
Abstract
To review post-mortem findings among deaths presenting as sudden and/or unexpected deaths in two centers in the UK during a 16-year period in order to identify those related to cardiovascular conditions. The post-mortem databases of two tertiary referral institutions were searched, and all reports were reviewed. Histological features and results of ancillary investigations were noted. All cases of sudden and/or unexpected cardiac deaths (SCD) between 2003 and 2018 were identified. The study was PRISMA compliant and approved by clinical governance. 68/1129 cases of SCD (6.0%) were identified in one center and 83/753 cases (11%) in the other. These 151 cases constituted the study cohort. The mean annual incidence of SCD was 0.3 per 100,000 persons/annum. The three most prevalent groups of cardiac pathology were cardiac malformations (51/151; 33.8%), cardiomyopathies (32/151; 21.2%), and myocarditis (31/151; 20.5%). Mean age at death was 3.4 years. Prematurity was predominantly associated with deaths related to cardiac malformations (p < 0.001). Symptoms had been present for a mean of 3.8, 3.0, and 3.5 days before death for myocarditis, cardiomyopathy, and cardiac malformations/complications post-surgery. This retrospective comparative study represents the largest autopsy series of SCD in infants and children in the UK. Some entities are very infrequent. Several diseases could have been identified earlier in life allowing for the possibility of intervention. Limitation includes the retrospective nature of the study and that, as arrhythmogenic gene mutations are not yet routinely performed in unexplained deaths, the incidence of SCD in infants and children is most likely underestimated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Each year, around 47,000 people under the age of 75 in the UK die from heart and circulatory diseases [1]. Sudden cardiac death (SCD) describes an unexpected death from a cardiac cause, in a person without any prior cardiac conditions [2]. The sudden death of a child or infant represents a devastating event for family members, often associated with significant psychological trauma. SCD constitutes a significant proportion of sudden deaths in infancy and childhood and must be identified, where possible, before the cause of death is labeled “undetermined,” “SIDS,” “sudden unexpected death in infancy (SUDI),” or “sudden unexpected death in childhood (SUDC).” Varying incidence rates of SCD in the young have been reported, mainly in developed countries, with the rates ranging from as few as 0.7 deaths per 100,000 population in Sweden [3], 1.8 per 100,000 in the UK [4], to as high as 10.1 per 100,000 in the USA [5]. A recent systematic review identified significant variation in the reporting of SCD in the young across different studies, with most studies reporting an incidence rate of 1–2 cases per 100,000 person years [6]. Because SCD is relatively rare, little is known about its pathogenesis and prevention.
Known causes of SCD in the young include channelopathies, cardiomyopathies, and myocarditis [5]. Cardiovascular pathology is likely to be underreported due to the challenging nature of its diagnosis post-mortem, especially in cases where there is no obvious morphological abnormality and where there is no molecular testing post-mortem. There is therefore a need for better identification of SCD cases and autopsy methods to characterize SCD. By understanding clinicopathological relations in SCD, further work can be done to prevent it.
In this study, we analyze the overall incidence of sudden cardiac death in infants and children in two referral institutions in the UK and delineate presentations of the most prevalent cardiovascular entities seen. We additionally consider cases of unexpected postoperative cardiac death in the context of congenital heart disease. For the purpose of the study, both groups were clustered as sudden and/or unexpected cardiovascular deaths (SUCVD).
Methods
The study period encompassed between 1 January 2003 to 31 December 2018, and all post-mortem (PM) coronial files in the electronic databases of Sheffield Children NHS Foundation Trust (SCH) and Barts Health NHS Trust (BHT) of children up to 17 years of age were scrutinized. Case demographics were collated including age, sex, ethnicity, and the circumstances surrounding each death. Clinical details such as prodromal symptoms, medical or surgical interventions, and records of familial pathology were recorded. Narrative descriptions of post-mortem findings were taken from the PM reports, including macroscopic and histologic features, and results of relevant ancillary investigations (for example, histochemistry, metabolic, molecular, and microbiological assays) were noted, permitting clinicopathological correlation. The three most prevalent groups of cardiac pathology seen in our institutions (cardiac malformations, myocarditis, and cardiomyopathies) represented 80% of the study population and were statistically compared, while other pathology was narratively described.
The Kruskal–Wallis test was used to identify statistically significant differences between mean ages at death and symptom duration of the three most prevalent groups of pathology seen in the two centers: myocarditis, cardiomyopathies, and cardiac malformations. Analysis of categorical variables including ethnicity, gender distribution, and prematurity was performed using a Chi-square test. Statistical significance was set at p = 0.05. GraphPad Prism and StatsDirect were used in statistical analysis and figure creation. STROBE guidelines used in cohort studies were followed.
This study was registered as a service evaluation with Clinical Governance (SE1295) and was PRISMA compliant.
Results
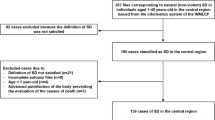
The retrospective scoping search of all cases of SUDI and SUDC from the pathology database of the two participating centers identified 1882 coronial pediatric post-mortems during the study period. In total, 68/1129 cases (6%) of SUCVD were identified at SCH, and 83/753 (11%) were seen at BHT. These 151 cases constituted the study group (Fig. 1). A breakdown of cases and pathological entities encountered over the 16-year period is included under broad sub-headings (Table 1). Gender distribution revealed a slight male predominance representing 55% of all cases. Mean age at death for all causes was 3.4 years for males and also for females.

Flow diagram illustrating the selection of cases for inclusion in the study. A total of 1882 post-mortems were identified from databases in the two centers, of which 151 cases were identified as SUCVD cases
The three most prevalent groups of cardiac pathology were cardiac malformations/complications post-surgery (51 cases), myocarditis (31 cases), and cardiomyopathies (32 cases). Demographics and mean age at death for each of these groups are provided in Table 2. As expected, the mean age of death for cardiac malformations was lower compared to myocarditis and cardiomyopathy cases (p = 0.01), and the babies were more likely to be premature (p = 0.001). The retrospective analysis also identified that symptoms had been present for a mean of 3.8, 3.0, and 3.5 days before death for myocarditis, cardiomyopathy, and cardiac malformations/complications post-surgery. According to the information provided by the coroner, myocarditis presented with the widest range of symptoms: dyspnea in 86.7% of cases, vomiting in 60%, pyrexia in 46.7%, and seizures in 40% of cases.
Case description
Macroscopic autopsy findings reported during tissue dissection, histological examination findings, and phenotypic associations are provided for the most prevalent and some of the unusual cases and disease entities seen (Table 3).
Inflammation
The nature of the inflammation in the majority of the myocarditis cases was lymphocytic, secondary to an acute viral infection. Enteroviridae were the most prevalent viruses demonstrated in myocarditis (15 cases) with Coxsackie-B sub-type (including types 3, 4) and Coxsackie-A (type 10) isolated from ventricular tissue following RT-PCR sequencing.
There was one case of an 18 months toddler with infective aortitis and endocarditis where human herpes virus-6 DNA was identified in association with Coxsackie-A (type 10) RNA from myocardial tissue. Macroscopic examination of the heart revealed a bacterial endocarditis obliterating the non-coronary leaflet with the formation of an abscess around the root of the aorta. The heart had a normal weight (52 gm). There was evidence of atrial inflammation which extended into the sinoatrial node. Staphylococcus aureus, cultured from the root of the aorta, resulted in aortic valvular destruction with vegetations, which on histology showed fibrin and neutrophilic infiltrate. Other viruses isolated included Parvovirus-B19 (3 cases). An unusual case corresponded to a 13 years old female, who has been admitted with dyspnea, abdominal pain, and pre-syncopal episodes. At post-mortem, the heart was globular and enlarged weighing 302 g (normal heart weight: 154 gm) with evidence of left ventricular subendocardial fibroelastosis, cardiac myocyte hypertrophy, and patchy ventricular fibrosis on histology. Symptoms in life were diagnosed as probable viral gastroenteritis 11 days prior to death. Although the histology did not identify a prominent lymphocytic myocarditis, Parvovirus-B19 infection was investigated with PCR, confirming its high viral load in myocardial tissue [7].
Parainfluenza type 3 infection was identified in myocardial tissue using PCR in 3 cases of dilated cardiomyopathy including one case in an 8-month-old infant with 2 days of central cyanotic episodes preceding death. This association has been rarely reported in pediatrics in the published literature [8].
In 4 cases of myocarditis, no infectious isolate was identified despite a lymphocytic histological picture post-mortem.
Bacterial isolates in myocarditis included one case of group A streptococcus and one case of mycoplasma pneumoniae myocarditis in a girl admitted with lower respiratory tract infection who deteriorated rapidly and was found to have a co-morbid atrioventricular septal defect at post-mortem.
Six cases of encephalomyocarditis were identified in the study, all of which were due to Enteroviridae (Coxsackievirus-B2), sequenced from cerebrospinal fluid and myocardial tissue. Macroscopically, the cases showed cerebral congestion, edema, and cerebral necrosis, as well as an enlarged heart with mottled endocardium (Fig. 2a). Histological findings included foci of necrosis associated with lymphoid inflammatory infiltrate in the myocardium, leptomeninges, and cerebrum, and particularly prominent in the brain stem. This latter is characteristically most severely affected in enterovirus encephalitis, predominantly the ventral, medial, and caudal areas [9] (Fig. 2b–e). All cases presented with seizures.

A few days old newborn who died suddenly. The post-mortem identified encephalomyocarditis caused by Coxsackie virus Type 3: a left ventricle with mottled appearances of the myocardium with pale and hemorrhagic areas; b myocardial necrosis associated to lymphocytic infiltrates and occasional eosinophils (H&E × 200); c C9 immunostain confirms extensive myocardial necrosis (× 100); d widespread encephalitis (H&E medulla × 200); e myelitis (H&E cervical spinal cord × 200)
Eosinophilic myocarditis was present in 4 cases with no previous medical history of note. The heart was morphologically normal; histology showed an eosinophilic-rich inflammatory infiltrate in the myocardium with myocyte necrosis and interstitial fibrosis. A specific preceding factor resulting in the hypersensitivity reaction was not found (Fig. 3).

Eosinophilic (hypersensitivity) myocarditis in a teenager male found dead in the morning. The heart showed myocardial necrosis, patchy areas of interstitial fibrosis, intercellular edema, and numerous eosinophilic infiltrates (left ventricle H&E × 200). No organisms were recovered from the myocardial tissue culture
Cardiomyopathies
In total, 32 cardiomyopathies presented as a sudden and unexpected death. Of these, 12 were hypertrophic, and 5 presented as dilated cardiomyopathy. Interestingly, molecular investigations in one of the cases with dilated cardiomyopathy unveiled a previous Parvovirus B19 infection as the primary etiology. There were a few examples of very rare cardiomyopathies (Table 1): histiocytoid, non-compaction, mitogenic, cardiomyopathy in association with GM1 gangliosidosis, Beckwith-Wiedemann Syndrome, and myotonic dystrophy. Four cases presented with subendocardial fibroelastosis as the main finding at post-mortem, and when electron microscopy was conducted, these cases were considered to be related to mitochondrial abnormalities. The post-mortem investigation did not include specific genetic analysis nor next-generation sequencing. In some cases, the presence of an associated condition allowed a diagnosis (i.e., GM1 gangliosidosis, Beckwith-Wiedemann Syndrome, or myotonic dystrophy). In other cases, the presence of specific histological features favored a specific cardiomyopathy (i.e., histiocytoid, mitogenic, or non-compaction cardiomyopathy). Some of the rarest cases are summarized below:
-
A 1-month male infant born at normal gestational age was found apneic lying in a prone position. PM findings showed left ventricular non-compaction, a mitochondrial X-linked heritable condition. The heart weight was normal (23.7 gm). Histological abnormalities included enlarged myocyte nuclei with expanded cytoplasm. This condition has been reported to have phenotypic associations with mosaic trisomy 22 [10].
-
A 19-month male infant born to consanguineous parents with a history of intrauterine growth restriction and a sudden movement deterioration was admitted to the hospital with bradycardia. Echocardiography revealed a dilated left atrium and ventricle, tricuspid regurgitation, and mitral regurgitation. The patient subsequently suffered a pulseless electric activity (PEA) arrest. Macroscopic and microscopic examination confirmed subendocardial fibroelastosis in the left ventricular endocardium. The heart weight (41.54 gm) was normal. There was patchy interstitial fibrosis with myocyte disarray and hypertrophy (Fig. 4). An excess of adipose tissue was seen with an oil red-O stain on the liver, which was in keeping with a probable metabolic disorder. Electron microscopy demonstrated increased connective tissue between myocytes, which appeared thinner than expected with diminished contractile apparatus. Mitochondrial numbers were noted to be increased, focally enlarged, and their cytoplasm filled with transverse cristae. Dilated cardiomyopathy with subendocardial fibroelastosis was demonstrated. Barth syndrome was thought to be the underlying cardiac pathology, as this entity is an infantile-onset, X-linked recessive mitochondrial disorder, frequently presenting with dilated cardiomyopathy, primarily affecting males and females due to variants in TAZ encoding for the cardiolipin transacylase tafazzin. It has been proposed that the increase in the number of mitochondria seen in Barth Syndrome is a compensatory mechanism that prevents a decrease of ATP synthesis [11] (Fig. 5).
-
A male infant born at term but small for gestational age had cyanotic episodes 12 h into life, poor feeding, and dyspnea. Respiratory function deteriorated, and cardiac arrest ensued. Autopsy showed an enlarged heart (52.9 g, expected 30 + / − 7 g) with a globular morphology. The right ventricle was dilated and hypertrophic. On histology, there were prominent myocyte hypertrophy, frequent mitoses including some giant forms, and subendocardial fibroelastosis in both the right and left ventricles. Findings were of mitogenic cardiomyopathy; Fig. 4 depicts this extremely rare entity only recently described in the literature [12].

a Myocyte hypertrophy and frequent mitosis characterizes mitogenic cardiomyopathy (H&E × 200); b Ki67 immunostain highlighted frequent mitotic figures (arrows) (× 200)

Electron microscopy of the myocardium showed moderate increased number of mitochondria in cardiac myocytes. Some are also enlarged (2.4 μm in diameter, white arrow), and others are filled with transverse cristae (grey arrow) (magnification 30,000 ×). Courtesy of Mr Bart Wagner, Head of Electron Microscopy at Sheffield Teaching NHS FT
Vascular lesions
Although these conditions are relatively rare in children, we observed 14 cases. The most frequent condition was the rupture of a clinically undiagnosed thoracoabdominal aortic aneurysm (4 cases). Some of the unusual cases are summarized below:
-
A 16-year-old female with a history of chest pain, asthma, and short intervals of central cyanosis when exposed to cold weather died suddenly. At PM, an anomalous coronary artery origin was noted, with the right coronary artery arising from the edge of the left coronary sinus and traversing the soft tissue plane between the pulmonary artery and aorta. The heart weight was normal (210 gm).
-
A 17-year-old male who died suddenly and unexpectedly had a thoracic aortic aneurysm due to an undiagnosed connective tissue disorder (suspected to be Loeys-Dietz syndrome). The weight of the heart (345 gm) was increased (expected for 60 kg male is 140–326 gm).
-
A 3-month-old female infant with a history of dyspnea and vomiting died from arterial calcification of infancy and subsequent papillary muscle rupture and acute myocardial infarction. The heart’s weight was increased (43 gm, expected 23.1–27.9 gm). Histology demonstrated subintimal fibrosis and calcification of the tunica media of numerous arteries within the heart, lungs, pancreas, periadrenal area, kidneys, thymus, and thyroid gland consistent with vascular calcification of infancy (Fig. 6a–d).
-
Other vascular processes seen included a case of fibromuscular dysplasia of the atrioventricular nodal artery in a white female infant with a history of ventricular septal defect, large patent ductus arteriosus (PDA) (3.5 mm in diameter), patent foramen ovale (PFO), and infantile respiratory distress syndrome (respiratory syncytial virus was isolated from the lungs and airway). She had several episodes of cardiac arrhythmias, cyanosis, and cardiac arrest. Autopsy findings included a muscular ventricular septal defect (VSD) of approximately 2 mm, and histopathology demonstrated normal myocardial and conduction system architecture, but focal and severe fibromuscular proliferation with near total luminal occlusion of the atrioventricular nodal artery. The PDA, PFO, and muscular VSD of approximately 2 mm diameter were confirmed at post-mortem. Fibromuscular dysplasia of intramyocardial arteries has been rarely described in the literature [13].
-
Many rarely described entities were identified in our study including long QT syndrome. One was a 3.5 years old female who developed sudden dyspnea over a period of 30 min and collapsed. She had a history of developmental delay, severe hearing impairment, anemia, and consanguineous parents. Macroscopic examination was unremarkable. The heart’s weight was normal (82.86 gm). The morphology of the sinoatrial nodes revealed abundant nerve fibers, an artery and fusiform fibers embedded in fibrous connective tissue. Due to her deafness, Jervell-Lange-Nielsen syndrome was suspected. This was confirmed by post-mortem mutational analysis, which revealed the presence of a nonsense mutation of KCNE1 at 21q22.12, thus confirming the diagnosis of Jervell-Lange-Nielsen syndrome.

A female infant with a history of dyspnoea and vomiting, whose post-mortem showed Idiopathic Arterial Calcification of Infancy, which caused secondary papillary muscle rupture and acute myocardial infarction: a coronary artery calcification (H&E × 200); b extensive myocardial infarction of the left ventricle; c calcification arteries in the thyroid (H&E × 200); and d pancreas among many other organs (H&E × 200)
Cardiac malformations/complications post-surgery
Undiagnosed cardiac malformations or complications after surgical repair were the most common etiology of sudden unexpected death in our cohort (51 cases). In 21/51 cases, deaths occurred after repaired malformations (Table 1). Atrioventricular septal defects, transposition of great vessels, and hypoplastic left heart were the most common conditions in this group. The more interesting cases are summarized below:
-
An 18 months old female toddler twin with a history of prematurity (corrected age: 14 months) and a large PDA underwent a transcatheter closure procedure. Three days later, she developed vomiting and reduced Glasgow Coma Scale (GCS). PM examination revealed the cause of death to be related to migration of the PDA closure device, which was found in the abdominal aorta. The device was surrounded by a thrombus, occluding the aorta. The heart’s weight was normal (63 gm) (Fig. 7a–c).
-
A 12 years old female with a history of cyanotic and complex congenital heart disease including pulmonary atresia, ventricular septal defect, multiple pulmonary aortopulmonary collateral arteries (MACPCAs), hypoplastic central pulmonary arteries with peripheral branching stenosis, and Alagille syndrome with early portal hypertension underwent cardiac catheterization, stenting, and ballooning of the right pulmonary artery. She presented 1 year later with a viral upper respiratory tract infection, a Staphylococcus aureus bacteremia, and subsequent bacterial endocarditis involving her right ventricular pulmonary artery conduit. She underwent surgery to replace the infected conduit with a Hancock pericardial xenograft valve; however, empyema, which was drained, was noted in the mediastinum. She had a poor postoperative recovery requiring intubation, ventilation, and inotropic support before suffering cardiac arrest. The autopsy revealed the repaired pulmonary trunk with the Hancock xenograft and a repaired peri-membranous ventricular septal defect. The heart’s weight was increased (350 gm, expected: 124 gm). Pulmonary arteries were occluded by thrombi, and there was aneurysmal dilatation extending into the hila of both lungs. Histology revealed occlusive thrombi and aneurysmal dilatation of both pulmonary arteries and a stent surrounded by bacterial colonies of P. putrida and infective endocarditis with dense neutrophilic infiltration extending up from the graft.

A female toddler with a large patent ductus arteriosus (PDA) underwent a transcatheter closure procedure. Three days later, she developed vomiting and reduced Glasgow Coma Scale (GCS). Cause of death was attributed to the migration of the PDA closure device, which was found in the abdominal aorta. a the PDA device was visualized on the X-ray conducted as part of the post-mortem at the level of T12 (arrow); b posterior view of the thoracoabdominal eviscerated block, demonstrating the migrated device in the abdominal aorta (note it below the diaphragm and just above both kidneys); c the PDA migrated device was surrounded by a fresh mural thrombus (H&E × 100)
Discussion
Among our 16-year caseload of sudden unexpected death in infancy and childhood, we identified several rare conditions including 6 cases of meningoencephalitis with myocarditis. All were between 5 and 11 days old, and virology studies identified Enteroviridae RNA by PCR assay, with Coxsackie B virus being the prominent serotype. The mothers of these neonates were asymptomatic, and enterovirus infection had not been suspected in any of the newborns prior to death. Importantly, three of the cases examined had histological evidence of myelitis. Meningoencephalitis with myocarditis secondary to enteroviral infection is likely to be acquired in utero via the transplacental route [14, 15]. It can also occur during delivery and lead to the so-called inflammatory cytokine storm with meningoencephalitis and imminent cardiac failure. Previous studies have reported Coxsackievirus B4 having tropism for neural tissues, myocardium, pancreas, adrenal glands, and renal tissues, but there is not yet an effective in utero vaccination for such a virus [16]. Coxsackievirus B is of the Picornaviridae family, is non-enveloped, and gains entry into endothelial cells and placental trophoblasts by endocytosis [17]. Studies have reported entry to placental trophoblasts via clathrin lipid rafts [15].
Other cardiotropic viruses including Parvovirus were encountered in our study. Interestingly, in a case presenting with an undiagnosed dilated cardiomyopathy, real-time PCR analysis identified Parvovirus B19, and this was interpreted as a late complication of a previous myocarditis (which had been undiagnosed). Myocardial endothelial cells, not cardiac myocytes, have been identified as parvovirus B19-specific target cell [18]. The molecular mechanisms responsible for the reactivation of latent parvovirus B19 infection and the development of chronic myocarditis have yet to be elucidated.
The significance of respiratory tract viral and bacterial isolates is unknown, but they were identified in several cases in our study, including human metapneumovirus, parainfluenza 3, and influenza A in cases of cardiomyopathies, as well as group A streptococcus and coronavirus in cases of cardiac death secondary to congenital malformations.
A few cases in our study demonstrated “left ventricular non-compaction” which is characterized by loss or absence of the compacted layer of myocardium and trabecular disturbance. This condition is poorly defined and is likely to be part of a spectrum of inherited disorders, some of which may be reversible and occur later in life [19].
In our study, we encountered a rare case of Jervell-Lange-Nielsen syndrome with a nonsense mutation of KCNE1 at 21q22.12. KCNE1 encodes for a minimal voltage-gated potassium ion channel protein, minK-related peptide 1 (MiRP1), a β subunit which is present in and regulates the potassium currents by forming ion channel complexes with pore-forming α subunits [20]. Loss of the minK protein results in the prolonged corrected QT interval of ventricular depolarization and atrial and ventricular repolarization and dysrhythmia and the associated sensorineural deafness characteristic of Jervell-Lange-Nielsen syndrome [21].
The role of rapid whole genome sequencing in pediatric autopsy practice (the so-called molecular autopsy) and the establishment of a molecular database may allow for the identification of pathogenic copy number variants in these conditions which are often poorly defined, helping to standardize diagnoses and allowing for genetic counseling and possible screening of family members with a high hereditary risk [22].
Conclusions and limitations
This study was small and retrospective in design, which only involved cases from two institutions in England; however, it remains the largest analysis of sudden and/or unexpected cardiovascular deaths in infancy and childhood in the UK.
Our study observed that cardiac malformations either undiagnosed or surgically repaired were the most prevalent cause of death in the young and resulted in the earliest age at death. Rarer conditions such as ion channelopathies and rare cardiomyopathies should be considered in a negative autopsy with appropriate selection of special tests as described in our study.
The increasing incidence of SCD in the young provides the impetus for enhanced diagnostic capabilities in SCD [23]. Further research studies are required to identify and map pathogenic copy number variants and single nucleotide polymorphisms resulting in fatal cardiac pathology to allow a genetic risk profile to be determined in life in select individuals to allow risk stratification, genetic counseling, and early medical or surgical intervention. Larger autopsy studies with data extracted and pooled from post-mortem tissue and the generation of epidemiological databases will allow for further understanding and better outcomes in cases of sudden cardiac death in the young through studies in morpho-molecular profiling, and decision-making algorithms incorporating clinical and genomic data will provide comprehensive genetic counseling and possible screening of family members at high hereditary risk of SCD.
In addition, investigation of arrhythmogenic gene mutations should be routinely undertaken in all post-mortems in which no definitive cause of death had been identified.
Key points
-
1.
A small percentage of sudden unexpected death in infants and children (8%) were due to cardiac conditions identified at gross and/or histological examination.
-
2.
Cardiac malformations, cardiomyopathies, and myocarditis were prevalent.
-
3.
Mean age at death—for both sexes—was 3.4 years.
-
4.
Symptoms had been present for a mean of 3.8, 3.0, and 3.5 days before death for myocarditis, cardiomyopathy, and cardiac malformations/complications post-surgery.
References
BHF CVD UK Fact Sheet. 2022. https://www.bhf.org.uk/what-we-do/our-research/heart-statistics (Accessed 26 Jun 22).
Zipes DP, Wellens HJJ. Sudden cardiac death. Circulation. 1998;98:2334–51. https://doi.org/10.1161/01.CIR.98.21.2334.
Molander N. Sudden natural death in later childhood and adolesce. Arch Dis Child. 1982;57:572–6. https://doi.org/10.1136/adc.57.8.572.
Papadakis M, Sharma S, Cox S, et al. The magnitude of sudden cardiac death in the young: a death certificate-based review in England and Wales. Europace. 2009;11:1353–8. https://doi.org/10.1093/europace/eup229.
Ha FJ, Han H-C, Sanders P, et al. Sudden cardiac death in the young. Circ Cardiovasc Qual Outcomes. 2020;13:745–55. https://doi.org/10.1161/CIRCOUTCOMES.119.006470.
Couper K, Putt O, Field R, et al. Incidence of sudden cardiac death in the young: a systematic review. BMJ Open. 2020;10. https://doi.org/10.1136/bmjopen-2020-040815.
Neagu O, Rodríguez AF, Callon D, et al. Myocarditis presenting as sudden death in infants and children: a single centre analysis by ESGFOR study group. Pediatr Dev Pathol. 2021;24(4):327–36. https://doi.org/10.1177/10935266211007262.
Romero-Gómez MP, Guereta L, Pareja-Grande J, et al. Myocarditis caused by human parainfluenza virus in an immunocompetent child initially associated with 2009 influenza A (H1N1) virus. J Clin Microbiol. 2011;49:2072–3. https://doi.org/10.1128/JCM.02638-10.
Hsueh C, Jung SM, Shih SR, et al. Acute encephalomyelitis during an outbreak of enterovirus type 71 infection in Taiwan: report of an autopsy case with pathologic, immunofluorescence, and molecular studies. Mod Pathol. 2000;13:1200–5.
Kalayinia S, Shahani T, Biglari A, et al. Mosaic trisomy 22 in a 4-year-old boy with congenital heart disease and general hypotrophy: a case report. J Clin Lab Anal. 2019;33:6–10. https://doi.org/10.1002/jcla.22663.
Finsterer J. Barth syndrome: mechanisms and management. Appl Clin Genet. 2019;5(12):95–106. https://doi.org/10.2147/TACG.S171481.
Chang KTE, Taylor GP, Meschino WS, et al. Mitogenic cardiomyopathy. A lethal neonatal familial dilated cardiomyopathy characterized by myocyte hyperplasia and proliferation. Hum Pathol. 2010;41:1002–8. https://doi.org/10.1016/j.humpath.2009.12.008.
Lee AH, Gray PB, Gallagher PJ. Sudden death and regional left ventricular fibrosis with fibromuscular dysplasia of small intramyocardial coronary arteries. Heart. 2000;83:101–2. https://doi.org/10.1136/heart.83.1.101.
Lauras B, Valla Y, Gaudin O, et al. [Coxsackie B5 infection in a newborn infant. Meningoencephalitis and myocarditis]. Pediatrie 39:561–5. http://www.ncbi.nlm.nih.gov/pubmed/6100130. Accessed 20 Oct 2021.
Delorme-Axford E, Sadovsky Y, Coyne CB. Lipid raft- and Src family kinase-dependent entry of coxsackievirus B into human placental trophoblasts. J Virol. 2013;87:8569–81. https://doi.org/10.1128/jvi.00708-13.
Bissel SJ, Winkler CC, DelTondo J, et al. Coxsackievirus B4 myocarditis and meningoencephalitis in newborn twins. Neuropathology. 2014;34:429–37. https://doi.org/10.1111/neup.12121.
Bozym RA, Morosky SA, Kim KS, et al. Release of intracellular calcium stores facilitates coxsackievirus entry into polarized endothelial cells. PLoS Pathog. 2010;6. https://doi.org/10.1371/journal.ppat.1001135.
Bock C-T, Klingel K, Kandolf R. Human parvovirus B19-associated myocarditis. N Engl J Med. 2010;362:1248–9. https://doi.org/10.1056/NEJMc0911362.
Arbustini E, Favalli V, Narula N, et al. Left ventricular noncompaction: a distinct genetic cardiomyopathy? J Am Coll Cardiol. 2016;68:949–66. https://doi.org/10.1016/j.jacc.2016.05.096.
Abbott GW, Goldstein SA. A superfamily of small potassium channel subunits: form and function of the MinK-related peptides (MiRPs). Q Rev Biophys. 1998;31:357–98. https://doi.org/10.1017/s0033583599003467.
Schulze-Bahr E, Wang Q, Wedekind H, et al. KCNE1 mutations cause jervell and Lange-Nielsen syndrome. Nat Genet. 1997;17:267–8. https://doi.org/10.1038/ng1197-267.
Boczek NJ, Tester DJ, Ackerman MJ. The molecular autopsy: an indispensable step following sudden cardiac death in the young? Herzschrittmacherther Elektrophysiol. 2012;23:167–73. https://doi.org/10.1007/s00399-012-0222-x.
Wong LCH, Behr ER. Sudden unexplained death in infants and children: the role of undiagnosed inherited cardiac conditions. Europace. 2014;16:1706–13. https://doi.org/10.1093/europace/euu037.
Author information
Authors and Affiliations
Contributions
FC Kon and M Haini acquired the data; FC Kon analyzed the data and drafted the report; I Scheimberg planned the study; M C Cohen analyzed and interpreted the data, planned the study, and oversaw the study and its report.
Corresponding author
Ethics declarations
Ethical approval
The study was a Service Evaluation approved by Clinical Governance (SE1295).
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kon, F.C., Scheimberg, I., Haini, M. et al. Cardiovascular-related death in infancy and childhood: a clinicopathological study of two referral institutions in England. Forensic Sci Med Pathol (2023). https://doi.org/10.1007/s12024-023-00630-5
Accepted:
Published:
DOI: https://doi.org/10.1007/s12024-023-00630-5