
Abstract
This work aimed to describe the dynamics of the Sf9 insect cells death and primary metabolism when this host is infected simultaneously by two recombinant baculoviruses (BV) expressing rabies glycoprotein (BVG) and matrix protein (BVM) genes to produce rabies virus-like particles (VLP) at different multiplicities of infection (MOI). Schott flasks essays covering a wide range of MOI for both BV were performed. Viable cell density, cell viability, glucose, glutamine, glutamate, lactate, ammonium, and rabies proteins concentrations were monitored over the infection phase. The expression of both recombinant proteins was not limited by glucose, glutamine, and glutamate in a broad MOI (pfu/cell) range of BVG (0.15–12.5) and BVM (0.1–5.0) using SF900 III serum free culture medium. Death phase initiation and the specific death rate depend on BV MOI. The wave pattern of nutrient/metabolite profiles throughout the viral infection phase is related to the baculovirus lytic cycle. The optimal MOIs ratio between BVG (2.5–4.5) and BVM (1.0–3.0) for maximum protein expression was defined. The produced rabies VLP sizes are close to 78 nm. In general, these work outputs bring a better understanding of the metabolic performance of Sf9 cells when infected by BV for producing VLP, and specifically, for progressing in a rabies VLP vaccine development.
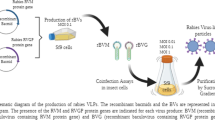
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rabies virus is a negative-sense RNA virus in the Rhabdoviridae family and is also included in the Lyssavirus genus. The virus genome length is around 12 kb, encoding five structural proteins: the nucleoprotein (N), the phosphoprotein (P), the large RNA polymerase protein (L), the matrix protein (M), and the glycoprotein (G). N, P, and L proteins form the nucleocapsid, and the envelope is composed of the M and G proteins [1]. As with other lyssaviruses, the rabies virus causes a fatal disease, affecting the central nervous system, causing severe encephalitis, which shows nervous signs of aggression, paresis, and paralysis, which if not treated with prophylaxis in time, results in death [2]. It is an anthropozoonosis transmitted through the saliva of infected animals by biting, licking open wounds, mucous membranes, and/or scratches. This virus is considered a threat to human health worldwide. Rabies kills close to 59,000 people annually, most of whom are children [3].
If there is no pre- or post-exposure prophylactic treatment with anti-rabies serum, the chance of death is close to 100% [4]. The biotechnological product available to prevent this disease is the vaccine. The licensed human rabies vaccines are primarily based on inactivated purified rabies virus grown in tissue culture or embryonated duck or chicken eggs. This vaccine technology is expensive for poor and developing countries, where the incidence of human rabies is high [5]. Some countries still use nerve tissue-based vaccines, which are less effective and pose a public health risk [6].
The new human rabies vaccines in the pipeline cover the following platforms: protein vaccines; genetically modified rabies virus; genetic, DNA, RNA, and viral vector vaccines; as well as the enhancing of adjuvants [7]. Specifically, the protein vaccine platform has been mainly based on G protein, that assembles as homotrimeric protein on the surface of the virion and most of the antigenic sites are conformational epitopes which rely on the proper quaternary protein structure. Different expressions systems have been utilized for this protein expression, such as mammalian cells, yeast, plants, and insect cells. The latter system shows a promising product (advanced clinical assays), a G protein nanoparticle vaccine generated by CPL Biologicals (India) using the platform Sf9 insect cells/recombinant baculovirus [7]. As a rule, the rabies protein or subunit vaccines have two main limitations, high cost because extensive downstream steps, and low immunogenicity to allow a 1-dose regime.
Therefore, it is essential to have new vaccine proposals aimed at decreasing costs, combined with immunogenicity and good protection. An alternative to improve the low immunogenicity of subunit vaccines is virus-like particles (VLP) platform. VLP are self-assembling complexes of capsid proteins that mimic the overall structure of their parental virus. The absence of genetic material makes these particles non-infectious, enhancing their safety profile when compared to live-attenuated vaccines [8]. Among the advantage of VLP over other particulate subunit vaccines are morphological similarity to their parent virus, a very repetitive immunogenic surface structure, and the retention of cell uptake and immune processing pathways associated with their parent virus. VLP can be produced using a variety of expression systems, such as bacteria, yeast, insect, and mammalian cells, in cell-free expression systems, in live organisms and plants [9]. One of the most employed hosts for enveloped VLP (much more complex in composition than non-enveloped VLP) production are the insect cells based on recombinant baculovirus. The scale up of this protein production system is feasible [10]. A virus-like particle has the appeal of being cheaper and having good biosecurity, both for those who manufacture it and for those who receive it. It is a vaccine preparation that does not use the native virus, not requiring a level 3 containment laboratory, reducing costs and risks. Currently, one of the main hurdles obstructing the vaccine industry's progression is the development of downstream processing technologies for VLP vaccine production. Therefore, several solutions have been explored to overcome this issue like aqueous two-phase systems and chromatographic techniques [11].
There are two expression strategies to produce protein complex in baculovirus/insect cell (B/IC) expression system: co-expression using single polycistronic baculovirus, and co-infection, using multiple monocistronic baculoviruses. Some of the important parameters for this VLP platform are the time of infection (implying a certain viable cell concentration), the multiplicity of infection (MOI, virus/cell), and harvest time [12]. Besides, the knowledge of insect cell metabolism is necessary for the rational design of cell culture medium, operation mode choice, and feed plan to produce VLP in B/IC expression system [13].
Thus, this work aimed to describe the Sf9 insect cell death and primary metabolism when infected by two monocistronic baculoviruses to produce rabies virus-like particles at different MOI of baculoviruses holding rabies G and M protein. In general, this study focused on understanding these upstream stage aspects, little explored in the literature, which are essential to suggest culture medium or cells feed strategies to produce recombinant proteins and VLP using the B/IC expression system; and specifically, to move forward in the development of rabies vaccine candidate based on virus-like particles.
Materials and Methods
Cell lines and Culture Media
The Sf9 cells used for virus inoculum, cell growth without infection and co-infection assays were kindly provided by Professor Dr. Paolo Marinho de Andrade Zanotto (Laboratory of Molecular Evolution and Bioinformatics, Department of Microbiology, Biomedical Sciences Institute, University of São Paulo) [14]. Besides, Sf9 Easy Titer (ET) cells were utilized for viral titration assays, donated by Professors Dr. Ralph Hopkins and Dr. Dominic Esposito, from the Protein Expression Laboratory of the National Cancer Institute of Frederick, Maryland, USA. Both cell lines were cultivated in SF900 III serum free medium (Thermo Fisher Scientific, Massachusetts, USA), though this medium for Sf9 ET cells was supplemented with 2.5% fetal bovine serum (FBS, HyClone®, Cytiva, USA). For all assays, including virus inoculum preparation, as well as host growth and co-infection runs for metabolism studies, frozen cells stored in liquid nitrogen were thawed and subcultured for three passages.
Viral Stock and Conservation
The recombinant baculoviruses bearing M and G proteins, named BVM and BVG, respectively, were generated by transfecting cloned bacmids with the genes encoding both rabies virus proteins into Sf9 cells, as previously described [15]. The production of viral stocks containing BVM and BVG were done through three infection passages, with a multiplicity of infection (MOI) of 0.1 at a viable cell concentration of about 1 × 106 cells mL−1. The supernatant was collected 96 h after infection and centrifuged at 500×g for 5 min. The clarified supernatant was then stored at 4 °C in a dark room.
Schott Flasks Assays
Five experimental blocks using three MOIs ratio (1.5; 2.0 and 2.5) between BVG and BVM were executed comprising BVM MOI from 0.1 to 5.0 (Table 1). A control assay was also done (cell growth without viral infection). All assays were carried out in triplicate.
Schott flask assays were performed in flasks with a total volume of 100 mL with 20 mL working volume. Cell cultures were maintained at 28 °C and 100 rpm in a rotating shaker incubator (Innova 4000, New Brunswick Scientific, Edson, NJ, USA). The initial concentration of the cells of the growth without infection and co-infection assays was around 1 × 106 cells mL−1. Daily samples were taken to determine cell concentration (Xv) and viability (CV), for quantifying nutrient and metabolite concentrations, virus titer and protein expression. Virus titers for experiments using 0.1 and 0.2 BVM MOIs was 1.6 × 107 and 1.5 × 106 pfu/mL, for BVG and BVM, respectively, while for experiments with 1.0 BVM MOI, the BVG and BVM titers were 6.7 × 107 pfu/mL and 6.9 × 107 pfu/mL, respectively; and assays with 3.0 and 5.0 BVM, the BVG and BVM titers were for both 9.0 × 108 pfu/mL.
Cell Density and Viability
The cell density and viability were determined by the trypan blue exclusion method (0.04% v/v) in a Neubauer-bright-line hemocytometer considering 8 quadrants. As a rule, cell samples were diluted tenfold in phosphate-buffered saline (PBS 1 ×).
Viral Titration
The titration assays of viral batches were performed according to the method described by Hopkins and Esposito [16]. This method is based on the activation of the eGFP gene, previously transfected into Sf9 ET cells, which fluoresces when infected. Briefly, cells were seeded in a 96-well plate at a concentration of 7.5 × 104 cells per well and kept for 1 h at 28 °C for sedimentation and adhesion. After that, the viral lot to be titrated was serially diluted to a 10–8 dilution. Diluted viruses were applied in octuplicate to columns 2 to 11. Column 12 was used as a negative control. The plate was incubated for 96 h at 28 °C. After this period, the cells were analyzed under an inverted fluorescence microscope to find fluorescence foci.
Dot Blotting
One hundred microliters samples were added to a nitrocellulose membrane (GE HealthCare Life Science) and incubated in blocking solution (non-fat milk powder 3% w/v) for 1 h. Subsequently, the membranes were incubated with the matching primary antibody; mouse anti-rabies glycoprotein antibody (LifeSpan BioSciences, LSBio – C75309, Seattle, USA) at 1:4000 dilution and antibody made in rabbit anti-rabies matrix protein (Cusabio Biotech, CSB-PA322192LA01RAI, Texas, USA) at 1:2000 dilution for G and M protein, respectively. Afterward, the membrane was incubated in secondary antibody conjugated with horseradish peroxidase (HRP) for 1 h; HRP-conjugated anti-mouse antibody (Thermo Fisher Scientific, G21040) and HRP-conjugated anti-rabbit antibody (Thermo Fisher Scientific, 656120), both at 1:5000 dilution for G and M protein, respectively. In the period separating both incubations, the membrane was washed 3-fold with PBS 1 × solution + 0.05% (v/v) Tween, incubating for 10 min between each wash. Finally, the membrane was revealed in an Alliance 2.7 photo documenter (Uvitec, Cambridge, UK) by means of a chemiluminescent reaction (ECL) using the PicoWest SuperSignal Chemiluminescent Substrate kit (Thermo Fisher Scientific).
Enzyme-Linked Immunosorbent Assay (ELISA) for Rabies G Protein
Rabies G protein quantification by ELISA was carried out using the Rabies Glycoprotein Enzyme Immunoassay kit from Pasteur Institute (Paris, France) and described in detail by Astray et al. [17]. One plate was sensitized with the primary antibody Mab-D1 (specific antibody for RVGP site III), diluted 1:2000 in carbonate buffer, incubated for 2 h at 37 °C and then incubated at 4 °C for 16 h. The samples and the standard curve (inactivated and lyophilized rabies virus) were added to the plate and incubated for 1 h at 37 °C. After incubation, the secondary antibody Mab-D1 conjugated with peroxidase was added to the plate, at a dilution of 1:2000, and the plate was incubated for another 1 h at 37 ºC. After this time, the plate was incubated with chromogenic substrate for 30 min. After this time, the reaction was stopped for further reading.
Transmission Electron Microscopy (TEM)
Coinfection samples holding rabies VLP were concentrated at 150,000×g and 4 ºC for ninety minutes by ultracentrifugation. The pellet formed was resuspended in PBS 1× and again ultracentrifuged in a 20% sucrose cushion (w/v) at 230,000×g and 4 ºC for 2 h. Afterward, the pellet was again resuspended in PBS 1× and applied to copper grids pre-coated with parlodium. The grids were added 1% aqueous uranyl acetate solution and they were observed in a transmission electron microscope (TEM) LEO 906E (Zeiss, Germany).
To perform immunostaining assays, primary antibodies (anti-G protein and anti-M protein) and secondary antibodies conjugated with colloidal gold (anti-mouse and anti-rabbit) were also added. Afterwards, samples were contrasted with uranyl acetate and observed in a TEM.
Nutrient and Metabolite Quantification
Glutamine (gln), glutamate (glu), glucose (gluc) and lactate (lac) concentrations were enzymatically quantified using a biochemical analyzer (Model 2900 Select Biochemistry Analyzer, YSI Inc., Yellow Springs, Ohio, USA). The ammonium (\({\text{NH}}_{4}^{+}\)) concentration was determined using the same device employing a proper Ion-Selective Electrode. For this purpose, clarified cell-free samples were placed in 96-well plates and read on the analyzer.
Biochemical Parameters Determination
The maximum specific growth rate (\({\upmu }_{\text{max}}, {\text{h}}^{-1}\)) was determined from linear regression of the logarithmic increase in viable cell density (\({X}_{\text{v}}, \frac{\text{cells}}{{\text{L}}}\)) with respect to time over the exponential growth phase. Besides, the specific death rate (\({k}_{\text{d}}, {\text{h}}^{-1}\)) throughout the infection phase was determined by Eq. 1. The term \(\frac{{\text{d}}{X}_{\text{v}}}{{\text{d}}t}\) (linear death rate) was determined as the slope of linear regression of \({X}_{\text{v}}\) vs time during the infection phase.
The uptake and release specific rates for substrates (\({q}_{\text{s}}\)) (glucose, glutamine, glutamate) and metabolites (\({q}_{\text{m}}\)) (lactate, ammonium) were quantified according to Eq. 2 and 3, respectively.
where S and M are the substrate and metabolite concentration (mmol/L), respectively. During the exponential growth phase, a plot of the S or M versus \({X}_{\mathrm{v}}\), yields a slope of \(\frac{{q}_{\mathrm{S}}}{{\upmu }_{\mathrm{max}}}\) or \(\frac{{q}_{\mathrm{m}}}{{\upmu }_{\mathrm{max}}}\). On the other hand, the post infection \({q}_{\mathrm{s}}\) and \({q}_{\mathrm{m}}\) were calculated in the middle (\({q}_{\mathrm{s}}\left(\overline{t }\right), {q}_{\mathrm{m}}\left(\overline{t }\right)\)) of two consecutive sampling points by Eq. 4 and 5.
where \(\overline{t }\) is the average of the two consecutive sampling times (\({t}_{n-1}, {t}_{n}\)); \({S}_{n}, {M}_{n}\) are the concentrations of substrate and metabolite at \({t}_{n}\), and \({S}_{n-1}, {M}_{n-1}\) are equivalent for \({t}_{n-1}\), \({\overline{\mathrm{X}} }_{\mathrm{v}}\) is the viable cell density average between \({t}_{n-1}, {t}_{n}\). For both culture systems, \({t}_{n-1}-{t}_{n}\) was kept close to 24 h.
Conversion yields (\(Y\)) in the exponential growth phase were calculated as a ratio between specific rates (\({{q}_{\mathrm{s}}, {q}_{\mathrm{M}},\upmu }_{\mathrm{max}}\)). Besides, protein expression-substrate yields were determined as the concentration variation ratio between protein and substrate.
Data Analysis
Raw data were arranged and stored in Excel spreadsheets, all calculations were performed in the same software (Office 365, Microsoft Corporation, Redmond, WA, USA). The graphs were plotted with Origin 2020 software (OriginLab Corporation, Northampton, MA, USA). Factorial ANOVA for determination of optimal conditions of multiplicity of infection and harvest time to ensure maximum protein expression as well as basic statistical analysis to assess VLP size were performed in Statistica version 14 (TIBCO software Inc, Palo Alto, CA, USA). All statistical decisions were taken with 95% confidence level (α = 0.05).
Results and Discussion
Sf9 cells are commonly used to produce recombinant proteins by transient expression due to this host achieves high protein yields as well as bears greater passaging stability than other insect cell lines. As is well known, the culture medium has a significant impact on protein output efficiency. However, the effect of modern insect cell culture medium on Sf9 cells has been scarcely assessed for different expression systems, specifically for producing VLP from baculovirus expression vector system (BEVS) [18]. This work aimed to understand the Sf9 cells' death and primary metabolism when using BEVS to make rabies VLP with SF900 III serum free culture medium in Schott flasks. The virus infections were carried out in wide range of multiplicities of infection of BVG and BVM.
Cell Growth and Metabolism Before Coinfection
As a reference, Sf9 cells were cultured in SF900 III without recombinant baculovirus infection. The cells entered the exponential from the inoculation at 1 × 106 cell/mL viable cell density. The maximum viable cell density (\({X}_{\mathrm{v}},\) 9.83 ± 3.48 × 106 cell/mL) was reached at 120 h and \({\upmu }_{\mathrm{max}}\) defines 31.7 ± 2.4 h doubling time (\({t}_{\mathrm{d}}=\frac{\mathrm{ln}2}{{\upmu }_{\mathrm{max}}}\)) (Supplementary material Fig. 1A; Table 2). The maximum \({X}_{\mathrm{v}}\) and \({t}_{\mathrm{d}}\) were comparable to the usual values for these parameters for Sf9 cells cultured in batch mode using SF900 III serum free medium (between 6–11 × 106 cell mL−1 and 24 h, respectively) [19].
Major nutrients (glucose, glutamine, glutamate, all used for energy production) and metabolites (lactate, ammonium) were monitored, as well as their yields and specific rates were also determined over the exponential growth phase (Table 2). The initial glucose, glutamine and glutamate masses were consumed 51, 86 and 57%, respectively (Supplementary material Fig. 1B, D, and E). In the case of glutamine, the spontaneous decomposition has been reported as a significant event (almost 30% in culture medium supplemented with fetal bovine serum), thus, part of its 86% depletion could not be utilized by the cells [20]. The \({q}_{\mathrm{gluc}}\) and \({q}_{\mathrm{gln}}\) confirmed in this work (Table 2) for SF900 III were within the same order as those reported for SF900 II during the growth phase, 8.64 × 10–11 and 3.96 × 10–11 mmol cell−1 h−1, respectively [21]. The difference between the current \({\mathrm{q}}_{\mathrm{gluc}}\) and \({\mathrm{q}}_{\mathrm{gln}}\) could be caused obviously because they are different culture media, but also by the absence of dissolved oxygen control in our culture system (Schott flasks).
The level of ammonium concentration throughout Sf9 growth was oscillating in narrow range (0.03–0.07 g/L), whereas lactate increase up to 0.2 g/L from initial concentration of 0.02 g/L (Supplementary material Fig. 1C and F). Nevertheless, the low \({Y}_{\frac{\mathrm{lac}}{\mathrm{gluc}}}\) value (0.08) (Table 2) confirmed the fully functional TCA cycle. The lactate production in Sf9 cells is only verified under conditions of anoxic stress. Besides, ammonia is not accumulated by Sf9 cells during growth if glucose is not a limiting substrate [21].
Cell Death and Metabolism After Coinfection
The Sf9 infection within a broad range of MOI of both baculovirus bearing genetic code for rabies virus proteins featured a linear death phase (zero-order death phase) with or without delay time respect to the infection moment. As a rule, the linear death phase began later at low MOI (Supplementary material Fig. 2–6A; Fig. 1A) and the linear death velocity (\(\frac{\mathrm{d}{X}_{\mathrm{v}}}{\mathrm{d}t}\)) was within − 6700–13500 cell/mL/h, except to BVM MOI 5, in which \(\frac{\mathrm{d}{X}_{\mathrm{v}}}{\mathrm{d}t}= - 15300-16300\) cell/mL/h (Supplementary Fig. 6A). The \({k}_{\mathrm{d}}\) increased over the infection in all assessed conditions, as time progressed, showing an exponential trend. This pattern has been reported and is triggered by the boost of virions titer [22]. High \({k}_{\mathrm{d}}\) values were confirmed at high values of MOI. Highest \({k}_{\mathrm{d}}\) values were close to 0.05 h−1 (Fig. 2). These \({k}_{\mathrm{d}}\) values were higher than those reported (0.007–0.029 h−1) for a culture medium supplemented by fetal bovine serum and silkworm hemolymph [23] and closer to other recombinant baculovirus using Sf9 cell host and SF900 II serum free culture medium (0.08 ± 0.01 h−1) [24].

Viable cell density monitoring over the cell death phase. A Linear death rate after beginning of the cell death caused by virus infection. B Initiation of cell death after virus infection

Specific death rate (\({\mathrm{k}}_{\mathrm{d}}, {\mathrm{h}}^{-1}\)) throughout the infection phase until 96 h at average BVG and BVM MOI ratio in each experimental block. A \({\mathrm{k}}_{\mathrm{d}}\) monitoring at relative low MOI values. B \({\mathrm{k}}_{\mathrm{d}}\) monitoring at relative high MOI values
Cells not being limited by glucose, glutamine and glutamate depletion were confirmed for protein expression with BVG-BVM MOI ratios of 0.15–0.1 to 12.5–5. Wave pattern was observed for these substrates over the infection phase (Supplementary material Fig. 2–6). For glucose and glutamate, their wave profiles were more pronounced (wave amplitude) since low MOI infections, whereas for glutamine profiles even within a wave pattern a slight decrease in its wave amplitude at low MOI was observed. However, the glutamine profiles are fairly ondulatory at the highest MOIs (5.0 BVM MOI) (Supplementary material Fig. 6E). The minimum (negative) and maximum (positive) numerical values of specific rates for these substrates reinforce these patterns (Table 3). The glucose specific rate profile over the infection revealed the higher amplitude wave (Table 3, Fig. 3). These patterns must be a consequence of the baculovirus lytic cycle (Supplementary material Fig. 2–6). The total cell density profiles support these nutrients and metabolites concentration oscillations (Fig. 3D–F). Up to date, the published works described a decrease in substrate specific consumption rates (71% and 45% decrease for glucose and glutamine, respectively) over the infection phase with respect to the growth phase [21], but detailed studies over the infection to describe the substrate specific rate profiles were not performed. Likely, this is the main contribution of this work. Thus, a nutrient feed strategy related to the primary metabolism is not necessary for improving VLP yield and productivity.

Specific nutrient and metabolite rates over the infection phase in Schott flasks at best conditions for maximum glycoprotein concentration by ELISA (Table 5) and corresponding total cell density profiles. A Specific rates for infection with 2.5 MOI of BVG and 1.0 MOI of BVM. B Specific rates for infection with 4.50 MOI of BVG and 3.0 MOI of BVM. C Specific rates for infection with 7.50 MOI of BVG and 5.0 MOI of BVM. Error bars represent standard deviation of three repetitions for experiments performed. D Total density (Xt) profile corresponding to conditions represented in A. E Xt profile corresponding to conditions represented in B. F Xt profile corresponding to conditions represented in C
The lactate was slightly accumulated at low MOIs during the infection phase, but also showed a wave pattern at high MOIs (Supplementary material Fig. 2–6D). Nevertheless, the wave pattern for ammonium or no accumulation was also confirmed over the entire assessed range of MOIs (Supplementary material Fig. 2–6G). Therefore, the inhibition by lactate and ammonium is not a significant event for VLP production using BV/IC system.
Protein Expression, VLP Production and Characterization
The rabies glycoprotein (monomer and trimer detection) and M protein expression quantified by Dot-blotting showed typical profiles for BEVS over the MOIs ratio considered in this work [25]. The optimal combination of MOIs ratio and harvest time for each experimental block (Table 1) to maximize Rabies glycoprotein expression by Dot-blotting (relative low-cost assay) was defined by factorial ANOVA, as a screening approach. Using this method, increasing levels of Rabies glycoprotein in supernatant was found until BVM MOI equal to 3.0 (1081 µg/L), when quantified by Dot-blotting, the 5.0 BVM MOI decreased the protein expression defined by this immunochemical technique. The Rabies M protein expression by Dot-blotting has a similar performance. However, the Rabies glycoprotein concentration by ELISA was featured by high expression (1014–1135 µg/L) at lowest and highest MOIs (Table 4). The optimal MOI depends on the cell line, baculovirus, culture medium, the operation mode, and the physiological state of the cells [26]. Besides, the cost of baculovirus as supply must be considered. Thus, the MOI optimization must be performed case to case. In our system BVG and BVM MOI ratios within 2.5:1 to 4.5:3 seem to be suitable. This finding is significant for progressing on the rabies VLP vaccine candidate development using scaling-up devices like stirred-tank bioreactors.
Another expression optimization was carried out focusing on maximization of glycoprotein production quantified by ELISA at relative high MOI experimental blocks was carried out. This optimization showed similar values for Rabies glycoprotein yields from 1.0 to 5.0 BVM MOIs (Table 1) by ELISA and M protein by Dot-blotting. Highest values for Rabies glycoprotein expression were within BVM MOI 1–3 range (Table 5). The protein expression yields were in harmony with others in advanced status of development (e.g., 1 mg/L Ebola VLP yield) [27]. The optimization of the MOI is significant for further bioprocess development, both technically and economically points of view.
The rabies glycoprotein-substrates yields were determined, even no effective substrate consumption was observed at high MOIs. The substrate variation was defined as the difference between substrate or initial or maximum concentration and final or minimal concentration over the virus infection/expression phase (Fig. 4). An increasing trend with increasing MOIs for this parameter was observed for glucose and glutamine.

Protein expression-substrate yields (YGPV/S) for optimal conditions using maximum G-glycoprotein concentration by ELISA as criterium at different high MOI experimental blocks (3–5). HT harvest time and MOI multiplicity of infection
Thus, a VLP size characterization was performed at BVG and BVM MOI ratio of 3:2 with 96 h harvest time. Rabies VLP carry on both rabies proteins according to immunostaining in transmission electron microscopy. Besides, the average particle size was 78 ± 17 nm (Fig. 5). The particles size is lower than that (128 ± 36 nm) determined for the same system using low MOI of both recombinant rabies baculovirus [15]. This VLP size pattern could be explained by the higher MOI after secondary infections, at low MOI, than those occurred at initial high MOI. However, the VLP heterogeneity could be affected at initial low MOI (more experiment should be performed to demonstrate this hypothesis). Nevertheless, this work's obtained rabies VLP size is inside the typical VLP size range [27].

Transmission electron microscopy micrographs and derived statistical analysis for VLP produced at BVG and BVM MOI ratio of 3:2 and 96 h harvest time. A Immunostaining with primary antibody anti-glycoprotein. Rabies VLP size: 103 nm. B Immunostaining with primary antibody anti-M protein. Rabies VLP size: 68 nm. C Frequency histogram for VLP size (n = 8), average size and standard deviation equal to 78 ± 17 nm and 50–103 nm particle size range
Conclusions
Considering the relevance that rabies still has, being considered a neglected tropical disease with high prevalence in poor and developing countries, that lack an effective and less expensive vaccine, this work aimed to study the primary metabolism of Sf9 cells coinfected with two recombinant baculoviruses carrying rabies glycoprotein (G) and matrix protein (M) at various MOI levels, aiming to produce virus-like particles (VLP) of this virus, as a potential vaccine candidate against this disease.
For the production of heterologous proteins it is essential to better understand the cells, their performance and metabolism, to increase production aiming at industrial/commercial scales. Therefore, the primary metabolism of Sf9 cells was analyzed for the production of rabies virus-like particles. The rabies protein expression using baculovirus expression vector system and Sf9 cells as a host was not limited by glucose, glutamine, and glutamate in a broad multiplicity of infection range of monocistronic baculovirus bearing gene of rabies glycoprotein (MOI 0.15–12.5) and M protein (MOI 0.1–5.0) using SF900 III serum free culture medium, thus, the nutritional need of cells was met in the parameters analyzed. The initiation of linear death phase after virus infection, and the associated specific death rate depend on BV MOI. The wave pattern of nutrient profile throughout the virus infection/protein expression phase is related to baculovirus lytic cycle. The optimization of rabies protein expression by this system suggests a MOIs ratio for BVG and BVM within 2.5–4.5 and 1.0–3.0 ranges, respectively. The produced rabies VLP sizes are close to 78 nm. These work outputs could be relevant as a numeric characterization of baculovirus infection death and primary metabolism using Sf9 insect cells and SF900III culture medium in virus-like particles production.
References
Yin, J., Wang, X., Mao, R., Zhang, Z., Gao, X., Luo, Y., & Yin, X. (2021). Research advances on the interactions between rabies virus structural proteins and host target cells: Accrued knowledge from the application of reverse genetics systems. Viruses, 13(11), 2288. https://doi.org/10.3390/v13112288
Yost, M. D., & Toledano, M. (2020). Severe infections of the central nervous system. In Neurological emergencies (pp. 121–149). Cham: Springer International Publishing. https://doi.org/10.1007/978-3-030-28072-7_7
Gold, S., Donnelly, C. A., Nouvellet, P., & Woodroffe, R. (2020). Rabies virus-neutralising antibodies in healthy, unvaccinated individuals: What do they mean for rabies epidemiology? PLOS Neglected Tropical Diseases, 14(2), e0007933. https://doi.org/10.1371/journal.pntd.0007933
Nadeem, M., & Panda, P. (2020). Survival in human rabies but left against medical advice and death followed: Community education is the need of the hour. Journal of Family Medicine and Primary Care, 9(3), 1736. https://doi.org/10.4103/jfmpc.jfmpc_1079_19
Ertl, H. C. J. (2019). New rabies vaccines for use in humans. Vaccines, 7(2), 54. https://doi.org/10.3390/vaccines7020054
Sreenivasan, N., Li, A., Shiferaw, M., Tran, C. H., Wallace, R., Blanton, J., Knopf, L., Abela-Ridder, B., Hyde, T., Siddiqi, U. R., Tahmina, S., Penjor, K., Sovann, L., Doeurn, Y., Sim, K., Houssiere, V., Tejiokem, M., Mindekem, R., Yu, L., … Nguyen, H. (2019). Overview of rabies post-exposure prophylaxis access, procurement and distribution in selected countries in Asia and Africa, 217–218. Vaccine, 37, A6–A13. https://doi.org/10.1016/j.vaccine.2019.04.024
Fooks, A. R., Banyard, A. C., & Ertl, H. C. J. (2019). New human rabies vaccines in the pipeline. Vaccine, 37, A140–A145. https://doi.org/10.1016/j.vaccine.2018.08.039
Charlton Hume, H. K., Vidigal, J., Carrondo, M. J. T., Middelberg, A. P. J., Roldão, A., & Lua, L. H. L. (2019). Synthetic biology for bioengineering virus-like particle vaccines. Biotechnology and Bioengineering, 116(4), 919–935. https://doi.org/10.1002/bit.26890
Donaldson, B., Lateef, Z., Walker, G. F., Young, S. L., & Ward, V. K. (2018). Virus-like particle vaccines: Immunology and formulation for clinical translation. Expert Review of Vaccines, 17(9), 833–849. https://doi.org/10.1080/14760584.2018.1516552
Deng, F. (2018). Advances and challenges in enveloped virus-like particle (VLP)-based vaccines. Journal of Immunological Sciences, 2(2), 36–41.
Leong, H. Y., Fu, X., Show, P. L., Yao, S., & Lin, D. (2022). Downstream processing of virus-like particles with aqueous two-phase systems: Applications and challenges. Journal of Separation Science, 45(12), 2064–2076. https://doi.org/10.1002/jssc.202100947
Sokolenko, S., George, S., Wagner, A., Tuladhar, A., Andrich, J. M. S., & Aucoin, M. G. (2012). Co-expression vs. co-infection using baculovirus expression vectors in insect cell culture: Benefits and drawbacks. Biotechnology Advances, 30(3), 766–781. https://doi.org/10.1016/j.biotechadv.2012.01.009
Cox, M. M. J. (2021). Innovations in the insect cell expression system for industrial recombinant vaccine antigen production. Vaccines, 9(12), 1504. https://doi.org/10.3390/vaccines9121504
Ventini-Monteiro, D., Dubois, S., Astray, R. M., Castillo, J., & Pereira, C. A. (2015). Insect cell entrapment, growth and recovering using a single-use fixed-bed bioreactor. Scaling up and recombinant protein production. Journal of Biotechnology, 216, 110–115. https://doi.org/10.1016/j.jbiotec.2015.10.013
Bernardino, T. C., Astray, R. M., Pereira, C. A., Boldorini, V. L., Antoniazzi, M. M., Jared, S. G. S., Núñez, E. G. F., & Jorge, S. A. C. (2021). Production of rabies VLPs in insect cells by two monocistronic baculoviruses approach. Molecular Biotechnology. https://doi.org/10.1007/s12033-021-00366-z
Hopkins, R. F., & Esposito, D. (2009). A rapid method for titrating baculovirus stocks using the Sf-9 Easy Titer cell line. BioTechniques, 47(3), 785–788. https://doi.org/10.2144/000113238
Astray, R. M., Augusto, E., Yokomizo, A. Y., & Pereira, C. A. (2008). Analytical approach for the extraction of recombinant membrane viral glycoprotein from stably transfected Drosophila melanogaster cells. Biotechnology Journal, 3(1), 98–103. https://doi.org/10.1002/biot.200700179
Käßer, L., Harnischfeger, J., Salzig, D., & Czermak, P. (2022). The effect of different insect cell culture media on the efficiency of protein production by Spodoptera frugiperda cells. Electronic Journal of Biotechnology, 56, 54–64. https://doi.org/10.1016/j.ejbt.2022.01.004
Chan, L. C. L., & Reid, S. (2016). Development of serum-free media for lepidopteran insect cell lines. In D. W. Murhammer (Ed.), Baculovirus and insect cell expression protocols, methods in molecular biology (3rd edn, Vol. 388, pp. 161–196). Humana Press. https://doi.org/10.1007/978-1-4939-3043-2_8
Palomares, L. A., & Ramirez, O. T. (1996). The effect of dissolved oxygen tension and the utility of oxygen uptake rate in insect cell culture. Cytotechnology, 22(1–3), 225–237. https://doi.org/10.1007/BF00353943
Ikonomou, L., Schneider, Y.-J., & Agathos, S. N. (2003). Insect cell culture for industrial production of recombinant proteins. Applied Microbiology and Biotechnology, 62(1), 1–20. https://doi.org/10.1007/s00253-003-1223-9
Saxena, A., Byram, P. K., Singh, S. K., Chakraborty, J., Murhammer, D., & Giri, L. (2018). A structured review of baculovirus infection process: Integration of mathematical models and biomolecular information on cell–virus interaction. Journal of General Virology, 99(9), 1151–1171. https://doi.org/10.1099/jgv.0.001108
Rhee, W. J., Kim, E. J., & Park, T. H. (1999). Kinetic effect of silkworm hemolymph on the delayed host cell death in an insect cell-baculovirus system. Biotechnology Progress, 15(6), 1028–1032. https://doi.org/10.1021/bp990093s
Power, J. F., Reid, S., Radford, K. M., Greenfield, P. F., & Nielsen, L. K. (1994). Modeling and optimization of the baculovirus expression vector system in batch suspension culture. Biotechnology and Bioengineering, 44(6), 710–719. https://doi.org/10.1002/bit.260440607
Jang, J. D., Sanderson, C. S., Chan, L. C. L., Barford, J. P., & Reid, S. (2000). Structured modeling of recombinant protein production in batch and fed-batch culture of baculovirus-infected insect cells. Cytotechnology, 34, 71–82. https://doi.org/10.1023/A:1008178029138
Contreras-Gómez, A., Sánchez-Mirón, A., García-Camacho, F., Molina-Grima, E., & Chisti, Y. (2014). Protein production using the baculovirus-insect cell expression system. Biotechnology Progress, 30(1), 1–18. https://doi.org/10.1002/btpr.1842
Vicente, T., Roldão, A., Peixoto, C., Carrondo, M. J. T., & Alves, P. M. (2011). Large-scale production and purification of VLP-based vaccines. Journal of Invertebrate Pathology, 107, S42–S48. https://doi.org/10.1016/j.jip.2011.05.004
Acknowledgements
This work was financially supported by the National Council of Technological and Scientific Development (CNPq) (Grants Nos. 2018-7/168539 and 401346/2020-0), the São Paulo Research Foundation (FAPESP) (Grants Nos. 2016/22780-6, 2018/10538-1) and Butantan Foundation. The research team is grateful to the Laboratory of Viral Vaccines of the Butantan Institute for its support in the biochemical quantifications.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Leme, J., Guardalini, L.G.O., Bernardino, T.C. et al. Sf9 Cells Metabolism and Viability When Coinfected with Two Monocistronic Baculoviruses to Produce Rabies Virus-like Particles. Mol Biotechnol 65, 970–982 (2023). https://doi.org/10.1007/s12033-022-00586-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-022-00586-x