://doi.org/10.1128/CMR.00046-08
Joffre O, Nolte MA, Sporri R, Reis e Sousa C (2009) Inflammatory signals in dendritic cell activation and the induction of adaptive immunity. Immunol Rev 227(1):234–247. https://doi.org/10.1111/j.1600-065X.2008.00718.x
Rohleder N (2014) Stimulation of systemic low-grade inflammation by psychosocial stress. Psychosom Med 76(3):181–189. https://doi.org/10.1097/PSY.0000000000000049
Onat A, Can G (2014) Enhanced proinflammatory state and autoimmune activation: A breakthrough to understanding chronic diseases. Curr Pharm Des 20(4):575–584
Lasselin J, Capuron L (2014) Chronic low-grade inflammation in metabolic disorders: relevance for behavioral symptoms. Neuroimmunomodulation 21(2–3):95–101. https://doi.org/10.1159/000356535
Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G (2009) The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10(3):241–247. https://doi.org/10.1038/ni.1703
Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13(6):397–411. https://doi.org/10.1038/nri3452
Goverman J (2009) Autoimmune T cell responses in the central nervous system. Nat Rev Immunol 9(6):393–407. https://doi.org/10.1038/nri2550
Taylor MW, Feng GS (1991) Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J 5(11):2516–2522
Wirleitner B, Neurauter G, Schrocksnadel K, Frick B, Fuchs D (2003) Interferon-gamma-induced conversion of tryptophan: immunologic and neuropsychiatric aspects. Curr Med Chem 10(16):1581–1591
Deans E (2016) Microbiome and mental health in the modern environment. J Physiol Anthropol 36(1):1. https://doi.org/10.1186/s40101-016-0101-y
Cryan JF, Dinan TG (2012) Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci 13(10):701–712. https://doi.org/10.1038/nrn3346
Grenham S, Clarke G, Cryan JF, Dinan TG (2011) Brain-gut-microbe communication in health and disease. Front Physiol 2:94. https://doi.org/10.3389/fphys.2011.00094
Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S (2012) Host-gut microbiota metabolic interactions. Science 336(6086):1262–1267. https://doi.org/10.1126/science.1223813
El Aidy S, Dinan TG, Cryan JF (2014) Immune modulation of the brain-gut-microbe axis. Front Microbiol 5:146. https://doi.org/10.3389/fmicb.2014.00146
Forsythe P, Bienenstock J, Kunze WA (2014) Vagal pathways for microbiome-brain-gut axis communication. Adv Exp Med Biol 817:115–133. https://doi.org/10.1007/978-1-4939-0897-4_5
Inserra A, Rogers GB, Licinio J, Wong ML (2018) The microbiota-inflammasome hypothesis of major depression. Bioessays 40(9):e1800027. https://doi.org/10.1002/bies.201800027
Dinan TG, Cryan JF (2013) Melancholic microbes: a link between gut microbiota and depression? Neurogastroenterol Motil 25(9):713–719. https://doi.org/10.1111/nmo.12198
Ianiro G, Bibbo S, Gasbarrini A, Cammarota G (2014) Therapeutic modulation of gut microbiota: current clinical applications and future perspectives. Curr Drug Targets 15(8):762–770
Alcocer-Gomez E, de Miguel M, Casas-Barquero N, Nunez-Vasco J, Sanchez-Alcazar JA, Fernandez-Rodriguez A, Cordero MD (2014) NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder. Brain Behav Immun 36:111–117. https://doi.org/10.1016/j.bbi.2013.10.017
Prossin AR, Koch AE, Campbell PL, McInnis MG, Zalcman SS, Zubieta JK (2011) Association of plasma interleukin-18 levels with emotion regulation and mu-opioid neurotransmitter function in major depression and healthy volunteers. Biol Psychiatry 69(8):808–812. https://doi.org/10.1016/j.biopsych.2010.10.014
Kokai M, Kashiwamura S, Okamura H, Ohara K, Morita Y (2002) Plasma interleukin-18 levels in patients with psychiatric disorders. J Immunother 25(Suppl 1):S68–S71
Haastrup E, Bukh JD, Bock C, Vinberg M, Thorner LW, Hansen T, Werge T, Kessing LV et al (2012) Promoter variants in IL18 are associated with onset of depression in patients previously exposed to stressful-life events. J Affect Disord 136(1–2):134–138. https://doi.org/10.1016/j.jad.2011.08.025
Kudinova AY, Deak T, Hueston CM, McGeary JE, Knopik VS, Palmer RH, Gibb BE (2016) Cross-species evidence for the role of interleukin-33 in depression risk. J Abnorm Psychol 125(4):482–494. https://doi.org/10.1037/abn0000158
Fung A, Vizcaychipi M, Lloyd D, Wan Y, Ma D (2012) Central nervous system inflammation in disease related conditions: mechanistic prospects. Brain Res 1446:144–155. https://doi.org/10.1016/j.brainres.2012.01.061
Sternberg EM (2006) Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol 6(4):318–328. https://doi.org/10.1038/nri1810
Cole SW (2010) Elevating the perspective on human stress genomics. Psychoneuroendocrinology 35(7):955–962. https://doi.org/10.1016/j.psyneuen.2010.06.008
Herbert J, Goodyer IM, Grossman AB, Hastings MH, de Kloet ER, Lightman SL, Lupien SJ, Roozendaal B et al (2006) Do corticosteroids damage the brain? J Neuroendocrinol 18(6):393–411. https://doi.org/10.1111/j.1365-2826.2006.01429.x
Hayashi R, Wada H, Ito K, Adcock IM (2004) Effects of glucocorticoids on gene transcription. Eur J Pharmacol 500(1–3):51–62. https://doi.org/10.1016/j.ejphar.2004.07.011
Turnbull AV, Rivier C (1995) Regulation of the HPA axis by cytokines. Brain Behav Immun 9(4):253–275. https://doi.org/10.1006/brbi.1995.1026
Dunn AJ (2000) Cytokine activation of the HPA axis. Ann N Y Acad Sci 917:608–617
Coutinho AE, Chapman KE (2011) The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol 335(1):2–13. https://doi.org/10.1016/j.mce.2010.04.005
Sudo N, Chida Y, Aiba Y, Sonoda J, Oyama N, Yu XN, Kubo C, Koga Y (2004) Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J Physiol 558(Pt 1):263–275. https://doi.org/10.1113/jphysiol.2004.063388
Borsini A, Zunszain PA, Thuret S, Pariante CM (2015) The role of inflammatory cytokines as key modulators of neurogenesis. Trends Neurosci 38(3):145–157. https://doi.org/10.1016/j.tins.2014.12.006
Felger JC, Lotrich FE (2013) Inflammatory cytokines in depression: neurobiological mechanisms and therapeutic implications. Neuroscience 246:199–229. https://doi.org/10.1016/j.neuroscience.2013.04.060
McKim DB, Weber MD, Niraula A, Sawicki CM, Liu X, Jarrett BL, Ramirez-Chan K, Wang Y et al (2017) Microglial recruitment of IL-1beta-producing monocytes to brain endothelium causes stress-induced anxiety. Mol Psychiatry. https://doi.org/10.1038/mp.2017.64
Nguyen MD, Julien JP, Rivest S (2002) Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci 3(3):216–227. https://doi.org/10.1038/nrn752
Erny D, Hrabe de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, Keren-Shaul H, Mahlakoiv T et al (2015) Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci 18(7):965–977. https://doi.org/10.1038/nn.4030
Kohler S, Cierpinsky K, Kronenberg G, Adli M (2016) The serotonergic system in the neurobiology of depression: relevance for novel antidepressants. J Psychopharmacol 30(1):13–22. https://doi.org/10.1177/0269881115609072
Nestler EJ, Carlezon WA Jr (2006) The mesolimbic dopamine reward circuit in depression. Biol Psychiatry 59(12):1151–1159. https://doi.org/10.1016/j.biopsych.2005.09.018
Strandwitz P (2018) Neurotransmitter modulation by the gut microbiota. Brain Res 1693(Pt B):128–133. https://doi.org/10.1016/j.brainres.2018.03.015
Herman JP, McKlveen JM, Solomon MB, Carvalho-Netto E, Myers B (2012) Neural regulation of the stress response: glucocorticoid feedback mechanisms. Braz J Med Biol Res 45(4):292–298
Gold PW, Goodwin FK, Chrousos GP (1988) Clinical and biochemical manifestations of depression. Relation to the neurobiology of stress (1). N Engl J Med 319(6):348–353. https://doi.org/10.1056/NEJM198808113190606
Holsboer F, Barden N (1996) Antidepressants and hypothalamic-pituitary-adrenocortical regulation. Endocr Rev 17(2):187–205. https://doi.org/10.1210/edrv-17-2-187
Nemeroff CB (1996) The corticotropin-releasing factor (CRF) hypothesis of depression: new findings and new directions. Mol Psychiatry 1(4):336–342
Owens MJ, Nemeroff CB (1993) The role of corticotropin-releasing factor in the pathophysiology of affective and anxiety disorders: laboratory and clinical studies. CIBA Found Symp 172:296–308 discussion 308-216
Pace TW, Miller AH (2009) Cytokines and glucocorticoid receptor signaling. Relevance to major depression. Ann N Y Acad Sci 1179:86–105. https://doi.org/10.1111/j.1749-6632.2009.04984.x
Pariante CM, Miller AH (2001) Glucocorticoid receptors in major depression: Relevance to pathophysiology and treatment. Biol Psychiatry 49(5):391–404
Fitzgerald P, O'Brien SM, Scully P, Rijkers K, Scott LV, Dinan TG (2006) Cutaneous glucocorticoid receptor sensitivity and pro-inflammatory cytokine levels in antidepressant-resistant depression. Psychol Med 36(1):37–43. https://doi.org/10.1017/S003329170500632X
Blatteis CM (1992) Role of the OVLT in the febrile response to circulating pyrogens. Prog Brain Res 91:409–412
Banks WA (2005) Blood-brain barrier transport of cytokines: a mechanism for neuropathology. Curr Pharm Des 11(8):973–984
Maier SF, Watkins LR (2003) Immune-to-central nervous system communication and its role in modulating pain and cognition: Implications for cancer and cancer treatment. Brain Behav Immun 17(Suppl 1):S125–S131
Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D et al (2015) Structural and functional features of central nervous system lymphatic vessels. Nature 523(7560):337–341. https://doi.org/10.1038/nature14432
Rivest S (1999) What is the cellular source of prostaglandins in the brain in response to systemic inflammation? Facts and controversies. Mol Psychiatry 4(6):500–507
Maier SF, Goehler LE, Fleshner M, Watkins LR (1998) The role of the vagus nerve in cytokine-to-brain communication. Ann N Y Acad Sci 840:289–300
Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, Ter Horst R, Jansen T et al (2016) Linking the human gut microbiome to inflammatory cytokine production capacity. Cell 167(4):1125–1136.e28. https://doi.org/10.1016/j.cell.2016.10.020
Maes M, Kubera M, Leunis JC, Berk M, Geffard M, Bosmans E (2013) In depression, bacterial translocation may drive inflammatory responses, oxidative and nitrosative stress (O&NS), and autoimmune responses directed against O&NS-damaged neoepitopes. Acta Psychiatr Scand 127(5):344–354. https://doi.org/10.1111/j.1600-0447.2012.01908.x
Gareau MG, Silva MA, Perdue MH (2008) Pathophysiological mechanisms of stress-induced intestinal damage. Curr Mol Med 8(4):274–281
Baumler AJ, Sperandio V (2016) Interactions between the microbiota and pathogenic bacteria in the gut. Nature 535(7610):85–93. https://doi.org/10.1038/nature18849
Grishin A, Bowling J, Bell B, Wang J, Ford HR (2016) Roles of nitric oxide and intestinal microbiota in the pathogenesis of necrotizing enterocolitis. J Pediatr Surg 51(1):13–17. https://doi.org/10.1016/j.jpedsurg.2015.10.006
Dopkins N, Nagarkatti PS, Nagarkatti M (2018) The role of gut microbiome and associated metabolome in the regulation of neuroinflammation in multiple sclerosis and its implications in attenuating chronic inflammation in other inflammatory and autoimmune disorders. Immunology 154(2):178–185. https://doi.org/10.1111/imm.12903
Weber MD, Frank MG, Tracey KJ, Watkins LR, Maier SF (2015) Stress induces the danger-associated molecular pattern HMGB-1 in the hippocampus of male Sprague Dawley rats: a priming stimulus of microglia and the NLRP3 inflammasome. J Neurosci 35(1):316–324. https://doi.org/10.1523/JNEUROSCI.3561-14.2015
Hanamsagar R, Hanke ML, Kielian T (2012) Toll-like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol 33(7):333–342. https://doi.org/10.1016/j.it.2012.03.001
Iwata M, Ota KT, Li XY, Sakaue F, Li N, Dutheil S, Banasr M, Duric V et al (2016) Psychological stress activates the Inflammasome via release of adenosine triphosphate and stimulation of the purinergic type 2X7 receptor. Biol Psychiatry 80(1):12–22. https://doi.org/10.1016/j.biopsych.2015.11.026
Ziemssen T, Kern S (2007) Psychoneuroimmunology--cross-talk between the immune and nervous systems. J Neurol 254(Suppl 2):II8–I11. https://doi.org/10.1007/s00415-007-2003-8
Schiepers OJ, Wichers MC, Maes M (2005) Cytokines and major depression. Prog Neuro-Psychopharmacol Biol Psychiatry 29(2):201–217. https://doi.org/10.1016/j.pnpbp.2004.11.003
Abbasi SH, Hosseini F, Modabbernia A, Ashrafi M, Akhondzadeh S (2012) Effect of celecoxib add-on treatment on symptoms and serum IL-6 concentrations in patients with major depressive disorder: randomized double-blind placebo-controlled study. J Affect Disord 141(2–3):308–314. https://doi.org/10.1016/j.jad.2012.03.033
Hannestad J, DellaGioia N, Bloch M (2011) The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis. Neuropsychopharmacology 36(12):2452–2459. https://doi.org/10.1038/npp.2011.132
Muller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Muller B, Spellmann I, Hetzel G et al (2006) The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry 11(7):680–684. https://doi.org/10.1038/sj.mp.4001805
Nery FG, Monkul ES, Hatch JP, Fonseca M, Zunta-Soares GB, Frey BN, Bowden CL, Soares JC (2008) Celecoxib as an adjunct in the treatment of depressive or mixed episodes of bipolar disorder: a double-blind, randomized, placebo-controlled study. Hum Psychopharmacol 23(2):87–94. https://doi.org/10.1002/hup.912
Foster JA, Rinaman L, Cryan JF (2017) Stress & the gut-brain axis: regulation by the microbiome. Neurobiol Stress 7:124–136. https://doi.org/10.1016/j.ynstr.2017.03.001
Hayley S, Poulter MO, Merali Z, Anisman H (2005) The pathogenesis of clinical depression: stressor- and cytokine-induced alterations of neuroplasticity. Neuroscience 135(3):659–678. https://doi.org/10.1016/j.neuroscience.2005.03.051
Kim YK, Jung HG, Myint AM, Kim H, Park SH (2007) Imbalance between pro-inflammatory and anti-inflammatory cytokines in bipolar disorder. J Affect Disord 104(1–3):91–95. https://doi.org/10.1016/j.jad.2007.02.018
Licinio J, Frost P (2000) The neuroimmune-endocrine axis: pathophysiological implications for the central nervous system cytokines and hypothalamus-pituitary-adrenal hormone dynamics. Braz J Med Biol Res 33(10):1141–1148
Licinio J, Wong ML (1999) The role of inflammatory mediators in the biology of major depression: central nervous system cytokines modulate the biological substrate of depressive symptoms, regulate stress-responsive systems, and contribute to neurotoxicity and neuroprotection. Mol Psychiatry 4(4):317–327
Drexhage RC, van der Heul-Nieuwenhuijsen L, Padmos RC, van Beveren N, Cohen D, Versnel MA, Nolen WA, Drexhage HA (2010) Inflammatory gene expression in monocytes of patients with schizophrenia: overlap and difference with bipolar disorder. A study in naturalistically treated patients. Int J Neuropsychopharmacol 13(10):1369–1381. https://doi.org/10.1017/S1461145710000799
Prolo P, Licinio J (1999) Cytokines in affective disorders and schizophrenia: new clinical and genetic findings. Mol Psychiatry 4(4):396
Saetre P, Emilsson L, Axelsson E, Kreuger J, Lindholm E, Jazin E (2007) Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry 7:46. https://doi.org/10.1186/1471-244X-7-46
Rausch JL (2005) Initial conditions of psychotropic drug response: studies of serotonin transporter long promoter region (5-HTTLPR), serotonin transporter efficiency, cytokine and kinase gene expression relevant to depression and antidepressant outcome. Prog Neuro-Psychopharmacol Biol Psychiatry 29(6):1046–1061. https://doi.org/10.1016/j.pnpbp.2005.03.011
Tourjman V, Kouassi E, Koue ME, Rocchetti M, Fortin-Fournier S, Fusar-Poli P, Potvin S (2013) Antipsychotics’ effects on blood levels of cytokines in schizophrenia: a meta-analysis. Schizophr Res 151(1–3):43–47. https://doi.org/10.1016/j.schres.2013.10.011
Raison CL, Miller AH (2013) Do cytokines really sing the blues? Cerebrum 2013:10
Rock KL, Latz E, Ontiveros F, Kono H (2010) The sterile inflammatory response. Annu Rev Immunol 28:321–342. https://doi.org/10.1146/annurev-immunol-030409-101311
Fleshner M (2013) Stress-evoked sterile inflammation, danger associated molecular patterns (DAMPs), microbial associated molecular patterns (MAMPs) and the inflammasome. Brain Behav Immun 27(1):1–7. https://doi.org/10.1016/j.bbi.2012.08.012
Gadek-Michalska A, Tadeusz J, Rachwalska P, Bugajski J (2013) Cytokines, prostaglandins and nitric oxide in the regulation of stress-response systems. Pharmacol Rep 65(6):1655–1662
Maes M (1995) Evidence for an immune response in major depression: a review and hypothesis. Prog Neuro-Psychopharmacol Biol Psychiatry 19(1):11–38
Maes M (1993) A review on the acute phase response in major depression. Rev Neurosci 4(4):407–416
Kling MA, Alesci S, Csako G, Costello R, Luckenbaugh DA, Bonne O, Duncko R, Drevets WC et al (2007) Sustained low-grade pro-inflammatory state in unmedicated, remitted women with major depressive disorder as evidenced by elevated serum levels of the acute phase proteins C-reactive protein and serum amyloid A. Biol Psychiatry 62(4):309–313. https://doi.org/10.1016/j.biopsych.2006.09.033
Gardner A, Boles RG (2011) Beyond the serotonin hypothesis: mitochondria, inflammation and neurodegeneration in major depression and affective spectrum disorders. Prog Neuro-Psychopharmacol Biol Psychiatry 35(3):730–743. https://doi.org/10.1016/j.pnpbp.2010.07.030
Smith RS (1991) The macrophage theory of depression. Med Hypotheses 35(4):298–306
Capuron L, Schroecksnadel S, Feart C, Aubert A, Higueret D, Barberger-Gateau P, Laye S, Fuchs D (2011) Chronic low-grade inflammation in elderly persons is associated with altered tryptophan and tyrosine metabolism: role in neuropsychiatric symptoms. Biol Psychiatry 70(2):175–182. https://doi.org/10.1016/j.biopsych.2010.12.006
Dieperink E, Willenbring M, Ho SB (2000) Neuropsychiatric symptoms associated with hepatitis C and interferon alpha: a review. Am J Psychiatry 157(6):867–876
Dantzer R, Kelley KW (2007) Twenty years of research on cytokine-induced sickness behavior. Brain Behav Immun 21(2):153–160. https://doi.org/10.1016/j.bbi.2006.09.006
Vergassola C, Pende A, Musso NR, Ioverno A, Lotti G, Criscuolo D (1990) Effects of interferon alpha-2a on catecholamines and lymphocyte beta 2 adrenoceptors in healthy humans. Int J Neurosci 51(3–4):211–213
Felger JC, Cole SW, Pace TW, Hu F, Woolwine BJ, Doho GH, Raison CL, Miller AH (2012) Molecular signatures of peripheral blood mononuclear cells during chronic interferon-alpha treatment: relationship with depression and fatigue. Psychol Med 42(8):1591–1603. https://doi.org/10.1017/S0033291711002868
Konsman JP, Parnet P, Dantzer R (2002) Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci 25(3):154–159
Leonard BE (2014) Impact of inflammation on neurotransmitter changes in major depression: an insight into the action of antidepressants. Prog Neuro-Psychopharmacol Biol Psychiatry 48:261–267. https://doi.org/10.1016/j.pnpbp.2013.10.018
Rawdin BJ, Mellon SH, Dhabhar FS, Epel ES, Puterman E, Su Y, Burke HM, Reus VI et al (2013) Dysregulated relationship of inflammation and oxidative stress in major depression. Brain Behav Immun 31:143–152. https://doi.org/10.1016/j.bbi.2012.11.011
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, Lanctot KL (2010) A meta-analysis of cytokines in major depression. Biol Psychiatry 67(5):446–457. https://doi.org/10.1016/j.biopsych.2009.09.033
Raison CL, Capuron L, Miller AH (2006) Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol 27(1):24–31. https://doi.org/10.1016/j.it.2005.11.006
Maes M, Song C, Lin A, De Jongh R, Van Gastel A, Kenis G, Bosmans E, De Meester I et al (1998) The effects of psychological stress on humans: increased production of pro-inflammatory cytokines and a Th1-like response in stress-induced anxiety. Cytokine 10(4):313–318
Myint AM, Leonard BE, Steinbusch HW, Kim YK (2005) Th1, Th2, and Th3 cytokine alterations in major depression. J Affect Disord 88(2):167–173. https://doi.org/10.1016/j.jad.2005.07.008
Song C, Halbreich U, Han C, Leonard BE, Luo H (2009) Imbalance between pro- and anti-inflammatory cytokines, and between Th1 and Th2 cytokines in depressed patients: the effect of electroacupuncture or fluoxetine treatment. Pharmacopsychiatry 42(5):182–188. https://doi.org/10.1055/s-0029-1202263
Huang TL, Lee CT (2007) T-helper 1/T-helper 2 cytokine imbalance and clinical phenotypes of acute-phase major depression. Psychiatry Clin Neurosci 61(4):415–420. https://doi.org/10.1111/j.1440-1819.2007.01686.x
Mahar I, Bambico FR, Mechawar N, Nobrega JN (2014) Stress, serotonin, and hippocampal neurogenesis in relation to depression and antidepressant effects. Neurosci Biobehav Rev 38:173–192. https://doi.org/10.1016/j.neubiorev.2013.11.009
Jiang H, Ling Z, Zhang Y, Mao H, Ma Z, Yin Y, Wang W, Tang W et al (2015) Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav Immun 48:186–194. https://doi.org/10.1016/j.bbi.2015.03.016
Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS (2000) Hippocampal volume reduction in major depression. Am J Psychiatry 157(1):115–118. https://doi.org/10.1176/ajp.157.1.115
Pannekoek JN, van der Werff SJ, van den Bulk BG, van Lang ND, Rombouts SA, van Buchem MA, Vermeiren RR, van der Wee NJ (2014) Reduced anterior cingulate gray matter volume in treatment-naive clinically depressed adolescents. Neuroimage Clin 4:336–342. https://doi.org/10.1016/j.nicl.2014.01.007
Lee BH, Kim YK (2010) The roles of BDNF in the pathophysiology of major depression and in antidepressant treatment. Psychiatry Investig 7(4):231–235. https://doi.org/10.4306/pi.2010.7.4.231
Czeh B, Michaelis T, Watanabe T, Frahm J, de Biurrun G, van Kampen M, Bartolomucci A, Fuchs E (2001) Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc Natl Acad Sci U S A 98(22):12796–12801. https://doi.org/10.1073/pnas.211427898
Dwivedi Y (2009) Brain-derived neurotrophic factor: role in depression and suicide. Neuropsychiatr Dis Treat 5:433–449
Piccinni A, Marazziti D, Catena M, Domenici L, Del Debbio A, Bianchi C, Mannari C, Martini C et al (2008) Plasma and serum brain-derived neurotrophic factor (BDNF) in depressed patients during 1 year of antidepressant treatments. J Affect Disord 105(1–3):279–283. https://doi.org/10.1016/j.jad.2007.05.005
Mohle L, Mattei D, Heimesaat MM, Bereswill S, Fischer A, Alutis M, French T, Hambardzumyan D et al (2016) Ly6C(hi) monocytes provide a link between antibiotic-induced changes in gut microbiota and adult hippocampal neurogenesis. Cell Rep 15(9):1945–1956. https://doi.org/10.1016/j.celrep.2016.04.074
Ogbonnaya ES, Clarke G, Shanahan F, Dinan TG, Cryan JF, O'Leary OF (2015) Adult hippocampal neurogenesis is regulated by the microbiome. Biol Psychiatry 78(4):e7–e9. https://doi.org/10.1016/j.biopsych.2014.12.023
Maes M, Galecki P, Chang YS, Berk M (2011) A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness. Prog Neuro-Psychopharmacol Biol Psychiatry 35(3):676–692. https://doi.org/10.1016/j.pnpbp.2010.05.004
Uttara B, Singh AV, Zamboni P, Mahajan RT (2009) Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol 7(1):65–74. https://doi.org/10.2174/157015909787602823
Black CN, Bot M, Scheffer PG, Cuijpers P, Penninx BW (2015) Is depression associated with increased oxidative stress? A systematic review and meta-analysis. Psychoneuroendocrinology 51:164–175. https://doi.org/10.1016/j.psyneuen.2014.09.025
Khanzode SD, Dakhale GN, Khanzode SS, Saoji A, Palasodkar R (2003) Oxidative damage and major depression: the potential antioxidant action of selective serotonin re-uptake inhibitors. Redox Rep 8(6):365–370. https://doi.org/10.1179/135100003225003393
Yager S, Forlenza MJ, Miller GE (2010) Depression and oxidative damage to lipids. Psychoneuroendocrinology 35(9):1356–1362. https://doi.org/10.1016/j.psyneuen.2010.03.010
Sarandol A, Sarandol E, Eker SS, Erdinc S, Vatansever E, Kirli S (2007) Major depressive disorder is accompanied with oxidative stress: short-term antidepressant treatment does not alter oxidative-antioxidative systems. Hum Psychopharmacol 22(2):67–73. https://doi.org/10.1002/hup.829
Forlenza MJ, Miller GE (2006) Increased serum levels of 8-hydroxy-2′-deoxyguanosine in clinical depression. Psychosom Med 68(1):1–7. https://doi.org/10.1097/01.psy.0000195780.37277.2a
Maes M, Mihaylova I, Leunis JC (2007) Increased serum IgM antibodies directed against phosphatidyl inositol (Pi) in chronic fatigue syndrome (CFS) and major depression: evidence that an IgM-mediated immune response against Pi is one factor underpinning the comorbidity between both CFS and depression. Neuro Endocrinol Lett 28(6):861–867
Maes M, Mihaylova I, Kubera M, Leunis JC (2008) An IgM-mediated immune response directed against nitro-bovine serum albumin (nitro-BSA) in chronic fatigue syndrome (CFS) and major depression: evidence that nitrosative stress is another factor underpinning the comorbidity between major depression and CFS. Neuro Endocrinol Lett 29(3):313–319
Kotan VO, Sarandol E, Kirhan E, Ozkaya G, Kirli S (2011) Effects of long-term antidepressant treatment on oxidative status in major depressive disorder: a 24-week follow-up study. Prog Neuro-Psychopharmacol Biol Psychiatry 35(5):1284–1290. https://doi.org/10.1016/j.pnpbp.2011.03.021
Tsuboi H, Shimoi K, Kinae N, Oguni I, Hori R, Kobayashi F (2004) Depressive symptoms are independently correlated with lipid peroxidation in a female population: comparison with vitamins and carotenoids. J Psychosom Res 56(1):53–58. https://doi.org/10.1016/S0022-3999(03)00567-1
Zafir A, Banu N (2007) Antioxidant potential of fluoxetine in comparison to Curcuma longa in restraint-stressed rats. Eur J Pharmacol 572(1):23–31. https://doi.org/10.1016/j.ejphar.2007.05.062
Gibson SA, Korade Z, Shelton RC (2012) Oxidative stress and glutathione response in tissue cultures from persons with major depression. J Psychiatr Res 46(10):1326–1332. https://doi.org/10.1016/j.jpsychires.2012.06.008
Scapagnini G, Davinelli S, Drago F, De Lorenzo A, Oriani G (2012) Antioxidants as antidepressants: fact or fiction? CNS Drugs 26(6):477–490. https://doi.org/10.2165/11633190-000000000-00000
Harkin A, Connor TJ, Burns MP, Kelly JP (2004) Nitric oxide synthase inhibitors augment the effects of serotonin re-uptake inhibitors in the forced swimming test. Eur Neuropsychopharmacol 14(4):274–281. https://doi.org/10.1016/j.euroneuro.2003.08.010
Joca SR, Guimaraes FS (2006) Inhibition of neuronal nitric oxide synthase in the rat hippocampus induces antidepressant-like effects. Psychopharmacology 185(3):298–305. https://doi.org/10.1007/s00213-006-0326-2
Lee BH, Lee SW, Yoon D, Lee HJ, Yang JC, Shim SH, Kim DH, Ryu SH et al (2006) Increased plasma nitric oxide metabolites in suicide attempters. Neuropsychobiology 53(3):127–132. https://doi.org/10.1159/000092542
Gandhi S, Abramov AY (2012) Mechanism of oxidative stress in neurodegeneration. Oxidative Med Cell Longev 2012:428010. https://doi.org/10.1155/2012/428010
Pan Y, Chen XY, Zhang QY, Kong LD (2014) Microglial NLRP3 inflammasome activation mediates IL-1beta-related inflammation in prefrontal cortex of depressive rats. Brain Behav Immun 41:90–100. https://doi.org/10.1016/j.bbi.2014.04.007
Zhang Y, Liu L, Peng YL, Liu YZ, Wu TY, Shen XL, Zhou JR, Sun DY et al (2014) Involvement of inflammasome activation in lipopolysaccharide-induced mice depressive-like behaviors. CNS Neurosci Ther 20(2):119–124. https://doi.org/10.1111/cns.12170
Zhang Y, Liu L, Liu YZ, Shen XL, Wu TY, Zhang T, Wang W, Wang YX et al (2015) NLRP3 inflammasome mediates chronic mild stress-induced depression in mice via neuroinflammation. Int J Neuropsychopharmacol 18(8). https://doi.org/10.1093/ijnp/pyv006
Goshen I, Kreisel T, Ben-Menachem-Zidon O, Licht T, Weidenfeld J, Ben-Hur T, Yirmiya R (2008) Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry 13(7):717–728. https://doi.org/10.1038/sj.mp.4002055
Koo JW, Duman RS (2008) IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci U S A 105(2):751–756. https://doi.org/10.1073/pnas.0708092105
Too LK, Mitchell AJ, Yau B, Ball HJ, McGregor IS, Hunt NH (2014) Interleukin-18 deficiency and its long-term behavioural and cognitive impacts in a murine model of pneumococcal meningitis. Behav Brain Res 263:176–189. https://doi.org/10.1016/j.bbr.2014.01.035
Sugama S, Wirz SA, Barr AM, Conti B, Bartfai T, Shibasaki T (2004) Interleukin-18 null mice show diminished microglial activation and reduced dopaminergic neuron loss following acute 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine treatment. Neuroscience 128(2):451–458. https://doi.org/10.1016/j.neuroscience.2004.07.020
Sugama S, Fujita M, Hashimoto M, Conti B (2007) Stress induced morphological microglial activation in the rodent brain: involvement of interleukin-18. Neuroscience 146(3):1388–1399. https://doi.org/10.1016/j.neuroscience.2007.02.043
Singhal G, Jaehne EJ, Corrigan F, Toben C, Baune BT (2014) Inflammasomes in neuroinflammation and changes in brain function: a focused review. Front Neurosci 8:315. https://doi.org/10.3389/fnins.2014.00315
Chen GY, Nunez G (2010) Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10(12):826–837. https://doi.org/10.1038/nri2873
Schaefer L (2014) Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem 289(51):35237–35245. https://doi.org/10.1074/jbc.R114.619304
Fleshner M, Campisi J, Amiri L, Diamond DM (2004) Cat exposure induces both intra- and extracellular Hsp72: the role of adrenal hormones. Psychoneuroendocrinology 29(9):1142–1152. https://doi.org/10.1016/j.psyneuen.2004.01.007
Creagh EM, O'Neill LA (2006) TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol 27(8):352–357. https://doi.org/10.1016/j.it.2006.06.003
Kigerl KA, de Rivero Vaccari JP, Dietrich WD, Popovich PG, Keane RW (2014) Pattern recognition receptors and central nervous system repair. Exp Neurol 258:5–16. https://doi.org/10.1016/j.expneurol.2014.01.001
Ganter MT, Ware LB, Howard M, Roux J, Gartland B, Matthay MA, Fleshner M, Pittet JF (2006) Extracellular heat shock protein 72 is a marker of the stress protein response in acute lung injury. Am J Physiol Lung Cell Mol Physiol 291(3):L354–L361. https://doi.org/10.1152/ajplung.00405.2005
Slavich GM, Cole SW (2013) The emerging field of human social genomics. Clin Psychol Sci 1(3):331–348
Dantzer R (2009) Cytokine, sickness behavior, and depression. Immunol Allergy Clin N Am 29(2):247–264. https://doi.org/10.1016/j.iac.2009.02.002
Guo H, Callaway JB, Ting JP (2015) Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat Med 21(7):677–687. https://doi.org/10.1038/nm.3893
Martinon F, Tschopp J (2004) Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 117(5):561–574. https://doi.org/10.1016/j.cell.2004.05.004
Dinarello CA (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27:519–550. https://doi.org/10.1146/annurev.immunol.021908.132612
Menu P, Vince JE (2011) The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol 166(1):1–15. https://doi.org/10.1111/j.1365-2249.2011.04440.x
Lawson MA, McCusker RH, Kelley KW (2013) Interleukin-1 beta converting enzyme is necessary for development of depression-like behavior following intracerebroventricular administration of lipopolysaccharide to mice. J Neuroinflammation 10:54. https://doi.org/10.1186/1742-2094-10-54
Mastronardi C, Whelan F, Yildiz OA, Hannestad J, Elashoff D, McCann SM, Licinio J, Wong ML (2007) Caspase 1 deficiency reduces inflammation-induced brain transcription. Proc Natl Acad Sci U S A 104(17):7205–7210. https://doi.org/10.1073/pnas.0701366104
Mastronardi CA, Paz-Filho G, Zanoni M, Molano-Gonzalez N, Arcos-Burgos M, Licinio J, Wong ML (2015) Temporal gene expression in the hippocampus and peripheral organs to endotoxin-induced systemic inflammatory response in caspase-1-deficient mice. Neuroimmunomodulation 22(4):263–273. https://doi.org/10.1159/000368310
Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M et al (1995) Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 80(3):401–411
Fung TC, Olson CA, Hsiao EY (2017) Interactions between the microbiota, immune and nervous systems in health and disease. Nat Neurosci 20(2):145–155. https://doi.org/10.1038/nn.4476
Weber A, Wasiliew P, Kracht M (2010) Interleukin-1 (IL-1) pathway. Sci Signal 3(105):cm1. https://doi.org/10.1126/scisignal.3105cm1
Kim YK, Paik JW, Lee SW, Yoon D, Han C, Lee BH (2006) Increased plasma nitric oxide level associated with suicide attempt in depressive patients. Prog Neuro-Psychopharmacol Biol Psychiatry 30(6):1091–1096. https://doi.org/10.1016/j.pnpbp.2006.04.008
Suzuki E, Yagi G, Nakaki T, Kanba S, Asai M (2001) Elevated plasma nitrate levels in depressive states. J Affect Disord 63(1–3):221–224
Galecki P, Galecka E, Maes M, Chamielec M, Orzechowska A, Bobinska K, Lewinski A, Szemraj J (2012) The expression of genes encoding for COX-2, MPO, iNOS, and sPLA2-IIA in patients with recurrent depressive disorder. J Affect Disord 138(3):360–366. https://doi.org/10.1016/j.jad.2012.01.016
Galecki P, Maes M, Florkowski A, Lewinski A, Galecka E, Bienkiewicz M, Szemraj J (2010) An inducible nitric oxide synthase polymorphism is associated with the risk of recurrent depressive disorder. Neurosci Lett 486(3):184–187. https://doi.org/10.1016/j.neulet.2010.09.048
Maes M, Kubera M, Mihaylova I, Geffard M, Galecki P, Leunis JC, Berk M (2013) Increased autoimmune responses against auto-epitopes modified by oxidative and nitrosative damage in depression: implications for the pathways to chronic depression and neuroprogression. J Affect Disord 149(1–3):23–29. https://doi.org/10.1016/j.jad.2012.06.039
Maes M, Mihaylova I, Kubera M, Leunis JC, Geffard M (2011) IgM-mediated autoimmune responses directed against multiple neoepitopes in depression: new pathways that underpin the inflammatory and neuroprogressive pathophysiology. J Affect Disord 135(1–3):414–418. https://doi.org/10.1016/j.jad.2011.08.023
Finkel MS, Laghrissi-Thode F, Pollock BG, Rong J (1996) Paroxetine is a novel nitric oxide synthase inhibitor. Psychopharmacol Bull 32(4):653–658
Di Paolo NC, Shayakhmetov DM (2016) Interleukin 1alpha and the inflammatory process. Nat Immunol 17(8):906–913. https://doi.org/10.1038/ni.3503
Kim B, Lee Y, Kim E, Kwak A, Ryoo S, Bae SH, Azam T, Kim S et al (2013) The interleukin-1alpha precursor is biologically active and is likely a key alarmin in the IL-1 family of cytokines. Front Immunol 4:391. https://doi.org/10.3389/fimmu.2013.00391
Kurt-Jones EA, Beller DI, Mizel SB, Unanue ER (1985) Identification of a membrane-associated interleukin 1 in macrophages. Proc Natl Acad Sci U S A 82(4):1204–1208
Bersudsky M, Luski L, Fishman D, White RM, Ziv-Sokolovskaya N, Dotan S, Rider P, Kaplanov I et al (2014) Non-redundant properties of IL-1alpha and IL-1beta during acute colon inflammation in mice. Gut 63(4):598–609. https://doi.org/10.1136/gutjnl-2012-303329
Wang X, Wu H, Miller AH (2004) Interleukin 1alpha (IL-1alpha) induced activation of p38 mitogen-activated protein kinase inhibits glucocorticoid receptor function. Mol Psychiatry 9(1):65–75. https://doi.org/10.1038/sj.mp.4001339
Arend WP, Palmer G, Gabay C (2008) IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev 223:20–38. https://doi.org/10.1111/j.1600-065X.2008.00624.x
Sugama S, Conti B (2008) Interleukin-18 and stress. Brain Res Rev 58(1):85–95. https://doi.org/10.1016/j.brainresrev.2007.11.003
Lee JK, Kim SH, Lewis EC, Azam T, Reznikov LL, Dinarello CA (2004) Differences in signaling pathways by IL-1beta and IL-18. Proc Natl Acad Sci U S A 101(23):8815–8820. https://doi.org/10.1073/pnas.0402800101
Smith DE (2011) The biological paths of IL-1 family members IL-18 and IL-33. J Leukoc Biol 89(3):383–392. https://doi.org/10.1189/jlb.0810470
Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, Okamura H, Nakanishi K et al (1998) Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 8(3):383–390
Yamamoto Y, Tanahashi T, Katsuura S, Kurokawa K, Nishida K, Kuwano Y, Kawai T, Teshima-Kondo S et al (2010) Interleukin-18 deficiency reduces neuropeptide gene expressions in the mouse amygdala related with behavioral change. J Neuroimmunol 229(1–2):129–139. https://doi.org/10.1016/j.jneuroim.2010.07.024
Sekiyama A, Ueda H, Kashiwamura S, Sekiyama R, Takeda M, Rokutan K, Okamura H (2005) A stress-induced, superoxide-mediated caspase-1 activation pathway causes plasma IL-18 upregulation. Immunity 22(6):669–677. https://doi.org/10.1016/j.immuni.2005.04.006
Montezuma K, Biojone C, Lisboa SF, Cunha FQ, Guimaraes FS, Joca SR (2012) Inhibition of iNOS induces antidepressant-like effects in mice: Pharmacological and genetic evidence. Neuropharmacology 62(1):485–491. https://doi.org/10.1016/j.neuropharm.2011.09.004
Dhir A, Kulkarni SK (2007) Involvement of nitric oxide (NO) signaling pathway in the antidepressant action of bupropion, a dopamine reuptake inhibitor. Eur J Pharmacol 568(1–3):177–185. https://doi.org/10.1016/j.ejphar.2007.04.028
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M et al (2005) IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23(5):479–490. https://doi.org/10.1016/j.immuni.2005.09.015
Haraldsen G, Balogh J, Pollheimer J, Sponheim J, Kuchler AM (2009) Interleukin-33 - cytokine of dual function or novel alarmin? Trends Immunol 30(5):227–233. https://doi.org/10.1016/j.it.2009.03.003
Maywald RL, Doerner SK, Pastorelli L, De Salvo C, Benton SM, Dawson EP, Lanza DG, Berger NA et al (2015) IL-33 activates tumor stroma to promote intestinal polyposis. Proc Natl Acad Sci U S A 112(19):E2487–E2496. https://doi.org/10.1073/pnas.1422445112
Esplugues JV (2002) NO as a signalling molecule in the nervous system. Br J Pharmacol 135(5):1079–1095. https://doi.org/10.1038/sj.bjp.0704569
Ankarali S, Ankarali HC, Marangoz C (2009) Further evidence for the role of nitric oxide in maternal aggression: effects of L-NAME on maternal aggression towards female intruders in Wistar rats. Physiol Res 58(4):591–598
Liu RP, Zou M, Wang JY, Zhu JJ, Lai JM, Zhou LL, Chen SF, Zhang X et al (2014) Paroxetine ameliorates lipopolysaccharide-induced microglia activation via differential regulation of MAPK signaling. J Neuroinflammation 11:47. https://doi.org/10.1186/1742-2094-11-47
Yun HY, Dawson VL, Dawson TM (1997) Nitric oxide in health and disease of the nervous system. Mol Psychiatry 2(4):300–310
Wong ML, Rettori V, al-Shekhlee A, Bongiorno PB, Canteros G, McCann SM, Gold PW, Licinio J (1996) Inducible nitric oxide synthase gene expression in the brain during systemic inflammation. Nat Med 2(5):581–584
Green SJ, Scheller LF, Marletta MA, Seguin MC, Klotz FW, Slayter M, Nelson BJ, Nacy CA (1994) Nitric oxide: cytokine-regulation of nitric oxide in host resistance to intracellular pathogens. Immunol Lett 43(1–2):87–94
Musial A, Eissa NT (2001) Inducible nitric-oxide synthase is regulated by the proteasome degradation pathway. J Biol Chem 276(26):24268–24273. https://doi.org/10.1074/jbc.M100725200
Karolewicz B, Paul IA, Antkiewicz-Michaluk L (2001) Effect of NOS inhibitor on forced swim test and neurotransmitters turnover in the mouse brain. Pol J Pharmacol 53(6):587–596
Moncada S, Higgs EA (1995) Molecular mechanisms and therapeutic strategies related to nitric oxide. FASEB J 9(13):1319–1330
Ikenouchi-Sugita A, Yoshimura R, Hori H, Umene-Nakano W, Ueda N, Nakamura J (2009) Effects of antidepressants on plasma metabolites of nitric oxide in major depressive disorder: comparison between milnacipran and paroxetine. Prog Neuro-Psychopharmacol Biol Psychiatry 33(8):1451–1453. https://doi.org/10.1016/j.pnpbp.2009.07.028
Sandor NT, Brassai A, Puskas A, Lendvai B (1995) Role of nitric oxide in modulating neurotransmitter release from rat striatum. Brain Res Bull 36(5):483–486
Dhir A, Kulkarni SK (2011) Nitric oxide and major depression. Nitric Oxide 24(3):125–131. https://doi.org/10.1016/j.niox.2011.02.002
Rivera-Chavez F, Zhang LF, Faber F, Lopez CA, Byndloss MX, Olsan EE, Xu G, Velazquez EM et al (2016) Depletion of butyrate-producing clostridia from the gut microbiota drives an aerobic luminal expansion of salmonella. Cell Host Microbe 19(4):443–454. https://doi.org/10.1016/j.chom.2016.03.004
Byndloss MX, Olsan EE, Rivera-Chavez F, Tiffany CR, Cevallos SA, Lokken KL, Torres TP, Byndloss AJ et al (2017) Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357(6351):570–575. https://doi.org/10.1126/science.aam9949
Klena J, Zhang P, Schwartz O, Hull S, Chen T (2005) The core lipopolysaccharide of Escherichia coli is a ligand for the dendritic-cell-specific intercellular adhesion molecule nonintegrin CD209 receptor. J Bacteriol 187(5):1710–1715. https://doi.org/10.1128/JB.187.5.1710-1715.2005
Tse JKY (2017) Gut microbiota, nitric oxide, and microglia as prerequisites for neurodegenerative disorders. ACS Chem Neurosci 8(7):1438–1447. https://doi.org/10.1021/acschemneuro.7b00176
Schroder K, Hertzog PJ, Ravasi T, Hume DA (2004) Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75(2):163–189. https://doi.org/10.1189/jlb.0603252
Meyer O (2009) Interferons and autoimmune disorders. Joint Bone Spine 76(5):464–473. https://doi.org/10.1016/j.jbspin.2009.03.012
Subramaniam PS, Torres BA, Johnson HM (2001) So many ligands, so few transcription factors: a new paradigm for signaling through the STAT transcription factors. Cytokine 15(4):175–187. https://doi.org/10.1006/cyto.2001.0905
Ramana CV, Gil MP, Schreiber RD, Stark GR (2002) Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol 23(2):96–101
Kim TK, Maniatis T (1996) Regulation of interferon-gamma-activated STAT1 by the ubiquitin-proteasome pathway. Science 273(5282):1717–1719
Boehm U, Klamp T, Groot M, Howard JC (1997) Cellular responses to interferon-gamma. Annu Rev Immunol 15:749–795. https://doi.org/10.1146/annurev.immunol.15.1.749
Schneider WM, Chevillotte MD, Rice CM (2014) Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. https://doi.org/10.1146/annurev-immunol-032713-120231
Samarajiwa SA, Forster S, Auchettl K, Hertzog PJ (2009) INTERFEROME: the database of interferon regulated genes. Nucleic Acids Res 37(Database issue):D852–D857. https://doi.org/10.1093/nar/gkn732
Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, Fleming MA, Hayashi N, Higashino K et al (1997) Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science 275(5297):206–209
Hu X, Ivashkiv LB (2009) Cross-regulation of signaling pathways by interferon-gamma: Implications for immune responses and autoimmune diseases. Immunity 31(4):539–550. https://doi.org/10.1016/j.immuni.2009.09.002
Hu X, Chen J, Wang L, Ivashkiv LB (2007) Crosstalk among Jak-STAT, toll-like receptor, and ITAM-dependent pathways in macrophage activation. J Leukoc Biol 82(2):237–243. https://doi.org/10.1189/jlb.1206763
Dai C, Krantz SB (1999) Interferon gamma induces upregulation and activation of caspases 1, 3, and 8 to produce apoptosis in human erythroid progenitor cells. Blood 93(10):3309–3316
Maes M, Scharpe S, Meltzer HY, Okayli G, Bosmans E, D'Hondt P, Vanden Bossche BV, Cosyns P (1994) Increased neopterin and interferon-gamma secretion and lower availability of L-tryptophan in major depression: further evidence for an immune response. Psychiatry Res 54(2):143–160
Kahl KG, Kruse N, Faller H, Weiss H, Rieckmann P (2002) Expression of tumor necrosis factor-alpha and interferon-gamma mRNA in blood cells correlates with depression scores during an acute attack in patients with multiple sclerosis. Psychoneuroendocrinology 27(6):671–681
Mohr DC, Goodkin DE, Islar J, Hauser SL, Genain CP (2001) Treatment of depression is associated with suppression of nonspecific and antigen-specific T(H)1 responses in multiple sclerosis. Arch Neurol 58(7):1081–1086
Maes M, Song C, Lin AH, Bonaccorso S, Kenis G, De Jongh R, Bosmans E, Scharpe S (1999) Negative immunoregulatory effects of antidepressants: inhibition of interferon-gamma and stimulation of interleukin-10 secretion. Neuropsychopharmacology 20(4):370–379. https://doi.org/10.1016/S0893-133X(98)00088-8
Myint AM, Bondy B, Baghai TC, Eser D, Nothdurfter C, Schule C, Zill P, Muller N et al (2013) Tryptophan metabolism and immunogenetics in major depression: a role for interferon-gamma gene. Brain Behav Immun 31:128–133. https://doi.org/10.1016/j.bbi.2013.04.003
Raitala A, Pertovaara M, Karjalainen J, Oja SS, Hurme M (2005) Association of interferon-gamma +874(T/a) single nucleotide polymorphism with the rate of tryptophan catabolism in healthy individuals. Scand J Immunol 61(4):387–390. https://doi.org/10.1111/j.1365-3083.2005.01586.x
Oxenkrug GF (2011) Interferon-gamma-inducible kynurenines/pteridines inflammation cascade: implications for aging and aging-associated psychiatric and medical disorders. J Neural Transm (Vienna) 118(1):75–85. https://doi.org/10.1007/s00702-010-0475-7
O'Connor JC, Andre C, Wang Y, Lawson MA, Szegedi SS, Lestage J, Castanon N, Kelley KW et al (2009) Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J Neurosci 29(13):4200–4209. https://doi.org/10.1523/JNEUROSCI.5032-08.2009
Konan KV, Taylor MW (1996) Importance of the two interferon-stimulated response element (ISRE) sequences in the regulation of the human indoleamine 2,3-dioxygenase gene. J Biol Chem 271(32):19140–19145
O'Garra A, Arai N (2000) The molecular basis of T helper 1 and T helper 2 cell differentiation. Trends Cell Biol 10(12):542–550
Smeltz RB, Chen J, Ehrhardt R, Shevach EM (2002) Role of IFN-gamma in Th1 differentiation: IFN-gamma regulates IL-18R alpha expression by preventing the negative effects of IL-4 and by inducing/maintaining IL-12 receptor beta 2 expression. J Immunol 168(12):6165–6172
Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM et al (1993) Immune response in mice that lack the interferon-gamma receptor. Science 259(5102):1742–1745
Litteljohn D, Cummings A, Brennan A, Gill A, Chunduri S, Anisman H, Hayley S (2010) Interferon-gamma deficiency modifies the effects of a chronic stressor in mice: implications for psychological pathology. Brain Behav Immun 24(3):462–473. https://doi.org/10.1016/j.bbi.2009.12.001
Campos AC, Vaz GN, Saito VM, Teixeira AL (2014) Further evidence for the role of interferon-gamma on anxiety- and depressive-like behaviors: involvement of hippocampal neurogenesis and NGF production. Neurosci Lett 578:100–105. https://doi.org/10.1016/j.neulet.2014.06.039
Kustova Y, Sei Y, Morse HC Jr, Basile AS (1998) The influence of a targeted deletion of the IFNgamma gene on emotional behaviors. Brain Behav Immun 12(4):308–324. https://doi.org/10.1006/brbi.1998.0546
Clark E, Hoare C, Tanianis-Hughes J, Carlson GL, Warhurst G (2005) Interferon gamma induces translocation of commensal Escherichia coli across gut epithelial cells via a lipid raft-mediated process. Gastroenterology 128(5):1258–1267
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Inserra, A., Mastronardi, C.A., Rogers, G. et al. Neuroimmunomodulation in Major Depressive Disorder: Focus on Caspase 1, Inducible Nitric Oxide Synthase, and Interferon-Gamma. Mol Neurobiol 56, 4288–4305 (2019). https://doi.org/10.1007/s12035-018-1359-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1359-3