
Abstract
Although the adult brain contains neural stem cells (NSCs) that generate new neurons throughout life, these astrocyte-like populations are restricted to two discrete niches. Despite their terminally differentiated phenotype, adult parenchymal astrocytes can re-acquire NSC-like characteristics following injury, and as such, these ‘reactive’ astrocytes offer an alternative source of cells for central nervous system (CNS) repair following injury or disease. At present, the mechanisms that regulate the potential of different types of astrocytes are poorly understood. We used in vitro and ex vivo astrocytes to identify candidate pathways important for regulation of astrocyte potential. Using in vitro neural progenitor cell (NPC)-derived astrocytes, we found that exposure of more lineage-restricted astrocytes to either tumor necrosis factor alpha (TNF-α) (via nuclear factor-κB (NFκB)) or the bone morphogenetic protein (BMP) inhibitor, noggin, led to re-acquisition of NPC properties accompanied by transcriptomic and epigenetic changes consistent with a more neurogenic, NPC-like state. Comparative analyses of microarray data from in vitro-derived and ex vivo postnatal parenchymal astrocytes identified several common pathways and upstream regulators associated with inflammation (including transforming growth factor (TGF)-β1 and peroxisome proliferator-activated receptor gamma (PPARγ)) and cell cycle control (including TP53) as candidate regulators of astrocyte phenotype and potential. We propose that inflammatory signalling may control the normal, progressive restriction in potential of differentiating astrocytes as well as under reactive conditions and represent future targets for therapies to harness the latent neurogenic capacity of parenchymal astrocytes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Astrocytes were historically seen as support cells in the central nervous system (CNS) and in fact discharge multiple functions including regulation of energy metabolism, calcium signalling, synaptic transmission and mediating inflammatory responses. This view has been expanded by the recognition that astrocytes are also highly heterogeneous [1–3]. Interestingly, neurogenic adult neural stem cells (aNSCs) that reside in two niches (the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone (SGZ) of the dentate gyrus [4, 5]) have an astrocyte-like phenotype [6]. Even more intriguingly, there is a growing body of evidence that some parenchymal astrocytes have a latent neurogenic capacity. These observations underlie the need to identify regulatory pathways that govern the ability of an astrocyte to express NSC properties.
Early evidence for the neurogenic potential of parenchymal astrocytes came from observations that immature astrocytes from neonatal mouse adopt a radial glia-like phenotype when cultured with embryonic day 14 (E14) cortical cells [7], whilst astrocytes from embryonic or neonatal brain can form multipotent neurospheres [8]. Furthermore, forced expression of neurogenic transcription factors including Mash1, Ngn2 or Dlx2 is capable of converting postnatal parenchymal astrocytes to functional neurons [9–12]. Collectively, these studies show that immature parenchymal astrocytes have a latent neurogenic potential that can be realised by manipulation of intrinsic transcriptional programmes or the cellular milieu. Importantly, astrocytes from more mature brain after postnatal day 10 (P10) are not capable of generating neurospheres, indicating that the ability to dedifferentiate is a unique property of immature astrocytes. Intriguingly, under inflammatory conditions or following injury, mature parenchymal astrocytes can become reactive and re-acquire more immature or neural progenitor cell (NPC)-like properties [6, 13, 14]. Furthermore, reactive astrocytes isolated from adult cortex can give rise to multipotent neurospheres [15]. These observations underlie the relevance of understanding the latent neurogenic capacity of astrocytes for regenerative medicine strategies designed to recruit astrocytes into repair of damaged brain. Reactive astrocytes are, by definition, present at the site of injury and therefore offer an advantage over niche aNSCs that may reside far from the injury site. Although these studies clearly demonstrate the latent neurogenic capacity of some parenchymal astrocytes, the signalling pathways and mechanisms that regulate the reprogramming of astrocytes to NPC-like states or directly to neurons remain largely unknown [16].
The drive to identify genes for ‘stemness’ was initially led by transcriptome studies [17, 18], but recently, the idea has emerged that epigenetic signatures (including post-translational modification to histones and DNA methylation) can provide an indicator of cellular potential [19–24]. Indeed, epigenetic reprogramming is key to the generation of induced pluripotent stem cells (iPSCs). Whilst we are beginning to unravel the role of epigenetics in NSCs and neuronal differentiation, understanding of its contribution to maturation and reactivation of astrocytes is poor.
Our aim was to explore the transcriptome and epigenetic profile of different astrocyte populations to identify molecular signatures of astrocyte plasticity. To do this, we used homogeneous populations of NPC-derived astrocytes that show a differential ability to revert to an NPC-like state. We identified two factors: the pro-inflammatory cytokine, tumor necrosis factor alpha (TNF-α), known to play a role in reactive gliosis and NPC proliferation [25–27] and the BMP antagonist, noggin, as key regulators that govern reprogramming of astrocytes to NPCs. We also show that changes in epigenetic profiles accompany changes in cellular potential. Importantly, by comparing in vitro and ex vivo astrocyte transcriptomes, we provide evidence that there are likely to be common pathways and regulators responsible for astrocyte identity and potential in normal and injured brain that include pro- and anti-inflammatory signalling. Further, we find that astrocyte potential is also reflected in their intrinsic epigenetic signatures. These data add to a growing repository that aids identification of regulatory pathways involved in maintenance or re-acquisition of neurogenic NPC potential that may allow recruitment of parenchymal astrocytes in repair strategies for treatment of brain injury and degeneration.
Materials and Methods
Mice
All UK animal handling and procedures were performed according to the UK Animals (Scientific Procedures) Act, 1986 under Home Office licence. All animal procedures in Luxembourg were performed according to Federation of European Laboratory Animal Science Associations (FELASA) guidelines for the use of animals in research. Transgenic mice were genotyped using standard protocols and specific primers (Table S1).
Cell Culture
Cell Line
The CTX12 cell line is a conditionally immortalised mouse NPC line derived from E12-5 mouse cortex that has been generated in-house at King’s College London by Dr. Bithell and Dr. Williams. Cells were grown on poly-d-lysine (PDL)/laminin-coated plastic (Sigma) in modified Sato’s medium (modified Sato’s medium: Dulbecco’s modified Eagle’s medium (DMEM)/F12 supplemented with 5.6 mg/ml glucose, 100 μg/ml bovine serum albumin (BSA), 16 μg/ml putrescine, 60 ng/ml progesterone, 400 ng/ml l-thyroxine, 300 ng/ml 3,3′,5-triiodothyronine, 5 μg/ml insulin, 5 μg/ml apo-transferrin, 5 ng/ml sodium selenite, 1× glutamine, 1× pen/strep) containing 10 ng/ml fibroblast growth factor 2 (FGF2), 20 ng/ml epidermal growth factor (EGF) and 100 nM 4-hydroxytamoxifen (4-OHT). Medium was changed every 2–3 days and cells passaged using trypsin-EDTA and trypsin inhibitor (Sigma). For astrocyte differentiation, CTX12 cells were plated at 0.5 × 105 cells/cm2 and cultured in modified Sato’s medium with 10 % foetal bovine serum (FBS) or 20 ng/ml bone morphogenetic protein 4 (BMP4) (Peprotech and R&D Systems). Where indicated, cells were treated with additional factors: noggin (500 ng/ml), TNF-α (50 ng/ml) (Peprotech) and JSH-23 nuclear factor-κB (NFκB) Activation Inhibitor II (JSH-23, 10 μM, Santa Cruz).
Primary Cells
Cortices were isolated from P21 Swiss Webster and collected in calcium/magnesium-free Hank’s balanced salt solution (HBSS), trypsinised and DNAseI treated (50 μg/ml, Sigma) for 20 min at 37 °C and mechanically dissociated into a homogenous cell suspension [28]. Following washes and centrifugation, mixed glial cells were plated onto PDL (Sigma) in DMEM/F12 (Invitrogen) with 100 U/ml penicillin/100 mg/ml streptomycin (Sigma) and 10 % FBS (Biosera). Once confluent, cultures were shaken overnight at 180 rpm to remove microglia and oligodendrocytes and treated for 4–7 days with 20 μM cytosine arabinoside (AraC) to kill remaining dividing cells and obtain essentially pure astrocytes (>98 %), determined by immunofluorescence using anti-glial fibrillary acidic protein (GFAP) (Millipore) and anti-S100β (Dako) for astrocytes, anti-Iba1 (Biocare, microglia), anti-O4 (Sigma, oligodendrocytes) and TuJ1 (Covance, neurons).
Preparation of Mouse Forebrain Cell Suspensions and Fluorescence-Activated Cell Sorting (FACS) of Astrocytes
Different developmental stages (P4, P10 or P21) of forebrains from Aldh1l1-EGFP (Fthfd) transgenic mice (GenSat/MMRRC) were collected in calcium/magnesium-free HBSS. Tissue was diced and papain digested at 33 °C for 90 min (20 U/ml, Sigma) in dissociation buffer [EBSS (Sigma), D(+)-glucose 22.5 mM, NaHCO3 26 mM and DNaseI 125 U/ml with EDTA 0.5 mM and l-cysteine–HCl 1 mM (Sigma)] and washed 3× in dissociation buffer with BSA (1.0 mg/ml, Sigma) and trypsin inhibitor (1.0 mg/ml, Sigma) before mechanical dissociation through 5 ml and fire-polished Pasteur pipettes to a single cell suspension. Cells were pelleted, resuspended in cold phosphate-buffered saline (PBS) with DNaseI at 1 × 106 cell/ml, passed through a 70-μm filter and 7-aminoactinomycin D (7-AAD, Sigma) added. FITC-positive/PE-Cy5-negative cells were sorted. FACS was performed using a FACSAria I SORP running FACSDiva6.3 software (BD Biosciences).
Immunocytochemistry
Cells were fixed for 10 min in 4 % paraformaldehyde, permeabilised and incubated with primary antibodies in 1× PBS with 10 % normal serum. Primary antibodies used were anti-Olig2 (1:500; Millipore), anti-GFAP (1:400; Millipore), anti-Ki67 (1:1,000; Abcam), anti-Nestin (1:1,000; Abcam), anti-Sox2 (1:200; Santa Cruz), anti-O4 (Sigma, 1:200), anti-Iba1 (1:200, Biocare), TuJ1 (1:1,000, Covance), anti-GFP (1:2,000, Abcam), anti-bromodeoxyuridine (BrdU) (1:250, Abcam) and anti-NFκB-p65 (1:500; Abcam). Primary antibodies were visualised using specific AlexaFluor secondary antibodies (Molecular Probes), and nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Coverslips were mounted in Prolong Gold anti-fade mounting medium (Molecular Probes) and analysed using Zeiss AxioImager Z1 microscopes and AxioVision software.
BrdU Labelling
Cells were pulsed with BrdU (100 μM, Sigma) for 24 h, fixed with 4 % paraformaldehyde and treated with 2 N HCl for 45 min and 0.1 M borax (pH 8.5) for 15 min before processing for immunocytochemistry (above).
RNA Isolation, Microarray Hybridisation and Data Analysis
Cells were pelleted and lysed with TRIzol reagent (Invitrogen). Total RNA was further purified using an RNeasy Mini Kit (Qiagen). Biological replicates were prepared for microarrays using an Illumina TotalPrep RNA Amplification Kit (Ambion) and amplified RNAs hybridised on Sentrix® Mouse Ref-8 Expression BeadChips (Illumina), washed and scanned with Illumina BeadStation according to the Illumina protocols. Raw data were analysed in R using BeadArray and the Limma package. Ingenuity Pathway Analysis was used to perform pathway analysis on geneset data (IPA, Ingenuity Systems Inc. at www.ingenuity.com).
RNA Isolation and Reverse-Transcription PCR (RT-PCR)
Total RNA was purified from cells using the Qiagen RNeasy Mini Kit (Qiagen) as per manufacturer’s instructions. First strand cDNA was synthesised from 1 to 2 μg of total RNA using M-MLV reverse transcriptase (Promega). RT-PCR was carried out on the Chromo4 System (Bio-Rad) using primers listed in Table S1. PCR conditions were as follows: 3 min at 95 °C and 40 cycles of 10 s at 95 °C, 30 s at 60 °C and 30 s at 72 °C followed by 10-s 70–95° melt curves. All experiments included three no-template controls and were performed on three biological replicates with three technical replicates for each sample and normalised to GAPDH. Results were analysed using Opticon Monitor software (Bio-Rad), and relative gene expression levels were calculated using the Pfaffl method [29].
Chromatin Immunoprecipitation (ChIP) Analysis
ChIP was performed as described previously [30] and detailed here briefly. Cells were crosslinked with 1 % formaldehyde in PBS, quenched with 125 mM glycine and washed 3× with cold PBS (containing protease inhibitors) before centrifugation and lysis in lysis buffer [5 mM PIPES pH 8.0, 85 mM KCl, 0.5 % NP-40] for 30 min on ice. Pelleted nuclei were resuspended in shearing buffer [50 mM Tris pH 8.1, 10 mM EDTA, 0.1 % sodium dodecyl sulphate (SDS), 0.5 % sodium deoxycholate] and sonicated in a Bioruptor (Diogenode) with sufficient cycles (30 s on, 30 s off) to obtain an average chromatin shear size of 200–500 bp. Ten micrograms of pre-cleared chromatin was immunoprecipitated in modified RIPA buffer [140 mM NaCl, 10 mM Tris pH 7.5, 1 mM EDTA, 0.5 mM EGTA, 1 % TX-100, 0.01 % SDS, 0.1 % sodium deoxycholate] with specific antibodies (2 μg), protease inhibitors and pre-blocked magnetic protein G beads (Active Motif) at 4 °C. Following washes [2× Wash Buffer 1–20 mM Tris pH 8.1, 50 mM NaCl, 2 mM EDTA, 1 % TX-100, 0.1 % SDS; 1× Wash Buffer 2–10 mM Tris pH 8.1, 150 mM NaCl, 1 mM EDTA, 1 % NP-40, 1 % sodium deoxycholate, 250 mM LiCl; 2× TE], the chromatin was eluted [0.1 M NaHCO3, 1 % SDS] and de-crosslinked for 4 h at 65 °C with RNase and 200 mM NaCl then treated with proteinase K for 2 h at 42 °C. ChIP DNA was purified using a QIAquick PCR purification kit (Qiagen, according to manufacturer’s instructions). ChIP-qPCR was performed using ChIP DNA with promoter-specific primers (see Table S1). Controls included non-specific IgG and H3 ChIPs and ChIP-qPCR with primers for non-specific regions of genomic DNA where enrichment is not expected. Enrichment was analysed using a standard curve to quantitate, and data were normalised to total H3 (unless specified otherwise). qPCRs were run using an iCycler and MyiQ software (Bio-Rad).
The antibodies used were as follows: anti-H3 (rabbit IgG, AbCam), anti-H3K4me3 (rabbit IgG, AbCam; rabbit serum, Active Motif) and anti-H3K27me3 (rabbit IgG, Upstate) with rabbit IgG as a non-specific negative control. For primer sequences, see Table S1.
Results
Generation of Phenotypically Distinct Astrocytes from NPCs
The mouse NPC line, CTX12, grown as a monolayer in the presence of EGF, FGF2 and 4-OHT (collectively ‘GFs’) expresses classic NPC markers including Nestin and Sox2 (Figs. 1a, b and S1A) and can differentiate into both neurons and astrocytes (Fig. S1B, C). CTX12s cultured without GFs in the presence of FBS or BMP4 for 3 days led to astrocyte differentiation. In both conditions, CTX12 cells ceased proliferation (loss of Ki67), down-regulated Nestin and Olig2 and up-regulated GFAP (Fig. 1a, b). However, each condition generated homogeneous astrocytes with distinct morphology: FBS astrocytes were flatter with few processes whilst BMP4 astrocytes were more ramified and stellate (Fig. 1a). To test whether either population retained plasticity in an NPC-permissive environment, FBS/BMP4 were removed and replaced by GFs for 3 days (‘dedifferentiation’ conditions). FBS astrocytes became morphologically NPC-like, re-entered cell cycle, down-regulated GFAP and up-regulated Nestin and Olig2 (Fig. 1a, b). In contrast, few BMP4 astrocytes became proliferative, and morphology and gene expression were relatively unchanged (Fig. 1a, b). We subjected dedifferentiated cells to a tripotential differentiation paradigm [31] to test their ability to generate neurons and glia. Both βIII-tubulin-positive neurons and GFAP-positive astrocytes were readily obtainable from FBS astrocytes, but almost none were generated from BMP4 astrocytes (Figs. 1c and S1D). Thus, only FBS astrocytes re-acquire an NPC state following dedifferentiation, suggesting that they differ in their neurogenic potential from the BMP4 astrocytes.

Molecular phenotype of CTX12 cells under proliferative conditions or in the presence of FBS or BMP4. a Immunocytochemistry showing GFAP (green) and Olig2 (red) expression (top panels) and Nestin (green) and Ki67 (red) expression (bottom panels). Nuclei are counterstained with DAPI (blue). CTX12s were differentiated for 3 days (D3) with either FBS or BMP4 followed by 3 days of dedifferentiation (D6). b Comparison of Mki67, Nes, GFAP and Olig2 gene expression in conditions shown in a. c Example of a βIII-tubulin-positive neuron (green) in FBS astrocyte cultures following dedifferentiation and tripotential differentiation (top panel). Bottom panel shows a representative image from BMP4 astrocytes under similar conditions (GFAP in magenta and DAPI in blue). Abbreviations: Astro, astrocyte; GF, growth factors (FGF2/EGF/4-OHT). Error bars in b show SEMs, n = 3
Identification of a Common Set of Astrocyte-Regulated Genes and Pathways
To identify pathways or factors that regulate astrocyte differentiation and potential in our cellular model, we performed microarray analysis on CTX12s, FBS and BMP4 astrocytes (GEO accession number, with the authors). We identified ~8,000 probes with significantly changed expression upon differentiation from NPCs to FBS or BMP4 astrocytes (6,326 and 6,256 genes, respectively, false discovery rate (FDR) < 0.05, Benjamini-Hochberg [32], Tables 1 and Table S2, qPCR validation in Fig. S2). We first focussed on changes and enriched pathways common to both astrocyte populations following differentiation. Classical astrocyte markers were highly up-regulated including GFAP, Aqp4 and Slc39a12 [33]. Pathway analysis (IPA, Ingenuity Systems, Inc.) following FBS or BMP4 differentiation revealed a remarkable overlap in the most significant results, including cell cycle and proliferation-associated functions (Table S3), several of which are known to be enriched in astrocytes [33]. IPA also permits prediction of upstream regulators and their activation state, with several common to BMP4 and FBS astrocytes (relative to CTX12) including activation of TP53 and NFκB-related regulators and inhibition of MYC (Table S3).
Identification of a Set of Differentially Regulated Astrocyte Genes
Next, we focussed on genes differentially expressed between BMP4 and FBS astrocytes and identified 1,775 probes (1,579 genes, Fig. 2a and Table S4, FDR < 0.05, Benjamini-Hochberg) including genes known to be enriched in specific astrocyte populations (Table S5) [33–35]. We first wanted to identify candidates for maintenance of NSC/NPC and neurogenic properties in astrocytes. We compared differential gene expression between BMP4 and FBS astrocytes with that of a second, well-characterised NSC line, NS5 [36], and NS5-derived FBS astrocytes (GEO accession number XXXX), which have a similar phenotype to CTX12-derived FBS astrocytes (Fig. S3). We identified 372 ‘differentiation’ candidate genes (changed upon differentiation in BMP4 astrocytes only) and 333 ‘plasticity’ candidate genes (changed in FBS but not BMP4 differentiation, Fig. 2b and Table S6, FDR < 0.05). The most significant differentiation candidate was Mmd2, recently shown to be important in astrogliogenesis in vivo downstream of NFIA and Sox9 [37]. ‘Differentiation’ and ‘plasticity’ lists were used to infer over-represented signalling pathways (FDR < 0.05) where we found 46 and 16 pathways, respectively (Fig. 2c and Table S6). ‘Differentiation’ pathways included several linked with inflammation and reactive astrocytes, including pro-inflammatory (interleukin (IL)-1/6/8 and TNF-related) and anti-inflammatory (peroxisome proliferator-activated receptor (PPAR)) signalling [38–42]. Upstream regulators included the NFκB complex, which can be activated by upstream TNF signalling. In addition, inhibition of transforming growth factor (TGF)-β1 was highly significant between BMP4 and FBS astrocytes. Interestingly, BMP4 astrocytes had lower expression of the reactive astrocyte marker, Serpina3n [40]. Despite few enriched pathways in the ‘plasticity’ list, these too included inflammation as well as NFκB- and TNF-linked pathways such as TWEAK and ILK signalling, the latter of which is activated in reactive astrocytes [40] (Table S6). Thus, pathway analysis results from in vitro astrocytes suggested that inflammatory pathways might regulate NPC potential in astrocytes.

Candidate genes and signalling pathways involved in phenotype and potential of specific astrocyte populations. a Venn diagram showing number of genes expressed at a significantly different level between BMP4 and FBS astrocytes. b Venn diagram showing genes whose expressions were significantly changed in FBS- or BMP4-derived astrocytes compared to undifferentiated CTX12 or NS5 cells (FDR < 0.05). c Top canonical pathways enriched in the differentiation candidate gene set ‘B’ and ‘plasticity’ candidate gene set ‘A’ both shown in b
Activation of NFκB by TNF-α Leads to Dedifferentiation of a Subset of BMP4 Astrocytes
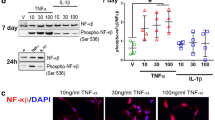
Under inflammatory conditions or following injury, mature parenchymal astrocytes can become reactive and re-acquire NPC-like properties [15]. Thus, we investigated the effect of activating the NFκB pathway, a common inflammatory signalling, on BMP4 astrocyte potential using TNF-α. The NFκB p65 subunit was largely cytoplasmic in BMP4 astrocytes but became nuclear following 2 h with TNF-α in proliferative conditions, confirming NFκB activation (Fig. S4A). BMP4 astrocytes in dedifferentiation conditions with TNF-α for 3 days showed a significant increase in mKi67, nestin and Olig2 and decrease in GFAP expression compared to growth factor (GF) alone and an increase in Ki67-positive cells (Fig. 3a–c), many of which became morphologically more NPC-like (Fig. 3a). A 24-h pulse of BrdU showed an increase in BrdU-positive cells with TNF-α (Fig. S4B, C). These effects were due to NFκB activation since they were reversed by the specific NFκB inhibitor, JSH-23 [43] (Fig. 3a, c). Cultures were subsequently tested for their neurogenic capacity, and a small number of ßIII-tubulin-positive neurons were observed with TNF-α (Fig. 3d). Therefore, NFκB activation through a TNF-α treatment induces a subset of BMP4 astrocytes to re-enter in the cell cycle and drives them to a more NPC-like state.

A subset of BMP4 astrocytes treated with TNF-α re-enter the cell cycle and re-acquire NPC characteristics. a Immunocytochemistry on BMP4 astrocytes cultured in dedifferentiation conditions for 3 days alone (GF), in the presence of TNF-α (+GF + TNFα) or with TNF-α and JSH23 (+GF + TNFα + JSH23). JSH23 was added 30 min before culturing astrocytes in dedifferentiation conditions in the presence of TNF-α. Top row shows GFAP (green) and Olig2 (red), and bottom row shows Nestin (green) and Ki67 (red). Nuclei were counterstained with DAPI (blue). b Percentage of Ki67-positive cells in cultures shown in a. c Gene expression levels of MKi67, Nestin (Nes), GFAP and Olig2 in BMP4 astrocytes in conditions shown in (a, b). Data are expressed as percentage of expression relative to D3 BMP4 astrocytes. d βIII-tubulin-positive neurons (green) and GFAP-positive astrocytes (magenta) with DAPI-labelled nuclei (blue) in BMP4 astrocyte cultures following dedifferentiation with TNF-α followed by tripotential differentiation. For b and c, n = 3. Scale bars in a and d: 20 μm. P values in b and c: *p < 0.05, **p < 0.01 (Student’s t test), error bars show SEMs
TNF-α-Treated BMP4 Astrocytes Have a More NPC-Like Transcriptome
For a global view of TNF-α effects on BMP4 astrocytes, we performed microarray analysis to compare BMP4 astrocytes (BMP) with those in dedifferentiation conditions (GF) with or without TNF-α (TNF) (GEO accession number XXXX). We identified 3,712, 2,734 and 2,285 significantly changed genes comparing ‘TNF + GF v BMP4’, ‘TNF + GF v GF’ and ‘GF v BMP4’, respectively (FDR < 0.05, Benjamini-Hochberg, Table S7, qPCR validation in Fig. S5). Interestingly, the most significant up-regulated TNF gene, Lcn2, is a marker of reactive astrocytes in vivo [40]. TNF-α regulated genes (compared to BMP4 or GF) were enriched for cell cycle and growth and proliferation functions with pathways including control of cell cycle, p53, cancer-related pathways and TNF-related pathways (Table S8), consistent with a change in proliferative potential. Predicted upstream regulators included TNF and NFκB, activation of MYC, WNT and other pro-inflammatory molecules (including IFNG, IL1/17, Table S8). One of the few predicted regulators in the ‘GF v BMP’ dataset was sonic hedgehog (SHH) (activated), also in the TNF-α dataset, which may reflect the roles of pro-inflammatory molecules and SHH signalling in astrocyte acquisition of NSC properties [44, 45]. Interestingly, in dedifferentiation conditions (GF or TNF + GF), Ascl1, encoding the proneural protein Mash1, was up-regulated whilst members of Notch (Notch1, 4), Id (Id1-3), Stat (Stat2) and Hes (Hes1, 5) families were down-regulated, particularly in TNF + GF conditions. This is consistent with a neurogenic NPC-like capability since many are involved in the neurogenic–gliogenic switch, including several regulated by BMP signalling [46]. Taken together, these transcriptomic results highlight that the expression levels of several NPC-associated genes/pathways are more highly re-activated in the presence of TNF-α when compared to the corresponding BMP4-derived astrocytes under normal or dedifferentiation conditions.
Mimicking CNS Damage Promotes NPC Properties in BMP4-Derived Astrocytes
In keeping with down-regulation of BMP-regulated genes, our analysis predicted inhibition of BMP itself under dedifferentiation conditions with and without TNF-α. This suggests that BMP4 astrocytes produce endogenous BMPs and that their inhibition is required for dedifferentiation. Interestingly, reactive astrocytes adjacent to penetrating CNS injuries in both spinal cord and brain up-regulate the BMP inhibitor, noggin [47], and BMP secreted from blood endothelial cells can induce reversible quiescence of NSC/NPCs in vitro, reversible with noggin [48]. We thus tested whether noggin could induce re-acquisition of NPC properties in BMP4 astrocytes. This led to a change from astrocyte to NPC-like morphology and a significant increase in proliferation (Fig. 4a, b), concomitant with down-regulation of GFAP and up-regulation of mKi67, Nestin and Olig2 (Fig. 4c) and down-regulation of genes enriched in parenchymal astrocytes, including Aqp4, Thrsp and Ngef (Table S5), to levels similar to dedifferentiated FBS-derived astrocytes (Fig. S6). Noggin-treated dedifferentiated astrocytes were also neurogenic, with a small number of ßIII-tubulin-positive neurons (Fig. 4d).

Inhibition of BMP signalling leads to re-acquisition of NPC characteristics in BMP4 astrocytes. a Immunocytochemistry comparing Ki67 (red) and GFAP (green) expression in BMP4 astrocytes in dedifferentiation conditions with and without noggin (+GF + noggin and +GF, respectively). Noggin was added simultaneously with GF. Nuclei are counterstained with DAPI (blue). b Percentage of Ki67-positive cells in the conditions shown in a. c Gene expression levels of Mki67, Nes, GFAP and Olig2 in the conditions shown in a. Expression levels are shown as percentage in +GF + noggin conditions relative to +GF conditions (100 %). d βIII-tubulin-positive neurons (green) and GFAP-positive astrocytes (magenta) with DAPI-labelled nuclei (blue) in BMP4 astrocyte cultures following dedifferentiation with noggin followed by tripotential differentiation. For b and c, n = 3. Scale bars in a and d, 20 μm. P values in b and c: *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test), error bars show SEMs
Histone Modifications at Key Promoters Distinguish Between Astrocyte Types
Differentiation, including acquisition and maintenance of a more restricted fate, is known to be associated with epigenetic changes [20, 21, 24, 49]. The ability of FBS astrocytes to dedifferentiate compared to BMP4 astrocytes may reflect intrinsic epigenetic as well as transcriptional differences, including histone modifications in promoters associated with active (e.g. H3K9ac and H3K4me3) or repressed (H3K27me3) chromatin states [21, 50].
To look specifically at histone modifications associated with cellular potential, we used ChIP-qPCR to examine relative levels of H3K4me3 and H3K27me3 at selected gene promoters: Ccnb1 (positive cell cycle regulator), Nestin (NPC-specific), Olig2 (neural-specific bHLH factor) and Sox2 (NSC/NPC marker). H3K4me3 was significantly decreased at Ccnb1, Nestin and Sox2 in BMP4 astrocytes compared to CTX12s and FBS astrocytes, whilst H3K27me3 increased at Ccnb1, Nestin and Olig2 in both astrocytes, corresponding with its down-regulation and cell cycle exit (Fig. 5a). Similar trends are shown at the Ascl1 locus, where H3K4me3 was significantly decreased in BMP4 astrocytes compared to CTX12s and FBS astrocytes along with H3K27me3 increase in both astrocytes (Fig. S7A, B). These data suggest that the epigenetic landscape reflects the differentiation state and may contribute to regulation of astrocyte plasticity. We therefore analysed H3K4me3 at the same promoters in TNF-α and noggin-treated BMP4 astrocytes in dedifferentiation conditions. With TNF-α, Ccnb1, Nestin and Olig2 promoters showed increased levels of H3K4me3, whilst noggin-mediated dedifferentiation was accompanied by an increase at all four loci (Fig. 5b), suggesting a more permissive chromatin state. We next asked whether cultured primary astrocytes are associated with a less permissive chromatin state. We cultured P21 mouse cortical astrocytes and compared relative H3K4me3 and H3K27me3 levels with our in vitro astrocytes. P21 astrocytes had significantly lower H3K4me3 enrichment at all four loci compared to FBS astrocytes and Sox2 in BMP4 astrocytes and higher H3K27me3 at Sox2 and Olig2 compared with both in vitro populations (Fig. 5c). Similar results were obtained at the Ascl1 locus, where H3K4me3 was significantly decreased, whilst H3K27me3 was increased in primary astrocytes compared to FBS- and BMP4-derived astrocytes (Fig. S7C, D). Although cultured parenchymal astrocytes may represent a more immature/reactive phenotype that non-cultured counterparts [33], our data are consistent with the restricted state of later postnatal astrocytes and suggest a more restricted state in BMP4 versus FBS astrocytes. Thus, astrocyte plasticity and neurogenic potential are reflected at the chromatin level.

Epigenetic comparison of phenotypically distinct astrocytes. a H3K4me3 (left) and H3K27me3 (right) enrichment relative to total H3 at specific gene promoters in CTX12 cells (grey), FBS astrocytes (red) and BMP4 astrocytes (blue). Results are expressed as a percentage of levels in CTX12 (100 %). b Left graph shows H3K4me3 enrichment relative to total H3 at specific gene promoters in CTX12 cells (grey) and BMP4 astrocytes after 3 days of dedifferentiation alone (+GF, yellow) or with the addition of noggin (+GF + noggin, green). Results are expressed as a percentage of levels in CTX12 (100 %). The right-hand graph shows a comparison between BMP4 astrocytes in dedifferentiation conditions alone (+GF, yellow) and with TNF-α (+GF + TNFα). c Comparison of H3K4me3 (left) and H3K27me3 (right) enrichment in FBS astrocytes (red), BMP4 astrocytes (dark blue) and primary cortical astrocytes (light blue). For a–c, n = 3. *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test), and error bars show SEMs
Temporal Changes in the Postnatal Astrocyte Transcriptome
To further identify regulatory pathways responsible for astrocyte plasticity associated with specific differentiation states, we isolated ex vivo postnatal astrocytes by FACS from Aldh1l1-EGFP mice (Fig. S8A–C) [33], and we performed microarray analysis of astrocytes obtained from periods before, during and after which they lose neurosphere-forming potential (GEO accession number XXXX) [8]. We identified significantly changed genes (FDR < 0.05, Benjamini-Hochberg, qPCR validation in Fig. S9) from P4 to 10, P10 to 21 and P4 to 21 (Table S9) and determined functionally enriched pathways and regulators (Table S10). Most changes occurred from P4 to P10 (1,650 genes, Fig. 6a) with significantly enriched cell cycle and neurological disorder functions and cell cycle pathways and regulators (Fig. S10). Up-regulated genes also included Ntsr2, Gjb6 and Mertk (Fig. 6b), all highly enriched in mature in vivo astrocytes [33]. The top pathway identified was cell cycle regulation of replication. Interestingly, this was enriched in TNF + GF gene sets from BMP4 astrocytes, where many genes down-regulated from P4 to P10 showed up-regulation with TNF-α in vitro (Fig. S9). Signalling involving janus tyrosine kinase (JAK) family kinases and IL-6 revealed down-regulation of several genes by P10 that were significantly lower in BMP4 versus FBS astrocytes (Fig. S11). P10–P21 analysis (357 genes) again showed enrichment for cell cycle-associated pathways (including GADD45) and P4–P21 analysis (2,732 genes) identified additional pathways, some of which were identified in BMP4 astrocytes exposed to TNF-α (Tables S10 and S9). These data suggest common pathways in vitro and ex vivo that regulate astrocyte phenotype, potential and differentiation. We therefore interrogated upstream regulators to identify regulators of astrocyte differentiation and NSC potential.

Developmental changes in gene expression in postnatal astrocytes. a Clustered heatmap showing relative gene expression levels in P4, P10 and P21 astrocytes from Aldh1l1-EGFP mice. Individual biological replicates are shown as individual columns for P4 (n = 3), P10 (n = 4) and P21 (n = 3). Relative expression levels are shown from low (blue) to high (yellow). b Graphs show Gadd45, Foxm1, Ccnd1, Ntsr2, TGF-β1, Gjb6, Mertk and PPARγ expression levels from FACSed Aldh1l1-EGFP astrocyte populations. Expression levels are shown in P10 (red) and P21 (blue) populations relative to P4 levels (x-axis). Data are the average of three biological replicates. Error bars show SEMs. P values: *p < 0.05, **p < 0.01, ns = not significant (Student’s t test). c Cell cycle diagram. The scheme shows activated (green) or inhibited (red) upstream regulators during in vivo astrocyte maturation as predicted by IPA
P4–P10 upstream regulators included PPARG (and PPARGC1A), TP53 and CDKN2A activation and MYC, E2F1, EP400 and vascular endothelial growth factor (VEGF) inhibition (Table S10) with additional regulators from P10-P21 including FOXM1 (inhibition). Analysing P4–P21 also predicted CDKN1A activation and TGF-β1 and CCND1 inhibition. Broadly, these data support progressive cell cycle arrest (Fig. 6c) accompanied by changes consistent with inhibition of inflammatory signalling and identified other candidates with potential roles during early postnatal stages.
Intriguingly, many candidates followed the same pattern of inhibition/activation as BMP4 compared to FBS astrocytes or the opposing activation status following TNF-α exposure (Fig. 7 and Table S11). This further suggests that there are common, upstream regulators that control astrocyte differentiation as well as reactive phenotypes and dedifferentiation events in our in vitro and ex vivo models.

Candidate upstream regulators in the astrocyte neurogenic/non-neurogenic shift. The scheme illustrates that selected potential regulators that are responsible for the shift of neurogenic astrocytes (FBS-derived and P4 astrocytes) towards non-neurogenic astrocytes (BMP4-derived and P21 astrocytes) are also responsible for the dedifferentiation process under a pro-inflammatory environment (BMP4-derived astrocytes + TNF-α)
Discussion
Adult neurogenesis occurs in two neurogenic niches from aNSCs with astrocyte-like characteristics [5, 51, 52]. Identification of glial progenitors outside these niches in healthy brain [53, 54] and reactive astrocytes with progenitor-like characteristics following injury [15] led to the notion that these cells may represent a source of progenitors for repair. In fact, recent studies have shown that stroke or excitotoxicity in mouse elicit a latent neurogenic program in striatal astrocytes [55, 56]. However, our current understanding of the key genes, pathways and mechanisms responsible for regulating astrocyte potential remains limited. Here, we describe two in vitro NSC-derived astrocytes populations that differ in their ability to dedifferentiate to an NPC-like state. We identified pathways associated with response to CNS injury and inflammation that regulate this differential potential and correlate with findings from ex vivo postnatal astrocytes. We also provide evidence for other putative regulators of parenchymal astrocyte development at postnatal stages when they acquire a terminally differentiated status and which may be involved in re-acquisition of NSC properties following injury. Finally, our data add to existing datasets from astrocyte populations that represent an invaluable resource to identify regulators of phenotype and potential towards developing therapeutic strategies for endogenous CNS repair.
Inflammatory Pathways Regulate Astrocyte Neurogenic Potential and Differentiation
Continued expression of neurogenic fate determinants by cortical astrocytes at early postnatal stages ensures a gradual change from neurogenic to gliogenic RGCs and astrocytes [57, 58]. Interestingly, these findings are supported by previous studies showing that early postnatal astrocytes can dedifferentiate and give rise to neurogenic neurospheres, whilst this capacity declines during the second postnatal week [8]. Conceptually, functional specialisation is likely to require cell cycle exit and acquisition of a postmitotic status rendering astrocytes refractory to mitogens [59]. However, under inflammatory conditions or following injury, mature parenchymal astrocytes can become reactive and re-acquire immature or NPC-like properties [15, 60]. Nevertheless, signalling pathways that modulate astrogliosis with respect to time after injury and the type of damage are complex and not fully known [16, 61].
Here, we have used BMP4- and FBS-derived astrocytes to identify regulators that control the balance between astrocyte quiescence and maintenance of latent neurogenic potential. We identified a likely role for pro-inflammatory signalling, showing that NFκB activation by TNF-α can facilitate the re-acquisition of NPC properties. Indeed, our results showing that TNF-α treatment of BMP4-derived astrocytes modulates several Notch members (such as a decrease in Notch1 expression levels), are in agreement with the recent discovery that attenuated Notch1 signalling is necessary for neurogenesis by striatal astrocytes [55]. Several other signalling molecules including SHH and VEGF were also candidate regulators, which is interesting in light of recent evidence for the likely importance of signals from the vasculature for reactive astrocyte proliferation [62] and that SHH is necessary and sufficient to induce NSC-like properties in astrocytes [45]. It is also known that during brain injury, pro-inflammatory molecules, such as IL-1 and TNF-α, are produced [63] and activate SHH signalling and subsequent reactive gliosis in astrocytes [44]. It is intriguing to speculate that our in vitro model may recapitulate the action of a pro-inflammatory niche environment to activate SHH and VEGF in BMP4 astrocytes, leading to re-acquisition of NPC properties in a sub-population of cells. This is supported by our ex vivo parenchymal astrocyte expression data before and after the ‘neurogenic’ period with candidate upstream regulators including VEGF (inhibition). SHH signalling was previously reported as a significantly enriched pathway in postnatal astrocytes [33]. Our ex vivo data are also consistent with the hypothesis that SHH, VEGF and inflammatory-associated signalling may be involved in control of NSC properties in parenchymal, non-niche astrocytes during normal development and reactive gliosis.
Other pro- and anti-inflammatory pathway candidates identified included TGF-β1 and PPAR signalling, respectively. Based on our expression analyses, a more restricted (non-neurogenic) astrocyte phenotype correlates with inhibition of TGF-β1 and activation of PPAR (particularly PPARγ), and both of these pathways are amongst those highly enriched in postnatal astrocytes [33]. PPAR proteins (α, δ and γ) are nuclear hormone receptors with roles including regulation of inflammation in CNS disorders and following injury [39]. PPARα agonists can inhibit glial activation by lipopolysaccharides (LPS) by inhibiting astrocyte (and microglial) induction of TNF-α, IL-1β and IL-6, and PPARγ can inhibit NFκB and JAK/STAT signalling [38, 39]. PPARγ agonists can also affect the proliferation and differentiation of NSCs [64] and thus appear to be bona fide candidates for further investigation. TGF-β1 is a known mediator of inflammation whose expression is increased in the CNS in association with many disorders [42]. Exposure of postnatal parenchymal astrocytes to TGF-β1 in vitro causes widespread gene expression changes including up-regulation of the reactive astrocyte marker Lcn2, enrichment of the PPARα/RXRα activation pathway and association with immune or inflammation signalling (including NFκB and TNF) when used in combination with LPS and IFNγ [42]. Together, these data lend weight to the possibility of a cross-regulatory role between pro- and anti-inflammatory regulators that may be common to normal astrocyte differentiation (accompanied by loss of NSC properties) and re-acquisition of these properties following injury. Moreover, we have shown the validity of our in vitro system for identification of physiologically relevant candidate pathways.
Changes in Cell Cycle Regulators Occur in the Postnatal ‘Neurogenic Window’
We identified few pathways or regulators linked with cell cycle as differentially represented between our two in vitro astrocytes; indeed, many were shared by both upon differentiation. For example, both showed inhibition of FOXM1 and activation of FOXO3—forkhead transcription factors with reciprocally antagonist actions implicated in many cancers and in cardiomyocyte proliferation [65, 66]. Furthermore, FOXO3 is also involved in regulation of NSCs, preventing premature neuronal differentiation [67, 68]. FOXM1 was significantly down-regulated during astrocyte maturation (from P4 to P21). Despite the heterogeneity of in vivo astrocyte populations, our ex vivo array data revealed a significant role for cell cycle-associated genes and regulators during maturation. This was particularly evident during the P4–P10 transition, after which the neurogenic capacity of parenchymal astrocytes is lost [8]. This included down-regulation of genes that have been previously shown to be decreased at P17 (Mcm2/5/6, Tgif2 and Uhrf1 [33]). Our observation that Stat3 and Jak2 are down-regulated between P4 and P10 (and are lower in BMP4 versus FBS astrocytes) implicate JAK kinases/IL-6 signalling in regulation of astrocyte plasticity. This is supported by their up-regulation during induction of astrogliosis [69].
Endogenous BMP Signalling Is Able to Maintain In Vitro Astrocyte Quiescence
Following CNS injury, reactive astrocytes can up-regulate noggin expression [47]. Exogenous noggin in our in vitro model increased proliferation and acquisition of a more NPC-like phenotype in BMP4 astrocytes, showing that endogenous BMP signalling may regulate their proliferative and neurogenic properties. Indeed, increased levels of secreted BMPs upon loss of p21 in adult NSCs lead to their premature differentiation into mature astrocytes [70]. However, expression of Bmp and noggin was not significantly different between BMP4 and FBS astrocytes (except Bmp1 is lower in BMP4 astrocytes). Serum is often used as a proxy for BMPs; however, different levels of BMPs can affect different cellular responses. For example, low BMP2 levels increase proliferation of embryonic NPCs, whilst higher levels induce differentiation [71]. Interestingly, this is due to the differential activation status of the BMP receptor BMPR1B (relative to BMPR1A), which at higher levels leads to cell cycle arrest (and apoptosis or terminal differentiation). Indeed, BMPR1A and 1B have directly opposing roles (positive and negative, respectively) in regulating astrogliosis in vivo [72]. BMP4 astrocytes express twofold higher Bmpr1b and significantly lower Bmpr1a than FBS astrocytes, a process that is reversed following dedifferentiation with TNF-α. Therefore, BMP4 and FBS astrocytes may have different responses to BMP signalling, and future work is required to test whether this explains noggin effects on astrocyte potential.
The Epigenetic Profile of Astrocytes Correlates with Their Potential
The chromatin landscape acts as a cellular memory that determines and permits long-lasting transcriptional programmes throughout development and differentiation [73]. Changes in H3K4me3 and H3K27me3 are coordinately controlled at genes activated or repressed, respectively, during differentiation [74, 75]. We have demonstrated that FBS astrocytes have more permissive chromatin at cell cycle- and NPC-related loci when compared to BMP4 astrocytes, correlating with their neurogenic potential. Moreover, TNF-α or noggin exposure induces the dedifferentiation of a subset of BMP4 astrocytes, and this is accompanied by epigenetic changes consistent with increased cellular potential. Of note, the importance of epigenetic regulation during neocortical development has been recently investigated, showing that the polycomb group complex (PcG) restricts neurogenic competence of NPCs and promotes the transition of NPC fate from neurogenic to astrogliogenic [76]. In this context, other studies in our group have shown that NSC-derived astrocytes retain an active epigenetic signature at promoters of neural lineage-specific genes, even though they are not expressed (unpublished data).
Our studies show that changes from neurogenic NPCs and astrocytes to non-neurogenic astrocytes are reflected at the transcriptional and epigenetic level. Further work will explore the relative contribution of identified inflammatory pathways and changes to the epigenome to astrocyte potential. Our data also add to existing datasets from astrocyte populations that represent an invaluable resource to identify regulators of phenotype and potential towards developing therapeutic strategies for endogenous CNS repair.
References
Zhang Y, Barres BA (2010) Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr Opin Neurobiol 20:588–594
Molofsky AV, Krenick R, Ullian EM, Tsai HH, Deneen B, Richardson WD, Barres BA, Rowitch DH (2012) Astrocytes and disease: a neurodevelopmental perspective. Genes Dev 26:891–907
Anderson MA, Ao Y, Sofroniew MV (2014) Heterogeneity of reactive astrocytes. Neurosci Lett 565:23–29
Doetsch F (2003) A niche for adult neural stem cells. Curr Opin Genet Dev 13:543–550
Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A (1999) Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97:703–716
Robel S, Berninger B, Gotz M (2011) The stem cell potential of glia: lessons from reactive gliosis. Nat Rev Neurosci 12:88–104
Hunter KE, Hatten ME (1995) Radial glial cell transformation to astrocytes is bidirectional: regulation by a diffusible factor in embryonic forebrain. Proc Natl Acad Sci U S A 92:2061–2065
Laywell ED, Rakic P, Kukekov VG, Holland EC, Steindler DA (2000) Identification of a multipotent astrocytic stem cell in the immature and adult mouse brain. Proc Natl Acad Sci U S A 97:13883–13888
Berninger B, Costa MR, Koch U, Schroeder T, Sutor B, Grothe B, Götz M (2007) Functional properties of neurons derived from in vitro reprogrammed postnatal astroglia. J Neurosci Off J Soc Neurosci 27:8654–8664
Heinrich C, Blum R, Gascon S, Masserdotti G, Tripathi P, Sánchez R, Tiedt S, Schroeder T et al (2010) Directing astroglia from the cerebral cortex into subtype specific functional neurons. PLoS Biol 8, e1000373
Heinrich C, Gascon S, Masserdotti G, Lepier A, Sanchez R, Simon-Ebert T, Schroeder T, Götz M et al (2011) Generation of subtype-specific neurons from postnatal astroglia of the mouse cerebral cortex. Nat Protoc 6:214–228
Blum R, Heinrich C, Sanchez R, Lepier A, Gundelfinger ED, Berninger B, Götz M (2011) Neuronal network formation from reprogrammed early postnatal rat cortical glial cells. Cereb Cortex 21:413–424
Pekny M, Nilsson M (2005) Astrocyte activation and reactive gliosis. Glia 50:427–434
Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35
Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Götz M (2008) Origin and progeny of reactive gliosis: a source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A 105:3581–3586
Buffo A, Rolando C, Ceruti S (2010) Astrocytes in the damaged brain: molecular and cellular insights into their reactive response and healing potential. Biochem Pharmacol 79:77–89
Ivanova NB, Dimos JT, Schaniel C, Hackney JA, Moore KA, Lemischka IR (2002) A stem cell molecular signature. Science 298:601–604
Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA (2002) "Stemness": transcriptional profiling of embryonic and adult stem cells. Science 298:597–600
Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A et al (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125:315–326
Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE et al (2008) Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454:766–770
Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W et al (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448:553–560
Azuara V, Perry P, Sauer S, Spivakov M, Jørgensen HF, John RM, Gouti M, Casanova M et al (2006) Chromatin signatures of pluripotent cell lines. Nat Cell Biol 8:532–538
Spivakov M, Fisher AG (2007) Epigenetic signatures of stem-cell identity. Nat Rev Genet 8:263–271
Burney MJ, Johnston C, Wong KY, Teng SW, Beglopoulos V, Stanton LW, Williams BP, Bithell A et al (2013) An epigenetic signature of developmental potential in neural stem cells and early neurons. Stem Cells 31:1868–1880
Widera D, Mikenberg I, Elvers M, Kaltschmidt C, Kaltschmidt B (2006) Tumor necrosis factor alpha triggers proliferation of adult neural stem cells via IKK/NF-kappaB signaling. BMC Neurosci 7:64
Peng H, Whitney N, Wu Y, Tian C, Dou H, Zhou Y, Zheng J (2008) HIV-1-infected and/or immune-activated macrophage-secreted TNF-alpha affects human fetal cortical neural progenitor cell proliferation and differentiation. Glia 56:903–916
Wu JP, Kuo JS, Liu YL, Tzeng SF (2000) Tumor necrosis factor-alpha modulates the proliferation of neural progenitors in the subventricular/ventricular zone of adult rat brain. Neurosci Lett 292:203–206
McCarthy KD, de Vellis J (1980) Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85:890–902
Pfaffl MW, Horgan GW, Dempfle L (2002) Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30, e36
Soldati C, Bithell A, Conforti P, Cattaneo E, Buckely NJ (2011) Rescue of gene expression by modified REST decoy oligonucleotides in a cellular model of Huntington’s disease. J Neurochem 116:415–425
Glaser T, Pollard SM, Smith A, Brüstle O (2007) Tripotential differentiation of adherently expandable neural stem (NS) cells. PLoS One 2, e298
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57:289–300
Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL et al (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci Off J Soc Neurosci 28:264–278
Beckervordersandforth R, Tripathi P, Ninkovic J, Bayam E, Lepier A, Stempfhuber B, Kirchhoff F, Hirrlinger J et al (2010) In vivo fate mapping and expression analysis reveals molecular hallmarks of prospectively isolated adult neural stem cells. Cell Stem Cell 7:744–758
Lovatt D, Sonnewald U, Waagepetersen HS, Schousboe A, He W, Lin JH, Han X, Takano T et al (2007) The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J Neurosci 27:12255–12266
Conti L, Pollard SM, Gorba T, Reitano E, Toselli M, Biella G, Sun Y, Sanzone S et al (2005) Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol 3, e283
Kang P, Lee HK, Glasgow SM, Finley M, Donti T, Gaber ZB, Graham BH, Foster AE et al (2012) Sox9 and NFIA coordinate a transcriptional regulatory cascade during the initiation of gliogenesis. Neuron 74:79–94
Xu J, Chavis JA, Racke MK, Drew PD (2006) Peroxisome proliferator-activated receptor-alpha and retinoid X receptor agonists inhibit inflammatory responses of astrocytes. J Neuroimmunol 176:95–105
Bright JJ, Kanakasabai S, Chearwae W, Chakraborty S (2008) PPAR regulation of inflammatory signaling in CNS diseases. PPAR Res 2008:658520
Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA (2012) Genomic analysis of reactive astrogliosis. J Neurosci 32:6391–6410
Drew PD, Xu J, Storer PD, Chavis JA, Racke MK (2006) Peroxisome proliferator-activated receptor agonist regulation of glial activation: relevance to CNS inflammatory disorders. Neurochem Int 49:183–189
Hamby ME, Coppola G, Ao Y, Geschwind DH, Khakh BS, Sofroniew MV (2012) Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple G-protein-coupled receptors. J Neurosci 32:14489–14510
Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS (2010) Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci U S A 107:2669–2674
Amankulor NM, Hambardzumyan D, Pyonteck SM, Becher OJ, Joyce JA, Holland EC (2009) Sonic hedgehog pathway activation is induced by acute brain injury and regulated by injury-related inflammation. J Neurosci 29:10299–10308
Sirko S, Behrendt G, Johansson PA, Tripathi P, Costa M, Bek S, Heinrich C, Tiedt S et al (2013) Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog [corrected]. Cell Stem Cell 12:426–439
Sauvageot CM, Stiles CD (2002) Molecular mechanisms controlling cortical gliogenesis. Curr Opin Neurobiol 12:244–249
Hampton DW, Steeves JD, Fawcett JW, Ramer MS (2007) Spinally upregulated noggin suppresses axonal and dendritic plasticity following dorsal rhizotomy. Exp Neurol 204:366–379
Mathieu C, Sii-Felice K, Fouchet P, Etienne O, Haton C, Mabondzo A, Boussin FD, Mouthon MA (2008) Endothelial cell-derived bone morphogenetic proteins control proliferation of neural stem/progenitor cells. Mol Cell Neurosci 38:569–577
Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schübeler D (2008) Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell 30:755–766
Bernstein BE, Meissner A, Lander ES (2007) The mammalian epigenome. Cell 128:669–681
Alvarez-Buylla A, Garcia-Verdugo JM, Tramontin AD (2001) A unified hypothesis on the lineage of neural stem cells. Nat Rev Neurosci 2:287–293
Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A (2001) Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci Off J Soc Neurosci 21:7153–7160
Dimou L, Simon C, Kirchhoff F, Takebayashi H, Götz M (2008) Progeny of Olig2-expressing progenitors in the gray and white matter of the adult mouse cerebral cortex. J Neurosci 28:10434–10442
Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD (2008) PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci 11:1392–1401
Magnusson JP, Göritz C, Tatarishvili J, Dias DO, Smith EM, Lindvall O, Kokaia Z, Frisén J (2014) A latent neurogenic program in astrocytes regulated by Notch signaling in the mouse. Science 346:237–241
Nato G, Caramello A, Trova S, Avataneo V, Rolando C, Taylor V, Buffo A, Peretto P et al (2015) Striatal astrocytes produce neuroblasts in an excitotoxic model of Huntington’s disease. Development 142:840–845
Hochstim C, Deneen B, Lukaszewicz A, Zhou Q, Anderson DJ (2008) Identification of positionally distinct astrocyte subtypes whose identities are specified by a homeodomain code. Cell 133:510–522
Sakurai K, Osumi N (2008) The neurogenesis-controlling factor, Pax6, inhibits proliferation and promotes maturation in murine astrocytes. J Neurosci Off J Soc Neurosci 28:4604–4612
Costa MR, Gotz M, Berninger B (2010) What determines neurogenic competence in glia? Brain Res Rev 63:47–59
Buffo A, Vosko MR, Erturk D, Hamann GF, Jucker M, Rowitch D, Götz M (2005) Expression pattern of the transcription factor Olig2 in response to brain injuries: implications for neuronal repair. Proc Natl Acad Sci U S A 102:18183–18188
Burda JE, Sofroniew MV (2014) Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81:229–248
Bardehle S, Kruger M, Buggenthin F, Schwausch J, Ninkovic J, Clevers H, Snippert HJ, Theis FJ et al (2013) Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci 16:580–586
Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, Borlongan CV (2015) Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat 11:97–106
Cimini A, Ceru MP (2008) Emerging roles of peroxisome proliferator-activated receptors (PPARs) in the regulation of neural stem cells proliferation and differentiation. Stem Cell Rev 4:293–303
Sengupta A, Kalinichenko VV, Yutzey KE (2013) FoxO1 and FoxM1 transcription factors have antagonistic functions in neonatal cardiomyocyte cell-cycle withdrawal and IGF1 gene regulation. Circ Res 112:267–277
Myatt SS, Lam EW (2007) The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer 7:847–859
Renault VM, Rafalski VA, Morgan AA, Salih DA, Brett JO, Webb AE, Villeda SA, Thekkat PU et al (2009) FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell 5:527–539
Webb AE, Pollina EA, Vierbuchen T, Urbán N, Ucar D, Leeman DS, Martynoga B, Sewak M et al (2013) FOXO3 shares common targets with ASCL1 genome-wide and inhibits ASCL1-dependent neurogenesis. Cell Rep 4:477–491
Sriram K, Benkovic SA, Hebert MA, Miller DB, O’Callaghan JP (2004) Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: key signaling pathway for astrogliosis in vivo? J Biol Chem 279:19936–19947
Porlan E, Morante-Redolat JM, Marques-Torrejon MA, Andreu-Agulló C, Carneiro C, Gómez-Ibarlucea E, Soto A, Vidal A et al (2013) Transcriptional repression of Bmp2 by p21(Waf1/Cip1) links quiescence to neural stem cell maintenance. Nat Neurosci 16:1567–1575
Panchision DM, Pickel JM, Studer L, Lee SH, Turner PA, Hazel TG, McKay RD (2001) Sequential actions of BMP receptors control neural precursor cell production and fate. Genes Dev 15:2094–2110
Sahni V, Mukhopadhyay A, Tysseling V, Hebert A, Birch D, Mcguire TL, Stupp SI, Kessler JA (2010) BMPR1a and BMPR1b signaling exert opposing effects on gliosis after spinal cord injury. J Neurosci 30:1839–1855
Orford K, Kharchenko P, Lai W, Dao MC, Woehunsky DJ, Ferro A, Janzen V, Park PJ et al (2008) Differential H3K4 methylation identifies developmentally poised hematopoietic genes. Dev Cell 14:798–809
Boyer LA, Mathur D, Jaenisch R (2006) Molecular control of pluripotency. Curr Opin Genet Dev 16:455–462
Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE et al (2006) Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125:301–313
Hirabayashi Y, Suzki N, Tsuboi M, Endo TA, Toyoda T, Shinga J, Koseki H, Vidal M et al (2009) Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 63:600–613
Acknowledgments
We thank Prof. Paul Heuschling for the laboratory and assistance with Aldh1L1-EGFP mice in Luxembourg as well as Dr. Tony Heurtaux and Annegrät Daujeumont for technical assistance with brain dissections and FACS, respectively.
Compliance with Ethical Standards
This work was supported by the Wellcome Trust, GIS, Fonds National de la Recherche Luxembourg (AFR PDR-09-003).
Conflict of interest
The authors declare that they have no competing interests.
All UK animal handling and procedures were performed according to the UK Animals (Scientific Procedures) Act, 1986 under Home Office licence. All animal procedures in Luxembourg were performed according to FELASA guidelines for the use of animals in research.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Alessandro Michelucci and Angela Bithell contributed equally to this work.
Electronic Supplementary Materials
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 38 kb)
Table S1
(XLSX 13 kb)
Table S2
(XLSX 7,604 kb)
Table S3
(XLS 1,683 kb)
Table S4
(XLSX 1,381 kb)
Table S5
(XLSX 43 kb)
Table S6
(XLSX 586 kb)
Table S7
(XLSX 1,525 kb)
Table S8
(XLS 2,217 kb)
Table S9
(XLSX 797 kb)
Table S10
(XLS 967 kb)
Table S11
(XLSX 12 kb)
Fig. S1
(GIF 148 kb)
Fig. S2
(GIF 34 kb)
Fig. S3
(GIF 206 kb)
Fig. S4
(GIF 152 kb)
Fig. S5
(GIF 48 kb)
Fig. S6
(GIF 44 kb)
Fig. S7
(GIF 53 kb)
Fig. S8
(GIF 287 kb)
Fig. S9
(GIF 37 kb)
Fig. S10
(GIF 104 kb)
Fig. S11
(GIF 64 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Michelucci, A., Bithell, A., Burney, M.J. et al. The Neurogenic Potential of Astrocytes Is Regulated by Inflammatory Signals. Mol Neurobiol 53, 3724–3739 (2016). https://doi.org/10.1007/s12035-015-9296-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9296-x