
Abstract
B lymphocytes are critical for effective immunity; they produce antibodies and cytokines, present antigens to T lymphocytes and regulate immune responses. However, because of the inherent randomness in the process of generating their vast repertoire of antigen-specific receptors, B cells can also cause diseases through recognizing and reacting to self. Therefore, B lymphocyte selection and responses require tight regulation at multiple levels and at all stages of their development and activation to avoid diseases. Indeed, newly generated B lymphocytes undergo rigorous tolerance mechanisms in the bone marrow and, subsequently, in the periphery after their migration. Furthermore, activation of mature B cells is regulated through controlled expression of co-stimulatory receptors and intracellular signalling thresholds. All these regulatory events determine whether and how B lymphocytes respond to antigens, by undergoing apoptosis or proliferation. However, defects that alter regulated co-stimulatory receptor expression or intracellular signalling thresholds can lead to diseases. For example, autoimmune diseases can result from altered regulation of B cell responses leading to the emergence of high-affinity autoreactive B cells, autoantibody production and tissue damage. The exact cause(s) of defective B cell responses in autoimmune diseases remains unknown. However, there is evidence that defects or mutations in genes that encode individual intracellular signalling proteins lead to autoimmune diseases, thus confirming that defects in intracellular pathways mediate autoimmune diseases. This review provides a synopsis of current knowledge of signalling proteins and pathways that regulate B lymphocyte responses and how defects in these could promote autoimmune diseases. Most of the evidence comes from studies of mouse models of disease and from genetically engineered mice. Some, however, also come from studying B lymphocytes from patients and from genome-wide association studies. Defining proteins and signalling pathways that underpin atypical B cell response in diseases will help in understanding disease mechanisms and provide new therapeutic avenues for precision therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Autoimmune diseases are pathological conditions in which defects in immunological tolerance to self lead to the initiation of effector immunity to self, chronic inflammation and tissue and organ damage. These diseases affect about 5–10% of human populations worldwide and cause significant degrees of morbidity and early death [1]. The cause of most autoimmune diseases remains largely unknown. However, susceptibility to develop these diseases is associated with a combination of genetic, environmental and hormonal factors [2]. These factors combine to cause defects in the survival and selection of self-reactive T and B lymphocytes. Evidence from the last 50 years of research indicates that T lymphocytes initiate autoimmune responses in conjunction with, or following incitement by, B lymphocytes. In addition to autoantibody production, there is compelling evidence that B lymphocytes also contribute to the development of the autoimmune diseases through mechanisms such as autoantigen presentation to activate autoreactive T cells and/or promote their polarization to produce disease-promoting/perpetuating cytokines. In this respect, it is perhaps revealing that a significant proportion of genetic susceptibility risk factors to develop autoimmune diseases corresponds with defects in the regulation of B cell responses, intracellular signalling and tolerance induction. These observations highlight changing perceptions about the role played by B cells in autoimmune diseases (Fig. 1; Tables 1 and 2). The importance of these roles is supported by the therapeutic benefit gained from depleting B cells in patients with a range of autoimmune diseases. For example, patients with diseases such rheumatoid arthritis (RA) [71], type 1 diabetes (T1D) [72], anti-neutrophil cytoplasmic antibody (ANCA) vasculitis [73], multiple sclerosis (MS) [74], systemic sclerosis (SSc) [75, 76], primary Sjögren’s syndrome [77,78,79,80] and systemic lupus erythematosus (SLE) [81,82,83] benefit from therapeutic depletion of B cells. Of note in this respect is that B cell-depleting therapy has a clinical benefit without significantly affecting autoantibody levels, suggesting that, perhaps, other B cell functions, including antigen presentation and cytokine production could be critical aspects of B cell involvement in the pathogenesis of the autoimmune diseases.

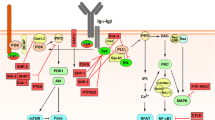
Signalling molecules and pathways in regulating B cell selection and responses. The diagram illustrates major signalling proteins/pathways involved in B cell physiology and whose regulation has been reported to be altered/defective in B cells in autoimmune disease. Proteins indicated in yellow are kinases, red for phosphatases, pink for proteins involved in ubiquitination, black for transcription factors and brown for adaptor proteins. Arrows indicate proteins that promote positive signalling, while blunt-ended lines indicate the protein negatively regulate signalling. Minus signs (encircled) indicate proteins/signalling pathways are reduced in mice and/or patients with autoimmune diseases or that reduction by genetic engineering promotes B lymphocyte hyperactivity and autoimmune disease. Positive signs (encircled) indicate enhanced activity of the proteins/signalling pathways in B lymphocytes from patients with autoimmune disease, mouse models or that their genetic manipulation promotes autoimmunity
The need for, and the ability to generate, a vast B cell repertoire to combat a universe of pathogens requires tolerance checkpoints and exquisite fine-tuning of B cell receptor (BCR) signalling to limit the emergence of pathogenic autoreactive B cells. Highly coordinated and integrated intracellular signalling transduced through the BCR and other co-stimulatory receptors, including innate pattern recognition receptors such as Toll-like receptors (TLRs), costimulatory/inhibitory molecules and cytokine receptors, are essential for regulating the outcome of BCR engagement by antigens. The available evidence indicates that minimal alterations in established thresholds of activating or inhibiting intracellular signalling can lead to a breakdown of immunological tolerance. This review provides a synopsis of current knowledge of signalling molecules and pathways involved in mediating and regulating B cell responses and how changes could lead to aggressive self-reactivity and autoimmune diseases.
Signals Controlling B Cell Development and Functions
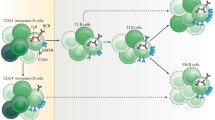
The BCR repertoire for antigens is vast, generated through random recombination of germline V(D)J mini genes, to provide broad immunity against pathogens. However, an intrinsic feature of generating this vast repertoire is the randomness with which germline V(D)J mini genes are recombined. This leads, in up to 80% of newly generated B cells, to the generation of BCRs that recognize self (Fig. 2). There is, therefore, a necessity for emerging B cells to undergo tolerance in the bone marrow and also subsequently in the periphery for B cells that escape bone marrow tolerance or those that emerge as a result of mutations in secondary lymphoid organs.

Pathways of B cell development and differentiation. B cells are generated from haematopoietic progenitor cells in the bone marrow. This process involves the expression of B lineage cell-specific proteins and the rearrangement of mini antibody V(D)J genes to generate the BCR repertoire. During the pro-B cell stage, antibody heavy chains are first generated by randomly rearranging and combining V, D and J mini genes. Pre-B cells express the pre-B cell antigen receptor (BCR) on the cell surface with the fully arranged heavy chain associated with the surrogate light chain (red). At later stages, light chain V and J mini genes are rearranged and a complete BCR is expressed in association with the Ig-α and Ig-β (green) subunits of the BCR complex. Immature B cells then undergo tolerance mechanisms with B cells recognizing self-protein undergoing light chain editing, apoptosis or functional inactivation (anergy). Surviving immature B cells then exit the bone marrow and migrate to secondary lymphoid organs where they develop into transitional (T) B cells. Transitional B cells can be subdivided into a number of developmental subsets. These include T1 B cells that express a high level of IgM and T2 B cells that express both IgM and IgD. These B cells undergo a range of tolerance checkpoint and cells that recognize self-antigens with high affinity are deleted. Cells with intermediate/low affinity to self-antigens and those that do not recognize self survive and circulate for about 3 weeks to survey the body for their target antigens. Transitional B cells develop into either marginal zone (MZ) B cells or follicular B cells. MZ B cells sample antigens and those that recognize antigens expand independently of T cell help. For their expansion, MZ B cells require TLR signalling to into short-lived plasma cells that produce antibodies with limited avidity for their target antigens. Follicular B cells are activated when they encounter their target antigens in the presence T cell help. Activated follicular B cells then migrate to B cell follicles and initiate somatic maturation in germinal centres. During this process, the cells proliferate, acquire somatic mutations, produce antibodies with higher avidity and class switch to IgG. Antigen-specific mature B cells then leave germinal centres and differentiate into either plasma cells or memory B cells. Plasma cells can either remain secondary lymphoid organs or travel to bone marrow to produce antibodies. B1 cells comprise a distinct subset of B cells that develop in the bone marrow and migrate to the periphery (peritoneal and pleural cavities in mice). B1 cells produce polyreactive IgM antibodies and partake in providing a first line of immunity against pathogens
Newly generated B cells first encounter self-antigens in the bone marrow, and their elimination or survival depends to a great extent on the strength with which their BCRs bind self-antigens and strength of the resulting intracellular signalling. Thus, recognition of self with high affinity initiates strong intracellular signalling, and as a result, B cells undergo receptor editing to replace nascent light chains that endow self-reactivity or, if this fails, apoptosis or anergy [84,85,86,87]. To facilitate B cell tolerance yet provide effective B cell immunity, intracellular signalling is regulated by highly refined thresholds. These thresholds regulate the magnitude and duration of intracellular B cell signalling and the outcome of BCR engagement by antigens. Subsequent to undergoing tolerance in the bone marrow, immature B cells migrate to the periphery where they will need tonic signalling for their transition to full maturity in readiness to respond to antigens. Signalling thresholds are also set for co-stimulatory receptors that modulate B cell responses following antigen recognition. These co-stimulatory receptors include CD40, TLR, BAFF and receptors for a range of other cytokines. Most of these signals involve the activation of phosphatidylinositol 3 kinase (PI3K) and the canonical pathway of NF-κB activation [88, 89].
BCR-Mediated Signalling
Proximal BCR Signalling Initiates B Cell Development and Responses
The BCR associates with a heterodimer of signalling proteins, Ig-α and Ig-β (also known as CD79a and CD79b), to form the BCR complex. The cytoplasmic domains of both Ig-α and Ig-β have immunoreceptor tyrosine-based activation motifs (ITAMs). ITAMs initiate signalling when their tyrosine residues are phosphorylated following BCR engagement and translocation to lipid raft signalling domains that contain the Src family tyrosine kinase Lyn. Downstream, activation of ITAMs generates a docking site for spleen tyrosine kinase (Syk) recruitment and phosphorylation [89, 90]. The activation of Syk is fundamental for initiating signalling cascades leading to lymphocyte activation [90,91,92]. Substrates of activated tyrosine kinases are adaptor molecules that, in turn, recruit other kinases to the BCR complex. B cell linker protein (BLNK) is a Syk substrate with nine tyrosine residues that are rapidly phosphorylated following engagement of the BCR [92, 93]. Phosphorylated BLNK is recruited to the plasma membrane and this requires its association with CIN85. The BLNK-CIN85 complex then coordinates the recruitment of growth factor receptor-bound protein 2 (Grb2) and phosphoinositide phospholipase C gamma (PLCγ) [94], a process essential for B cell development and responses.
Defective Regulation of BCR-Mediated Signalling Leads to Aberrant B Cell Responses and Autoimmune Diseases
Src Family Tyrosine Kinase Lyn
Lyn is a key dual activity kinase. It initiates BCR-mediated signalling by phosphorylating Ig-α/Ig-β ITAMs but then regulates this signalling by phosphorylating immunoreceptor tyrosine-based inhibition motifs (ITIMs) in CD5, CD22 and FcγRIIB [95]. Thus, Lyn-deficient mice develop spontaneous lupus-like autoimmune disease, splenomegaly and glomerulonephritis and produce anti-dsDNA autoantibodies [96, 97]. In addition, BCR-mediated calcium (Ca2+) influx is enhanced in B lymphocytes in Lyn−/−, mice and there is accelerated class switching of anti-dsDNA and anti-RNA autoantibodies [97]. Interestingly, however, deletion of myeloid differentiation primary response gene 88 (MyD88) in Lyn−/− mice, both globally or selectively in B lymphocytes, suppresses B cell activation and class switching of autoantibodies and ameliorates lupus disease [98]. This finding suggests that aberrant B cell responses in Lyn−/− mice are likely to be influenced not only by BCR-mediated signalling bust also by signalling through TLRs.
In humans, there is evidence for reduced Lyn expression in B lymphocytes from patients with SLE and that this reduction impacts B cell responses. For example, B cells from Lyn-insufficient SLE patients produce IgG autoantibodies to dsDNA and disease-promoting cytokines in vitro [4]. The association between Lyn insufficiency and SLE is supported by genetic studies. Thus, single nucleotide polymorphism (SNP) analyses and genome-wide association studies have revealed that polymorphisms in LYN, as well as other Src family tyrosine kinases including BLK, are risk factors for susceptibility to SLE [56, 99].
CD45 Tyrosine Phosphatase
CD45 is a membrane protein tyrosine phosphatase that positively regulates Lyn activation by dephosphorylating a tyrosine residue at position 507 (Y-507). This causes a conformational change that exposes the catalytic domain of Lyn and promotes autophosphorylation of the positive regulatory tyrosine at position 396 (Y-396) [100]. In addition to Lyn, CD45 regulates the activation of other kinases, such as Janus kinases (JAKs) and, thus, influences cytokine signalling [101], Src kinases involved in cell adhesion [102], TLR signalling [103] and apoptosis [104]. Dysregulation of CD45, therefore, can affect multiple B lymphocyte functions leading to autoimmune-like diseases, but the precise impact of changes of each of the multiple pathways that CD45 regulates in promoting autoimmune disease remains unclear. In genetically engineered mice, a single nucleotide replacement in the dimerization wedge of the CD45 molecule was shown to lead to autoantibody production and the development of lupus-like disease [11]. However, it is established that CD45 also influences apoptosis and defects in its expression have been shown to promote lupus disease in Fas ligand-mutant (Faslgld/gld) mice. In this setting, reduced CD45 expression enhanced B lymphocyte hyperactivity and auto-Ab production [105]. Furthermore, defects in CD45 regulation has been shown to affect B lymphocyte tolerance. For example, CD45−/− mice and CD45−/− B cell lines show reduced CD22 activation, SHP-1 recruitment, increased Syk activation [106] and Ca2+ influx [107]. Of note, however, is that loss of function mutations in sialic acid acetyl esterase (SIAE), which is required for the inhibitory function of CD22, has been shown to also create a significant risk for developing RA, T1D and SLE [108]. In hen egg lysozyme (HEL) transgenic mice, HEL induced tolerance in mature CD45+/+ B lymphocytes but led to the activation and accumulation of long-lived CD45−/− HEL-reactive B lymphocytes [109].
Studies of B lymphocytes in patients with SLE in our laboratory revealed that Lyn insufficiency was associated with increased CD45 translocation to lipid raft signalling domains and, ultimately, to reduced cellular expression of this phosphatase [12]. The noted increase in the translocation of CD45 to lipid raft signalling domains is likely to be relevant to reduced Lyn expression since CD45 promotes Lyn activation, ultimately its degradation in the proteasome [4].
Downstream Kinases
Defects in the regulation of BCR-associated signalling molecules downstream of Lyn have also been reported and shown to promote aberrant B cell responses and autoimmune diseases. For example, defective regulation of the adaptor protein B cell adaptor protein with ankyrin repeats 1 (BANK1) which initiates BCR-mediated Ca2+ signalling after Lyn-mediated phosphorylation of inositol 1,4,5-trisphosphate receptor (IP3R) causes autoimmune-like disease in mice. Thus, while BCR-mediated Ca2+ influx was shown to be normal in Bank1−/− primary B cells, this deficiency led to enhanced CD40-mediated proliferation, survival, increased Akt activation, and enhanced T-dependent antibody production and formation of germinal centres [110]. In humans, genetic studies have revealed that two variants of BANK1, R61H and A383T, are strongly associated with susceptibility to SLE [54]. The molecular basis for this association, however, remains to be determined. Nevertheless, the increase in CD40-mediated Akt activation in Bank1−/− B cells suggests that the allelic variants may promote autoimmunity through affecting cognate B-T cell interactions.
More recent studies of B cells from patients with SLE carried out in our laboratory revealed that the extent of defects in intercellular signalling is more complex and extensive than previously thought with each of the many identified defects likely to impact different B cell responses and clinical symptoms differently [28]. For example, these studies identified defective regulation of PI3K, MAPK, cyclin-dependent kinase1 (CDK1) and PKC to varying degrees in B cells from patients with SLE compared with matched healthy controls. These studies also revealed that the activity of Rho, a serine/threonine kinase involved in cell motility, was reduced in B cells from patients with SLE. Although as stated above, the relevance of many of these defects remains to be determined, it is likely that reduced activity of Rho can lead to defective migration of B lymphocytes. In addition to the above defects, reduced activity of the cell cycle kinase ATR was noted in the SLE patients. ATR is involved in activating the DNA damage response pathway, which leads either to cell cycle arrest or apoptosis and is, therefore, a key checkpoint in regulating cell responses to DNA damage.
Protein Tyrosine Phosphatases
In addition to kinases that positively regulate BCR-mediated signalling, defects in phosphatases that control the activation of kinases downstream of Lyn have also been reported to be involved in promoting aberrant B cell responses in autoimmune diseases. For example, defects in LYP tyrosine phosphatase, which is encoded by the protein tyrosine phosphatase non-receptor 22 (PTPN22), were shown to be sufficient to promote systemic autoimmunity. GWAS studies also revealed that a SNP in PTPN22, 1858C/T that resulted in R620W amino acid substitution is associated with increased risk of SLE, TID and RA [111,112,113]. Interestingly, expression of the R619W LYP variant in B cells alone was shown to be sufficient to develop splenomegaly, spontaneous germinal centre formation, glomerulonephritis and anti-dsDNA autoantibody production [8].
Calcium and Diacylglycerol Signalling
In Transcriptional Activation and B Cell Survival
The recruitment of PLCγ to the BCR signalling complex following engagement by antigens initiates phosphatidylinositol 4,5 biphosphate (PIP2) hydrolysis leading to the generation of inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG) [94, 114]. DAG binds to the cysteine-rich domain of Ras/Rap guanyl-releasing protein and this activates rat sarcoma (Ras) and ras-related protein (Rap) GTPases, the serine/threonine protein kinase C (PKC) and protein kinase D (PKD). IP3, in contrast, binds to IP3 receptors on the endoplasmic reticulum (ER) to release Ca2+ from its stores and, thus, increase cytosolic Ca2+ concentration [114]. The depletion of Ca2+ stores in the ER is sensed by stromal interaction molecules 1 and 2 (STIM1 and STIM2). As a result, these proteins relocate to the ER-plasma membrane junction where they bind to the Ca2+-release activated channel (CRAC) protein Orail and/or canonical transient receptor potential 1 (TRPC1) channels allowing extracellular Ca2+ entry to increase its intracellular level. As a consequence, the sustained increase in intracellular Ca2+ triggers the activation of the Ca2+/calmodulin-dependent protein kinase kinases (CaMKKs), serine threonine kinases involved in the regulation of important cellular processes such as survival and cytoskeletal reorganization [115]. The BCR-induced increase in intracellular Ca2+ levels also activates calcineurin (also known as protein phosphatase 2B, PP2B), a protein phosphatase that controls intracellular localization of nuclear factor of activated T cells (NFAT) family of transcription factors [116, 117]. In resting B cells, NFATs are constitutively phosphorylated by casein kinase 1 and glycogen synthase kinase-3 (GSK3) and are sequestered in the cytosol as a result of binding to 14-3-3 proteins. The BCR-induced activation of calcineurin leads to dephosphorylation of NFATs, thus permitting their translocation to the nucleus. In the nucleus, NFATs form complexes with other transcription factors to regulate the transcription of target genes including IL-2, IL-4, IL-10, tumour-necrosis factor alpha (TNFα) and interferon gamma (IFNγ) [117, 118].
Defective Regulation of Ca2+ Signalling Promotes Defective B Cell Tolerance
Numerous studies have examined molecular mechanisms leading to aberrant Ca2+ signalling in B lymphocytes with special emphasis on how BCR and co-receptors CD19 and CD21 mediate PLCγ2-IP3-Ca2+ signalling. CD21-mediated Ca2+ signalling plays an important role in breaching B lymphocyte tolerance leading to autoantibody production [119]. Furthermore, changes in the regulation of Ca2+ signalling are recognized as a major signalling event that contributes to the loss of B lymphocyte tolerance. Noteworthy, and perhaps paradoxically, is that an elevation in the baseline level of Ca2+ and reduced BCR-mediated elevation have been noted in tolerized/anergic B cells [84, 120]. Elevated baseline level of Ca2+ is likely to be due to persistent but low level engagement of the BCR by self-antigens and a recognized characteristic of anergic B cells in experimental models [84, 121]. In response to antigen engagement, naïve B cells show a rapid increase in intracellular Ca2+ followed by a drop to reach a plateau within minutes. This plateau is similar to the basal level seen in anergic B cells and continues as long as the BCR is engaged by the antigen. Thus, anergic B cells represent the physiological equivalent of chronically antigen-stimulated naïve B cells. Modulating BCR-mediated mechanisms of Ca2+ signalling could, therefore, provide a potential therapeutic approach for treating autoimmune diseases. Indeed, treatment of B cells with 1,4-benzodiazepine Bz-423 which increases sensitivity to BCR engagement by causing sustained high level of Ca2+ promotes apoptosis [122]. Since hyperactivation and altered Ca2+ signalling are distinguishing features of autoreactive B cells, treatment with Bz-423 has been suggested to be a useful approach for eliminating autoreactive B cells in autoimmune diseases.
Phosphatidylinositol 3 Kinase Signalling
PI3K Signalling in B Cell Development, Survival and Activation
PI3K signalling is important for B cell development, survival and activation. PI3Ks represent a family of lipid and protein kinases that function mainly through phosphorylation of phosphoinositide [123, 124]. Based on molecular structure and functions, PI3Ks are divided into four classes: I, II, III and IV. Members of class I PI3Ks are the ones whose altered activation is implicated in autoimmunity and inflammation. This class of PI3Ks is subdivided into two distinct subgroups, IA and IB. In mammals, the IA subgroup includes three members: PI3Kα, PI3Kβ and PI3Kδ [125]. All three kinase members of the IA subgroup are heterodimers consisting of p110 catalytic subunits (p110α, p110β and p110δ) and a regulatory subunit, usually referred to as p85 [125]. Subgroup IB, in contrast, consists of one catalytic subunit, p110γ, associated with either a p101 or p84 regulatory subunit [123]. These different PI3Ks function in different signalling pathways in lymphocytes with p110δ expression been restricted to haematopoietic cells. Upon receptor activation, PI3Ks phosphorylate PIP2 leading to the production of PIP3 [126]. The production of PIP3 requires recruitment of PI3Ks to the plasma membrane either through binding of the SH2 domain of their regulatory units to the phosphorylated tyrosine residues in receptor signalling complex domains and adaptors, or through direct recruitment by Ras. Signalling through PI3K is negatively regulated by the lipid phosphatase, SH2 domain-containing inositol phosphatase (SHIP) and phosphatase and tensin homologue deleted on chromosome ten (PTEN) [127]. Upon engagement of the BCR, CD19 recruits PI3K to the plasma membrane through binding of p85 to its tyrosine-phosphorylated cytoplasmic domain. In B lymphocytes, the B cell activating factor (BAFF) and low basal signalling by un-engaged BCR maintains low PIP3 levels [87]. The level of PIP3 increases dramatically following BCR engagement by antigens and co-stimulation through CD19, IL-4 receptor and/or TLRs. The recruitment and binding of the key downstream target of PI3K, Akt [also known as protein kinase B (PKB)], to the PIP3 through its pleckstrin homology (PH) domain causes conformational changes to Akt and, as a result, permits PIP3-dependent kinase 1 (PDK1)-mediated phosphorylation of Akt at threonine 304 within its catalytic domain. PDK1 has a PH domain that binds PIP3 and promotes its translocation to the plasma membrane to co-localize with Akt [128]. Once activated, Akt phosphorylates important downstream targets including Rheb GAP TSC2, FOX1/3 and Fox4A. Akt-induced phosphorylation of Ras homologue enriched in brain (Rheb) GAP TSC2 that leads to the accumulation of Rheb-GTP complex results in the activation of mammalian target of rapamycin complex 1 (mTORc1) [129]. The Faxo family of transcription factors is active and located in the nucleus in resting cells; however, when phosphorylated by Akt, they translocate to the cytosol where their transcriptional activities are terminated. Akt is, therefore, important for metabolism and cell survival in peripheral B lymphocytes [87]. Additionally, Akt/Foxo pathway plays a critical role in regulating the expression of recombinase activating genes (RAGs) that are responsible for antigen-receptor rearrangement in B cells [130]. When Akt is inactive in quiescent B lymphocytes, Foxo1, Foxo3 and Foxo4A drive transcription of genes encoding IL-7, an essential homeostatic cytokine for lymphocytes, as well as for Kruppel-like factor 2 (KLF2) transcription factor [131]. KLF2 directly regulates the expression of adhesion molecules and chemokine receptors responsible for controlling B lymphocyte entry into and exit from secondary lymphoid organs.
Dysregulated PI3K Signalling Alters Normal B Cell Development and Differentiation to Plasma Cells in Autoimmune Diseases
The involvement of aberrant PI3K signalling in the pathogenesis of autoimmune diseases is intriguing as all leukocytes express all members of class I PI3Ks. However, evidence for the involvement of defective regulation of PI3K signalling has mainly emerged from studying pathways involving PI3Kγ and PI3Kδ as these two class I PI3Ks are exclusively expressed in immune cells. In contrast to PI3Kα and PI3Kβ where ablation of their genes leads to embryonic lethality [132], PI3Kγ- and PI3Kδ-deficient mice are viable but are immunodeficient [133,134,135,136,137]. Furthermore, enhanced activity of either PI3Kγ or PI3Kδ has been implicated in promoting autoimmunity [138,139,140]. In murine models of lupus and in SLE patients, the activity of PI3K is increased [141]. The exact cause(s) and impact of enhanced PI3K activity on SLE and autoimmune diseases in general remains to be determined. However, PI3K promotes B cell survival and the generation of short-lived plasma cells and suppresses class switch recombination through activating Akt, which, in turn, represses Foxo transcription factors [142, 143]. Of note, is that PI3Kδ is the main PI3K family member that is involved in regulating B lymphocyte responses. Thus, mice lacking PI3Kδ show reduced development of pro-B to pre-B cells in the bone marrow and impaired responses of mature B cells [136, 144]. Additionally, PI3Kδ is involved in regulating marginal zone (MZ) and B-1 B cell responses including antibody production [145]. Interestingly, B cell development in PI3Kγ−/−δ−/− mice is similar to PI3Kδ−/− mice, whereas no defects are seen in B cell development in PI3Kγ−/− mice [146]. These observations indicate that PI3Kγ does not play a notable role in B cell development. Indeed, genetically engineered mice expressing a catalytically inactive PI3Kδ manifest impaired BCR signalling and reduced IgM and IgG antibody production [144]. Similarly, heterozygous deletion of PI3Kδ diminishes autoantibody production, ameliorates nephritis and improves survival in Lyn-deficient mice that develop lupus-like disease. In contrast, mice expressing constitutively active PI3Kδ show a reduced ability to eliminate autoreactive B lymphocytes [140]. In addition to its involvement in regulating BCR-mediated signalling, PI3Kδ is involved in mediating inflammation triggered by the engagement of TLRs [147]. These observations suggest that targeting of PI3Kδ could be an attractive therapeutic option for treating patients with autoimmune diseases and chronic inflammation [148,149,150]. Of note in this respect is that studies using mouse models of lupus have shown that inhibiting PI3K blocked glomerulonephritis and extended survival [139].
In addition to direct evidence for the role of dysregulated PI3K signalling in promoting autoimmune diseases in mice, there is indirect evidence for its involvement in promoting disease in patients. For example, there is evidence for decreased expression of PTEN, a lipid phosphatase that negatively regulates PI3K signalling in B cell subsets, except in memory B cells, in patients with SLE [31]. Furthermore, the level of PTEN in B cells from patients with SLE is inversely related to disease activity. Decreased levels of PTEN also concur with the upregulation of microRNA (miR-7) that downregulates PTEN expression. These findings suggest that defective miR-7 regulation of PTEN could contribute to B cell hyperresponsiveness in SLE [31]. Functional screening of a microRNA library also revealed that another miR, miR-148a, is a potent regulator of B cell tolerance [151]. Furthermore, increased expression levels of miR-148a were reported in patients with lupus and also in lupus-prone mice [151]. Elevated miR-148a levels impair B cell tolerance through enhancing the survival of immature B cells following BCR engagement by self-antigens [151]. Molecular studies revealed that miR-148a functions by suppressing the expression of Gadd45α, PTEN and the pro-apoptotic protein Bim. Furthermore, increased expression of miR-148a leads to lethal autoimmune disease in a mouse model of lupus [151]. Using adoptive transfer of anergic B cells, a recent study revealed that continuous signalling through the inhibitory molecules SHP-1 and SHIP-1 was required to maintain B cell anergy. Furthermore, reducing signalling through either of these two signalling pathways leads to rapid B cell activation, proliferation and the generation of short-lived plasma cells [152].
Ubiquitination-Regulated Signalling
Ubiquitination Regulation of BCR-Mediated Signalling and Antigen Processing
Ubiquitination is an important posttranslational modification process that regulates signal transduction through covalent attachment of ubiquitin (Ub) moieties, a 76-amino acid peptide, to targeted proteins. The process involves at least three enzymes, Ub-activating enzyme (E1) that activates Ub, Ub-conjugating enzyme (E2) and Ub ligase (E3). E3 enzymes, such as Cbl, catalyse ligation of the C-terminal residue of Ub to a lysine residue on the target protein [153]. Lysine residues K6, K11, K27, K29, K33, K48 and K63 of Ub can potentially form seven different types of linkages in branched poly Ub chains, whereas a linear form of the Ub chain can be formed when only one lysine in each Ub in involved in linkage formation [153, 154]. Mono ubiquitination promotes endocytic trafficking and DNA repair and the K48-linked Ub moieties tag proteins for degradation via the proteasomal system. In contrast, K63-linked and linearly linked Ub chains provide docking sites for downstream effectors and promote protein-protein interactions and signalling [155]. Ubiquitination can also be regulated through deubiquitinating enzymes, proteases that remove mono-ubiquitins and poly-ubiquitins from proteins. In this regard, A20 acts as a deubiquitinating as well as an ubiquitin-editing enzyme. A20 inhibits the activation of NF-κB. It also restricts apoptosis induced by TNFα [156]. The following section will review data on two key effector enzymes involved in the ubiquitination cycle, Cbl and A20, since there is an abundance of evidence for their involvement in autoimmunity.
In mammals, the Casitas B lineage lymphoma (Cbl) family of proteins has three members: c-Cbl, Cbl-b, and Cbl-3. c-Cbl and Cbl-b are expressed in B cells [157] and function as prominent substrates for tyrosine phosphorylation and regulators of the threshold of signalling [157,158,159,160]. c-Cbl effectively inhibits B cell responses through downregulating Syk kinase [161]. c-Cbl and Cbl-b interact with several BCR-associated signalling molecules such as PLCγ2, BLNK, PI3 kinase, Lyn, Vav and Syk [42, 162, 163]. Subsequent to binding to ITAMs, Syk is phosphorylated on tyrosine 323 and this creates a binding site for c-Cbl [164]. c-Cbl recruits components of the ubiquitin conjugation pathway and acts as an ubiquitin ligase [165]. Binding of c-Cbl results in Syk ubiquitination and downregulation of BCR signalling [164]. Apart from regulating BCR signalling, c-Cbl mediates BCR ubiquitination, a process crucial for facilitating antigen processing and presentation by B cells through the internalization of antigen-BCR complexes and guiding them to multi-vesicular body-like MIIC. In these multi-vesicular body-like MIICs, antigen-BCR complexes are processed into peptides and loaded onto MHC class II for presentation to T cells [166,167,168,169]. The recruitment of Cbl-b to clustered BCRs is also required for the entry of endocytosed BCRs into late endosomes. Recruitment of Cbl-b is also required for the entry of TLR9 into endosomes as has been noted after in vitro activation of TLR9 by BCR-captured antigens [170].
In contrast to Cbl, A20 is a widely expressed cytoplasmic protein that inhibits NF-κB activation and signalling downstream of interleukin-1 receptor (IL-1R), TNF receptor 1 (TNFR1), CD40 as well as signalling through innate-type receptors such as TLRs and NOD-like receptors (NLRs) [171,172,173,174]. In addition, A20 promotes cell survival through which it can regulate immune responses [174]. By destabilizing E2 enzymes, A20 can disrupt the interaction between E2 and E3 and, therefore, restrict ubiquitination of target proteins [175]. To achieve its critical biochemical functions, A20 interacts with key effectors including the receptor interacting kinase-1 (RIPK1), a key player in inflammation and cell death, E2, E3, ABIN-1 (ubiquitin sensors) and NEMO/IKKγ, a key player in NF-κB signalling [176,177,178,179,180,181]. Additionally, A20 binds directly to ubiquitin chains [177, 179] and modifies ubiquitinated protein substrates in multiple ways. For example, A20 cleaves poly-ubiquitin chains, thereby, exhibiting a deubiquitinating activity. In addition, A20 works with E1 and E2 proteins to build ubiquitin chains, thus displaying E3-like activity [171, 182]. Through its Ub-editing functions, A20 also removes K63-linked poly-ubiquitin chains from substrates and builds K48-linked ubiquitin chains [182].
Altered Ubiquitination in Defective in B Cell Tolerance
Inappropriate ubiquitination has been associated with the development of autoimmune diseases. A large body of evidence implicates defects in the level and regulation of Cbl and A20 in the pathogenesis of autoimmune diseases. Thus, Cbl-b-deficient mice develop autoimmune diseases and highlight a connection between Cbl-b-mediated protein degradation and the regulation of BCR signalling thresholds [158]. These mice produce high levels of autoantibodies to double-stranded DNA and develop signs of spontaneous lupus-like disease [158]. Another study revealed that Cbl-b-deficient mice had an enhanced susceptibility to develop experimental autoimmune encephalitis (EAE) [183]. B cells from Cbl-b-deficient mice showed an enhanced ability to proliferate in response to BCR and CD40 engagements [158]. The lowering of BCR thresholds caused by the loss of Cbl-b correlated with increased susceptibility to develop autoimmune disease.
Many signalling proteins associate with Cbl-b, including PLCγ, PI3K, Syk and the adaptor proteins Slp-76 and Vav. However, Cbl-b-deficient cells have a selective enhancement of Vav phosphorylation, indicating that Cbl-b is a negative regulator of Vav phosphorylation. Vav is a key guanine nucleotide exchange factor for the Rho family of GTP-binding proteins [184], and mice with B cell-specific ablation of c-Cbl and Cbl-b manifest lupus-like disease and have a significant increase in MZ and B1 B cell numbers [184]. Interestingly, however, c-Cbl/Cbl-b-deficient B cells were not hyperresponsive to BCR engagement, did not proliferate extensively nor produced antibodies but tolerance induction was impaired [42]. Apart from attenuated BLNK phosphorylation, these mutant B cells showed enhanced phosphorylation of BCR-proximal signalling proteins including Syk, PLCγ-2 and Vav and increased Ca2+ mobilization. These results, therefore, indicate that Cbl proteins regulate B cell tolerance possibly through fine-tuning of BCR-mediated signalling thresholds [42].
In contrast to Cbl proteins, as cited earlier, A20 is expressed in all cell types and regulates the canonical pathway of NF-κB activation and promote cell survival. The regulation of these signals by A20 is important for preventing autoimmune diseases and defects could lead to autoimmune inflammatory diseases. For example, A20-deficient mice were shown to develop multi-organ inflammation and perinatal lethality which prevented detailed studies of A20 functions in adult mice [173]. However, mice lacking A20 expression specifically in B cells provided better insights into how A20 regulates B cell development and functions. These mice spontaneously developed a lupus-like disease characterized by increased plasma cell and germinal centre B cell numbers, elevated levels of IgM and IgG autoantibodies and immunoglobulin deposits in the kidney [37,38,39]. The increase in germinal centre B cell numbers could be due to resistance to FAS-mediated apoptosis [39] and/or enhanced expression of NF-κB-dependent anti-apoptotic proteins including Bcl-X. Of note, however, is that these mice did not develop renal failure but severe nephritis when lupus-prone mice were used. Furthermore, heterozygous mice which expressed reduced levels of A20 specifically in B cells manifested increased numbers of germinal centre B cells and produced autoantibodies [38]. In addition to enhanced BCR-mediated signalling, A20-deficient mice were hyperresponsive to TLR and CD40 engagement. Furthermore, when stimulated, A20-deficient B cells produced higher levels of IL-6 compared with wild-type B cells [39]. Enhanced IL-6 production in A20-deficient B cells may account for the moderate increase in T cell numbers in mice lacking A20 expression in B cells [39].
In humans, GWAS and SNP analyses of TNFAIP3, the gene encoding A20, revealed a potential role for A20 in susceptibility to autoimmune diseases in humans (Table 2) [185]. Subsequent studies confirmed an association with a number of autoimmune diseases including SLE [186], RA [187], psoriasis [188], T1D [189, 190] and SSc [191, 192]. Since mice expressing low levels of A20 develop spontaneous inflammation and autoimmune diseases [37,38,39], TNFAIP3 SNPs might affect its function or expression. Indeed, reduced A20 functions in patients with SLE were associated with a SNP in the coding region of TNFAIP3 that caused a substitution in residue 127 from phenylalanine to cysteine. In contrast, reduced A20 level was associated with a SNP at the 3′ enhancer region of TNFAIP3 [193]. Additionally, it was suggested that SNPs located outside of the coding regions of TNFAIP3 may confer susceptibility to diseases by reducing A20 expression [194, 195]. Polymorphisms could also have prognostic and therapeutic values. Thus, TNFAIP3 polymorphisms and altered A20 expression levels were associated with therapeutic responses to RA patient treated with anti-TNFα agents [196]. The association of TNFAIP3 polymorphisms with lymphoma in patients with Sjögren’s also highlights the potential role of A20 in regulating B cell hyperactivity and malignant transformation leading to lymphomagenesis [197]. Moreover, the presence of certain TNFAIP3 SNPs was associated with the risk of severe renal or haematological complications in patients with SLE [193].
The NF-κB-associated signalling cascade is regulated by an E2 enzyme, UBE2L3 (also called UBCH7). UBE2L3 participates in the ubiquitination of p53, c-Fos and the NF-κB precursor p105, and defects are associated with increased susceptibility to many autoimmune diseases including RA and SLE [198, 199]. A single haplotype spanning UBE2L3, rs140490, was associated with increased UBE2L3 expression in B cells and aligned across multiple autoimmune diseases. Additionally, the UBE2L3 risk allele correlated with increased numbers of plasmablasts and plasma cells in patients with SLE suggesting a role for UBE2L3 in plasmablast and plasmacyte development [67, 200].
Innate Immune Receptor-Mediated Signalling
Innate Immune Receptor-Mediated Signalling and B Cell Tolerance
Innate immune receptors, such as TLRs, are pattern recognition molecules that bind conserved pathogen-associated molecular patterns (PAMPs) on pathogens. Ten TLRs are expressed in human cells, whereas in murine cells, there are 13 such receptors. Naïve human B cells express TLR1, 2, 3, 4, 6, 7 and 9, while plasma cells only express TLR3 and 4 [201]. TLR7 and TLR9 are known to be able to directly influence B cell tolerance. Engagement of TLR2 and TLR4, in contrast, has been implicated in promoting autoimmune diseases in mice although there is no direct evidence to support how, or indeed if, these two receptors modulate B cell. TLR7 and TLR9 are intracellular receptors that bind their ligands in endosomes. TLR7 binds ssRNA while TLR9 binds CpG DNA in viruses and bacteria. Interestingly, these receptors can be stimulated in self-reactive B cells by RNA and/or DNA-containing immune complexes. The two receptors dimerize upon ligand binding and recruit the adaptor protein myeloid differentiation primary response gene 88 (MyD88). The IL-1 receptor-associated kinase 4 (IRAK4) binds to MyD88 and activates IRAK1 and IRAK2. The resulting signalling complexes initiate the activation of NF-κB, MAPK and IFN-regulatory factor 1 (IRF1) and IRF5 signalling pathways and regulate the production of pro-inflammatory cytokine [202,203,204,205].
Dysregulated Innate Immune Receptor-Mediated Signalling Promotes B Cell Autoreactivity
A key feature of immunological abnormality in patients with autoimmune diseases is the production of autoantibodies, such as autoantibodies with specificity for nuclear antigens including DNA and proteins. There is evidence that crosstalk between signalling mediated by the BCR and TLRs could play an important role in the loss of B cell tolerance to these antigens [206, 207]. For example, BCR engagement by nucleic acid associated with self-antigens facilitates trafficking to endosomal compartments where TLRs reside leading to their engagement and B cell activation [208, 209]. In this respect, both TLR7 and TLR9 which initiate MYD88-dependent signalling pathways have been implicated in the pathogenesis of animal models of lupus and the production of anti-nuclear autoantibodies. Indeed, deletion of the TLR7 gene in lupus mice suppresses the production of autoantibodies to RNA-associated proteins and ameliorate systemic autoimmunity. Paradoxically, however, deletion of the TLR9 gene abolishes anti-dsDNA and anti-chromatin autoantibody production but exacerbates clinical symptoms [210, 211]. Since both TLR7 and TLR9 are expressed in B cells and myeloid cells, it is unclear whether the phenotype seen in these mice could be attributed to the effect of the two receptors on myeloid and/or B cells. However, deletion of TLR7 in Wiskott-Aldrich syndrome protein (WASp) in mice inhibited systemic autoimmunity, whereas deletion of TLR9 promoted systemic autoimmunity which recapitulates the phenotype seen in TLR7/9-deficient lupus mice [212,213,214]. WASp is expressed in haematopoietic cells and is implicated in BCR- and TLR-mediated signalling. Mutations in the WASp gene in humans cause Wiskott-Aldrich syndrome, an X-linked recessive disease characterized by primary immunodeficiency and high levels of autoantibodies [215]. In contrast to attenuating T cell receptor (TCR)-mediated signalling, WASp-deficient B cells are hyperresponsive to both BCR and TLR engagement leading to enhanced signalling adequate to mediate autoimmune disease even in the autoimmune-resistant B6 mouse [216]. In addition, WASp-deficient B cells are capable of activating wild-type CD4+ T cells and inducing spontaneous germinal centre formation, glomerulonephritis and the production of class-switched autoantibodies in mixed bone marrow chimeras in mice [216]. These effects were all MyD88-dependent since deletion of MyD88 in B cells abrogated T cell activation and spontaneous germinal centre formation [98, 217,218,219]. The pivotal role of TLR7 signalling in the pathogenesis of lupus was confirmed in several mouse models with the Y-chromosome-linked genomic-modifier Yaa in which there is duplication of the Tlr7 gene [220]. In Yaa mouse models, duplication of the Tlr7 gene was reported to be the sole requirement for accelerated autoimmunity and that reduction of Tlr7 gene dosage abolished the autoimmune phenotype. Furthermore, in TLR7 transgenic mice, B cells preferentially homed to spontaneous germinal centres in competitive chimeras suggesting a key role for TLR7-expressing B cells in driving the formation of autoreactive germinal centres [221]. Of note, overexpression of soluble RNAase ameliorated autoimmunity in TLR7-transgenic mice suggesting an important role for RNA in the pathogenesis of disease in these mice [222]. In genetic studies in humans, SNPs within Tlr7 and polymorphisms in genes encoding proteins and transcription factors downstream of TLR signalling, including TNFAIP3, TNIP1 and IRF5, associate with susceptibility to SLE [53, 223,224,225,226]. In addition, variants of SLC15A4, a histidine transporter involved in lysosomal TLR signalling, also associate with susceptibility to SLE. Furthermore, deletion of SLC15A4 in B cells limits autoimmunity in murine models of the disease [61]. Noteworthy in this respect is that humans deficient in either IRAK4 or MYD88, downstream effectors of TLR signalling, show increased autoreactivity within the naïve B cell compartment suggesting a pivotal role for TLR signalling in regulating tolerance in B cells [227, 228].
Co-stimulatory Receptor-Mediated Signalling
Co-stimulatory Receptor Signalling and B Lymphocyte Responses
The outcome of BCR engagement is influenced by signalling generated through a number of co-stimulatory receptors including CD5, CD19, CD21, CD22, CD40, CD45, CD72 and FcγRIIB. Signalling through these molecules upregulate and/or downregulate BCR-mediated signalling to fine-tune B cell responses. Any imbalance, or dysregulation, in signalling mediated through these co-receptors can either mediate autoimmune responses, or limit the ability of the immune system to mount an effective humoral response.
One of the key co-receptors involved in modulating BCR-mediated signalling is CD19. The cytoplasmic domain of CD19 has nine tyrosine residues which, when phosphorylated, act as docking sites for SH2-containing adaptors and kinases including PI3Ks, Vav-family guanosine exchange factors (GEFs) and growth factor receptor-bound protein 2 (Grb2). The engagement of CD40 by its ligand, CD40L, in contrast, initiates signalling through TNFR-associated factors (TRAFs) leading to the activation of downstream signalling pathways including MAPKs and NF-κB.
The activation of BCR-mediated signalling is also regulated by protein tyrosine phosphatases (PTPs), some of which, such as CD45, play dual positive and negative roles as cited earlier. Cytoplasmic phosphatases are recruited to the BCR complex through ITIM-containing co-receptors, such as CD5, CD22 and the low-affinity Fcγ receptors, specifically FcγRIIB [229]. Altered expression and/or activation either of kinases or phosphatases can lead to defective BCR-mediated signalling which, in turn, alters B-lymphocyte responses [230,231,232].
Co-ligation of the BCR and the FcγRIIB by antigen-antibody complexes leads to tyrosine phosphorylation of ITIMs [233], which, in turn, recruit SHIP, a lipid phosphatase with specificity for 5′-phosphate of PIP3 [234] through SH2-domain-mediated binding. SHIP dephosphorylates PIP3 to produce PI(3,4)P2 and, thus, diminish BCR-mediated elevation of PIP3. B lymphocytes also express the siglec family member CD22, an ITIM-containing receptor which interacts with ligands carrying a 2–6-linked sialic acids [235]. CD22 modulates BCR signalling threshold and inhibits signalling by recruiting SHP-1, a tyrosine phosphatase.
CD72 is constitutively expressed on B cells at all stages of their development except on plasma cells. CD72 negatively regulates BCR-induced signalling by recruiting SHP-1 through its cytoplasmic ITIM motif [236]. CD72 plays an essential regulatory role in modulating BCR-mediated signalling in autoreactive B cells [237]. In anergic B cells, CD72 downregulates BCR-mediated signalling by limiting antigen-induced Ca2+ influx and the activation of NFATc1, NF-κB, MAPK and Akt. Noteworthy, CD72 associates with SHP-1 and Cbl-b, suggesting a role for SHP-1 and Cbl-b in CD72-mediated inhibitory effects on BCR-mediated signalling in anergic B cells. CD100, a ligand for CD72, can turn off the negative effect of CD72 by inhibiting the phosphorylation of CD72 and, consequently, disrupting the interaction between SHP-1 and CD72 [238, 239].
Dysregulated Co-stimulatory Receptor-Mediated Signalling Promotes Autoreactive B Cell Expansion and Autoantibody Production
Cognate interactions between B and T cells involving co-stimulatory receptors such as CD40 and CD40L are critical for thymus-dependent humoral immunity. Ligation of CD40 induces B cell proliferation, class switching and somatic mutations. In lupus disease, loss or blockade of cognate B-T cell interactions involving CD40-CD40L ameliorates disease and prolongs survival in the NZB/NZW F1 and MRL-lpr spontaneous models of lupus [240,241,242]. In addition, the use of agonist anti-CD40 antibodies inhibits apoptosis of rheumatoid factor (RF) precursor B cells in arthritic mice, while blockade of CD40-CD40L abolishes RF production in transgenic mice [243, 244]. Furthermore, cd40l gene-deficient mice or treatment of neonatal NOD mice with anti-CD40L antibodies suppresses autoimmune diabetes [245,246,247,248]. Moreover, treatment of EAE mice with anti-CD40L antibody improved disease [249, 250]. Preclinical assessment of anti-CD40 antibody in a model of multiple sclerosis (MS) in monkeys provided additional support for the importance of CD40-CD40L interaction in autoimmune diseases [251,252,253]. However, clinical trials of anti-CD40L in patients with lupus had mixed outcomes, and in addition, some patients developed thromboembolism [254,255,256].
As cited above, in addition to CD40, defective regulation of engagement or signalling through other co-stimulatory receptors such as CD5, CD22 and FcγRIIB can also promote autoimmune diseases. CD5 and CD22 negatively regulate BCR-mediated signalling through ITIMs in their intracellular domains and PTPs. The PTPs can have dual inhibitory and activating effects. In autoimmune diseases, there is substantive evidence that defects in the regulation of co-stimulatory receptors and associated PTPs promote lupus disease, both in animal models and in patients. For example, there is evidence for altered expression of CD22 and SHP-1 in patients with SLE [4, 6, 257]. In genetically engineered mice, deletion of cd22, FcγRIIB or PTPN6, which encodes SHP-1, leads to B lymphocyte hyperactivity, auto-Ab production and lupus-like disease [5, 96, 258]. However, it remains unclear whether defects in the regulation of these co-stimulatory receptors and associated PTPs in patients are inherent or result from the disease process or, indeed, if they have a causal relationship with the disease.
In addition to the role of dysregulated kinases and phosphatases that regulate proximal BCR signalling, defects in co-stimulatory receptors that regulate downstream signalling have been associated with the development of autoimmune diseases. For example, dysregulation of CD72 has been shown to promote autoimmune diseases. In anergic B cells, CD72 constitutively regulates BCR-mediated signalling and limits proliferation and survival through suppressing cyclin D2 expression and Rb phosphorylation, key regulators of the cell cycle. Indeed, CD72-deficient mice spontaneously produce autoantibodies and develop lupus-like disease. Furthermore, CD72-deficient B cells proliferate and survive when their BCRs are engaged by self-antigens. The proliferative response of anergic B cells to BCR engagement by self-antigens results in the loss of immunological self-tolerance, upregulation of cyclin D2 and Bcl-xL, proliferation and survival of autoreactive B cells [259]. In contrast to anergic B cells where calcineurin/NFAT and NF-κB signalling pathways are defective [260, 261], self-antigen binding to anergic CD72−/− B cells leads to the activation of both calcineurin/NFAT and NF-κB [259]. Both calcineurin/NFAT and NF-κB are required for the induction of cyclin D2 [262, 263] and activation of MAPK and Akt, key regulators of cell cycle and survival.
Cytokine-Mediated Signalling
Cytokine Signalling in B Cell Differentiation
Dynamic regulation of cytokine production and cytokine receptor expression is required for B cell development, differentiation and efficient immune responses. Cytokines are involved in cellular communications and signalling and initiate a wide range of effects including cell differentiation, proliferation and regulation. Cytokines involved in B cell differentiation and responses include interferons (IFNs), interleukins (ILs) and members of the TNF family of ligands and receptors. Almost 40 cytokine receptors are known to initiate intracellular signalling mostly through JAKs and signal transducers and activators of transcription (STATs) [264, 265]. In addition to activating JAKs and STATs, cytokines can also initiate other signalling pathways such as activating Ras and PI3Ks [266, 267]. The binding of cytokines to their receptors induces dimerization or polymerization of the receptors and this activates associated JAKs. Activated JAKs induce phosphorylation and homo- and hetero-dimerization of STATs. Dimerized STATs translocate to the nucleus where they induce transcription of their target genes. There are four JAKs (JAK1, JAK2, JAK3 and TYK2) and seven known STATs (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6). Receptors for type 1 IFN signal via JAK1 and Tyk2, whereas receptors for IL-12 and IL-23 signal through JAK2 and Tyk2. Receptors for IL-2, IL-4, IL-7, IL-9, IL-15 and IL-23 signal through JAK1 and JAK3, whereas IFNγ receptor signals via JAK1 and JAK2 [268].
BAFF, which is a member of the TNF family of ligands and receptors, is crucial for B cell survival and development [269, 270]. The cytokine can bind to three different receptors: BAFF-R, B cell maturation antigen (BCMA) and transmembrane activator and Ca2+ modulator and cyclophilin ligand interactor (TACI) [271, 272]. Binding of BAFF to BAFF-R plays a predominant role in B cell maturation and survival, and mice deficient in either BAFF or BAFF-R exhibit a B cell developmental block at the T2 stage of transitional B cell maturation. In contrast, B cells in BCMA- or TACI-deficient mice develop normally into mature cells [273]. BAFF-R is linked to TRAFs and signals through both the canonical and alternative NF-κB pathways as well as through MAPK and PI3K pathways [274, 275]. Activation of the NF-κB pathway by engagement of the BAFF-R rescues transitional B cells receiving BCR signals from apoptosis in response to engagement by self-antigens, possibly through increased transcription of anti-apoptotic proteins and posttranscriptional modifications of pro-apoptotic proteins [276,277,278]. Interestingly, recent studies have revealed that there is crosstalk between BAFF-R and BCR signals that can induce survival signals independent of NF-κB activation [279, 280]. Thus, BAFF promotes rapid phosphorylation of proximal BCR signalling involving Ig-α and Syk. Deletion of Syk impairs survival and renders B cells non-responsive to BAFF. Survival in this setting can partially be restored by ectopic activation of MAPK or PI3K suggesting that BAFF-R signalling is likely to facilitate BCR-induced survival through activation of MAPK and PI3K. In this respect, BCR and CD19 have been implicated in regulating BAFF-R levels [86, 273].
Altered Cytokine Profiles Impacts Intracellular B Cell Signalling and Responses
Studies over the last few years have suggested that B cells can be subdivided into effector subsets based on the profile of cytokines they produce. Thus, B cells have been subdivided, in a manner akin to the subdivision of Th1 and Th2 cells, into B effector 1 cells that produce IFNγ and IL-12 and B effector 2 cells that produce IL-2, IL-4 and IL-6. More recent studies identified another effector B cell subset, B-regulatory cells (Bregs), characterized by their ability to produce IL-10, TGFβ and IL-35 and with immunosuppressive functions [281]. However, the available evidence indicates that, in contrast to effector T cell subsets, effector B cell subsets do not fulfil requirements of classic immune lineages such as defining transcription factors and may also exhibit plasticity depending on their microenvironmental settings. Nevertheless, the evidence provides support for a differential profile of cytokine production in B cells in different pathophysiological conditions. For example, altered profiles of cytokine production have been implicated in aberrant B cell responses in autoimmune diseases. Furthermore, in addition to the role of cognate T-B cell interactions in diverging cytokine production in B cells, TLR co-engagement with CD40 has been shown to synergize in promoting IL-10 and IL-35 production [282,283,284]. Both cytokines have important regulatory functions including limiting the generation of autoreactive germinal centres [284]. Interestingly, IL-10 can have dual effects on autoimmune diseases: acting as a B cell stimulator and also suppressor of T cell activations [285]. In this respect, IL-10 impacts autoimmune disease pathology differently depending on which cell and/or mechanism drives a disease. For example, while the transfer of IL-10-producing Bregs drives Treg cell expansion and modulates arthritis in mice, treatment of SLE patients with IL-10-specific monoclonal antibodies ameliorates disease [286, 287]. This outcome is consistent with evidence showing that high levels of IL-10 correlate with lupus disease activity in patients [288]. Interestingly, however, there is also evidence for defective signalling that regulate IL-10 production by B cells in patients with SLE [281, 282]. Similar to IL-10, IL-35 production by B cells has been shown to have immune regulatory functions and essential for recovery from EAE in mice [283].
In addition to the differentiation of B cells to distinct effector subsets with different cytokine profiles that impact autoimmune diseases differently, altered regulation of cytokine signalling in B cells can promote or enhance autoimmune disease pathology. For example, lupus is associated with high levels of IFNα and INFγ production with both altering B lymphocyte responses and autoantibody isotype production [289,290,291]. IFNα lowers BCR activation thresholds and promotes B cell differentiation through activating IRF5 transcription factor [292]. Indeed, polymorphism in the IRF5 gene has been associated with susceptibility to SLE [7]. Interestingly, excess production of IFNα can be induced by immune complexes suggesting a positive feedback circuit between IFNα and autoreactive B cells in lupus [290]. High levels of IFNγ, in contrast, enhance the production of complement-fixing IgG subclass of autoantibodies in lupus mice and promote lupus disease in patients with RA when treated with the cytokine [293]. Further evidence for the harmful role of excess IFNγ production in promoting B cell abnormalities and lupus diseases comes from studies in which deletion of IFNγ receptor in B cells abrogates spontaneous germinal centre formation, class switching of autoantibodies and nephritis in lupus-prone mice [294].
Studies in humans and mice have also revealed an important role for excess IL-21 production in promoting B cells to differentiate to plasma cells [295]. Thus, IL-21−/− mice have a diminished ability to produce IgG1 in response to immunization, whereas transgenic mice with enhanced expression of IL-21 develop hypergammaglobulinemia [295]. In contrast, IL-21 blockade successfully ameliorates lupus symptoms in lupus-prone MRL mice. Furthermore, knocking out il-21r gene suppresses lupus manifestations in the BXSB Yaa mouse model [295].
Overproduction of other cytokines, such as BAFF, a cytokine crucial for peripheral B cell development, has also been implicated in promoting autoimmune diseases [269, 270]. As cited earlier, BAFF binds to BAFF-R, BCMA and TACI. BAFF-R plays a key role in B cell maturation and survival and excess BAFF production is noted in many autoimmune diseases including SLE, RA and MS [296]. Indeed, transgenic mice expressing a high level of BAFF (BAFF-Tg) display B cell hyperplasia and develop lupus-like disease [297]. Of note in this respect is that high levels of BAFF rescue low-affinity self-reactive transitional B cells from negative selection at tolerance checkpoints and allows them to become mature B cells [298, 299]. These observations suggest that signalling through BAFF-R synergizes with BCR-mediated signalling in autoreactive B cells to override tolerance in these cells and permit the generation of pathogenic autoreactive B cells. Interestingly, MyD88, essential for TLR signalling, is crucial for autoantibody production in BAFF-Tg mice, suggesting that there is an interplay between BAFF-R and TLR signalling in promoting autoimmune diseases [297].
In addition to overt changes in cytokine production and signalling abnormalities in B cells, subtle changes in the regulation of signalling pathways activated by cytokine binding can also promote humoral autoimmunity. For example, heterozygous mutations in the stat3 gene leading to replacements of key amino acids, such as K392R, M394T and K658N, and enhanced STAT3 binding, or dimerization are associated with multi-organ autoimmunity, lymphoproliferation and hypogammaglobulinemia with terminal B cell maturational arrest [300]. Similarly, SNPs in stat3 and stat4 genes have been associated with autoimmune thyroid diseases [301]. However, the mechanism involved in promoting altered B cell responses in individuals with these polymorphisms and how B cells promote disease are yet to be determined.
It is noteworthy that several JAK inhibitors have been recently developed as new therapies for treating patients with inflammatory autoimmune diseases such as RA. Thus, tofacitinib that inhibits JAK1, JAK2, JAK3 and, to a lesser extent, TYK2 is used in the clinic for treating RA patients in many countries (Table 3). JAKs are a family of non-receptor tyrosine kinases that are critical for signalling through cytokine receptors. At present, a number of other JAK inhibitors are in clinical trials for treating patients with diseases including RA and SLE (Table 3).
Conclusions
It is now established that B cells play a key role in initiating and driving pathogenesis in many autoimmune diseases including SLE, RA, MS and SSc beyond their role in producing autoantibodies. Pathogenic roles played by B cells depend on tolerance status of the cells, which co-receptors are co-engaged with the BCR, the cytokine milieu and, ultimately, the nature and extent of intracellular signalling generated within B cells. The outcome of BCR engagement by antigens, self or exogenous, is determined by optimal levels or engagement and activation of kinases and phosphatases that regulate the strength and duration of intracellular signalling. Therefore, abnormal or incongruous engagement/regulation of signalling proteins, co-receptors or cytokines/TLRs can override B cell tolerance and lead to autoimmune disease development. Indeed, the available evidence indicates that regulated and coordinated engagement of co-receptors and signalling pathways ensures immune response specificity and efficiency and prevents the development of autoimmune diseases. This article has provided an overview of some of the key B cell signalling pathways and how defects in these could impact pathophysiology (Fig. 1). The available functional evidence on how changes in the level or function of signalling proteins and pathways impact B cell responses provides molecular mechanisms for how GWAS and SNP association with diseases such as SLE, RA, MS and TID could predispose to these diseases (Table 2). Noteworthy is that even in healthy individuals, autoreactive B cells, perhaps with low affinity for self-antigens, persist in the naïve B cell repertoire, yet these fail to become high-affinity pathogenic B cells. Thus, defining the molecular mechanisms that constrain the activation and/or regulation of self-reactive B cells will help understanding disease mechanisms and could also pave the way for new therapeutic strategies in precision medicine. Although B cell depletion therapy has proved to be highly effective in treating a number of autoimmune diseases including RA, MS and SSc, total ablation of B cells carries its own risks including life-threatening infections. In this respect, some recent studies have revealed that treatment of lupus mice with inhibitors of Bruton’s tyrosine kinase (BTK) can ameliorate disease [310, 311]. Thus, targeting dysregulated signalling effectors associated with proximal or downstream of BCR, CD40, TLR or cytokine receptors could prove an effective therapeutic strategy. However, despite substantial progress in the past few years in defining the role of altered B cell signalling in autoimmune diseases many questions and challenges remain.
Take-Home Points
-
B lymphocytes are essential for effective immunity to pathogens, but they can also cause diseases through producing autoantibodies, disease-promoting cytokines and presenting antigens to autoreactive T lymphocytes.
-
The potential of B lymphocyte to cause diseases is prevented by tolerance and by a tight control of intracellular signalling pathways.
-
Defects in intracellular signalling, however, can occur and these lead to autoimmune, lymphoproliferative or immunodeficient diseases.
-
Defects in proximal BCR signalling due to reduced Lyn and/or CD45 levels promote autoreactive B cell activation, class switching to IgG autoantibodies and disease.
-
Defects in downstream BCR signalling, e.g. Ca+2 or diacylglycerol pathways, lead to increased activation of NFAT transcription factor, tolerance defects, B lymphocyte hyperactivity and autoimmune diseases. Defects in PI3K lead to apoptosis defects and the generation of short-lived autoreactive plasma cells.
-
Defects in ubiquitination, which regulates signalling in B lymphocytes through controlling protein levels and transcription factors, enhance activation leading to enhanced B lymphocyte proliferation to BCR and CD40 engagements and autoantigen presentation to T lymphocytes and lead to autoimmune diseases.
-
Defects in innate immune receptor signalling, such as TLRs, exaggerate defects in BCR signalling and enhance autoreactive B cell expansion and responses.
-
Dysregulated expression of co-stimulatory receptors, stimulatory or inhibitory, including CD5, CD19, CD21, CD22, CD40, CD45, CD72 and FcγRIIB, leads to B lymphocyte hyperactivity, auto-Ab production and autoimmune diseases.
-
Altered cytokine production, cytokine receptor expression and/or cytokine signalling help rescue low-affinity self-reactive B lymphocytes, their differentiation and IgG isotype autoantibody production.
-
Defects in the level and activation of JAKs and STATs enhance B lymphocyte differentiation leading to multi-organ autoimmunity, lymphoproliferation or hypogammaglobulinemia.
References
Yu X (2015) Autoimmune diseases: what have we learned from mice? Curr Pharm Des 21(18):2306–2307. doi:10.1038/mp.2016.165
Konsta OD, Le Dantec C, Brooks WH, Renaudineau Y (2015) Genetics and epigenetics of autoimmune diseases. eLS 1–9; Brooks Wh, J Autoimmunity, 2010
Hibbs ML, Tarlinton DM, Armes J, Grail D, Hodgson G, Maglitto R, Stacker SA, Dunn AR (1995) Multiple defects in the immune system of Lyn-deficient mice, culminating in autoimmune disease. Cell 83(2):301–311
Flores-Borja F, Kabouridis PS, Jury EC, Isenberg DA, Mageed RA (2005) Decreased Lyn expression and translocation to lipid raft signaling domains in B lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum 52(12):3955–3965. doi:10.1002/art.21416
Mary C, Laporte C, Parzy D, Santiago ML, Stefani F, Lajaunias F, Parkhouse RM, O’Keefe TL, Neuberger MS, Izui S et al (2000) Dysregulated expression of the Cd22 gene as a result of a short interspersed nucleotide element insertion in Cd22a lupus-prone mice. J Immunol 165(6):2987–2996
Liossis SN, Solomou EE, Dimopoulos MA, Panayiotidis P, Mavrikakis MM, Sfikakis PP (2001) B-cell kinase lyn deficiency in patients with systemic lupus erythematosus. J Investig Med 49(2):157–165. doi:10.2310/6650.2001.34042
Alarcón-Riquelme ME, Ziegler JT, Molineros J, Howard TD, Moreno-Estrada A, Sánchez-Rodríguez E, Ainsworth HC, Ortiz-Tello P, Comeau ME, Rasmussen A et al (2016) Genome-wide association study in an Amerindian ancestry population reveals novel systemic lupus erythematosus risk loci and the role of European admixture. Arthritis Rheumatol 68(4):932–943. doi:10.1002/art.39504
Dai X, James RG, Habib T, Singh S, Jackson S, Khim S, Moon RT, Liggitt D, Wolf-Yadlin A, Buckner JH et al (2013) A disease-associated PTPN22 variant promotes systemic autoimmunity in murine models. J Clin Invest 123(5):2024–2036. doi:10.1172/JCI66963
Miyagawa H, Yamai M, Sakaguchi D, Kiyohara C, Tsukamoto H, Kimoto Y, Nakamura T, Lee JH, Tsai CY, Chiang BL et al (2008) Association of polymorphisms in complement component C3 gene with susceptibility to systemic lupus erythematosus. Rheumatol 47(2):158–164. doi:10.1093/rheumatology/kem321
Vang T, Miletic AV, Bottini N, Mustelin T (2007) Protein tyrosine phosphatase PTPN22 in human autoimmunity. Autoimmunity 40(6):453–461. doi:10.1080/08916930701464897
Hermiston ML, Tan AL, Gupta VA, Majeti R, Weiss A (2005) The juxtamembrane wedge negatively regulates CD45 function in B cells. Immunity 23(6):635–647. doi:10.1016/j.immuni.2005.11.001
Flores-Borja F, Kabouridis PS, Jury EC, Isenberg DA, Mageed RA (2007) Altered lipid raft-associated proximal signaling and translocation of CD45 tyrosine phosphatase in B lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum 56(1):291–302. doi:10.1002/art.22309
Halcomb KE, Musuka S, Gutierrez T, Wright HL, Satterthwaite AB (2008) Btk regulates localization, in vivo activation, and class switching of anti-DNA B cells. Mol Immunol 46(2):233–241. doi:10.1016/j.molimm.2008.08.278
Kil LP, de Bruijn MJ, van Nimwegen M, Corneth OB, van Hamburg JP, Dingjan GM, Thaiss F, Rimmelzwaan GF, Elewaut D, Delsing D et al (2012) Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood 119(16):3744–3756. doi:10.1182/blood-2011-12-397919
Maas A, Hendriks RW (2001) Role of Bruton’s tyrosine kinase in B cell development. Dev Immunol 8(3–4):171–181
Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA et al (2010) The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A 107(29):13075–13080. doi:10.1073/pnas.1004594107
O’Keefe TL, Williams GT, Batista FD, Neuberger MS (1999) Deficiency in CD22, a B cell-specific inhibitory receptor, is sufficient to predispose to development of high affinity autoantibodies. J Exp Med 189(8):1307–1313
O’Keefe TL, Williams GT, Davies SL, Neuberger MS (1996) Hyperresponsive B cells in CD22-deficient mice. Science 274(5288):798–801
Uckun FM, Goodman P, Ma H, Dibirdik I, Qazi S (2010) CD22 EXON 12 deletion as a pathogenic mechanism of human B-precursor leukemia. Proc Natl Acad Sci U S A 107(39):16852–16857. doi:10.1073/pnas.1007896107
Saito E, Fujimoto M, Hasegawa M, Komura K, Hamaguchi Y, Kaburagi Y, Nagaoka T, Takehara K, Tedder TF, Sato S (2002) CD19-dependent B lymphocyte signaling thresholds influence skin fibrosis and autoimmunity in the tight-skin mouse. J Clin Invest 109(11):1453–1462. doi:10.1172/JCI15078
Sato S, Hasegawa M, Fujimoto M, Tedder TF, Takehara K (2000) Quantitative genetic variation in CD19 expression correlates with autoimmunity. J Immunol 165(11):6635–6643
Kuroki K, Tsuchiya N, Tsao BP, Grossman JM, Fukazawa T, Hagiwara K, Kano H, Takazoe M, Iwata T, Hashimoto H et al (2002) Polymorphisms of human CD19 gene: possible association with susceptibility to systemic lupus erythematosus in Japanese. Genes Immun 3(Suppl 1):S21–S30. doi:10.1038/sj.gene.6363906
Yoshizaki A (2016) B lymphocytes in systemic sclerosis: abnormalities and therapeutic targets. J Dermatol 43(1):39–45. doi:10.1111/1346-8138.13184
Bolland S, Ravetch JV (2000) Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity 13(2):277–285
Yuasa T, Kubo S, Yoshino T, Ujike A, Matsumura K, Ono M, Ravetch JV, Takai T (1999) Deletion of fcgamma receptor IIB renders H-2(b) mice susceptible to collagen-induced arthritis. J Exp Med 189(1):187–194
Mackay M, Stanevsky A, Wang T, Aranow C, Li M, Koenig S, Ravetch JV, Diamond B (2006) Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. J Exp Med 203(9):2157–2164. doi:10.1084/jem.20051503
Maxwell MJ, Duan M, Armes JE, Anderson GP, Tarlinton DM, Hibbs ML (2011) Genetic segregation of inflammatory lung disease and autoimmune disease severity in SHIP-1−/− mice. J Immunol 186(12):7164–7175. doi:10.4049/jimmunol.1004185
Taher TE, Parikh K, Flores-Borja F, Mletzko S, Isenberg DA, Peppelenbosch MP, Mageed RA (2010) Protein phosphorylation and kinome profiling reveal altered regulation of multiple signaling pathways in B lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum 62(8):2412–2423. doi:10.1002/art.27505
Anzelon AN, Wu H, Rickert RC (2003) Pten inactivation alters peripheral B lymphocyte fate and reconstitutes CD19 function. Nat Immunol 4(3):287–294. doi:10.1038/ni892
Suzuki A, Kaisho T, Ohishi M, Tsukio-Yamaguchi M, Tsubata T, Koni PA, Sasaki T, Mak TW, Nakano T (2003) Critical roles of Pten in B cell homeostasis and immunoglobulin class switch recombination. J Exp Med 197(5):657–667
Wu XN, Ye YX, Niu JW, Li Y, Li X, You X, Chen H, Zhao LD, Zeng XF, Zhang FC et al (2014) Defective PTEN regulation contributes to B cell hyperresponsiveness in systemic lupus erythematosus. Sci Transl Med 6(2446):246ra299. doi:10.1126/scitranslmed.3009131
Medgyesi D, Hobeika E, Biesen R, Kollert F, Taddeo A, Voll RE, Hiepe F, Reth M (2014) The protein tyrosine phosphatase PTP1B is a negative regulator of CD40 and BAFF-R signaling and controls B cell autoimmunity. J Exp Med 211(3):427–440. doi:10.1084/jem.20131196
Qian Y, Giltiay N, Xiao J, Wang Y, Tian J, Han S, Scott M, Carter R, Jorgensen TN, Li X (2008) Deficiency of Act1, a critical modulator of B cell function, leads to development of Sjogren’s syndrome. Eur J Immunol 38(8):2219–2228. doi:10.1002/eji.200738113
Huffmeier U, Uebe S, Ekici AB, Bowes J, Giardina E, Korendowych E, Juneblad K, Apel M, McManus R, Ho P et al (2010) Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat Genet 42(11):996–999. doi:10.1038/ng.688
Perricone C, Ciccacci C, Ceccarelli F, Di Fusco D, Spinelli FR, Cipriano E, Novelli G, Valesini G, Conti F, Borgiani P (2013) TRAF3IP2 gene and systemic lupus erythematosus: association with disease susceptibility and pericarditis development. Immunogenetics 65(10):703–709. doi:10.1007/s00251-013-0717-6
Potter C, Eyre S, Cope A, Worthington J, Barton A (2007) Investigation of association between the TRAF family genes and RA susceptibility. Ann Rheum Dis 66(10):1322–1326. doi:10.1136/ard.2006.065706
Chu Y, Vahl JC, Kumar D, Heger K, Bertossi A, Wojtowicz E, Soberon V, Schenten D, Mack B, Reutelshofer M et al (2011) B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood 117(7):2227–2236. doi:10.1182/blood-2010-09-306019
Hovelmeyer N, Reissig S, Xuan NT, Adams-Quack P, Lukas D, Nikolaev A, Schluter D, Waisman A (2011) A20 deficiency in B cells enhances B-cell proliferation and results in the development of autoantibodies. Eur J Immunol 41(3):595–601. doi:10.1002/eji.201041313
Tavares RM, Turer EE, Liu CL, Advincula R, Scapini P, Rhee L, Barrera J, Lowell CA, Utz PJ et al (2010) The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity 33(2):181–191. doi:10.1016/j.immuni.2010.07.017
Musone SL, Taylor KE, Lu TT, Nititham J, Ferreira RC, Ortmann W, Shifrin N, Petri MA, Kamboh MI, Manzi S et al (2008) Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat Genet 40(9):1062–1064. doi:10.1038/ng.202
Plenge RM, Cotsapas C, Davies L, Price AL, de Bakker PI, Maller J, Pe’er I, Burtt NP, Blumenstiel B, DeFelice M et al (2007) Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet 39(12):1477–1482. doi:10.1038/ng.2007.27
Kitaura Y, Jang IK, Wang Y, Han YC, Inazu T, Cadera EJ, Schlissel M, Hardy RR, Gu H (2007) Control of the B cell-intrinsic tolerance programs by ubiquitin ligases Cbl and Cbl-b. Immunity 26(5):567–578. doi:10.1016/j.immuni.2007.03.015
Bergholdt R, Taxvig C, Eising S, Nerup J, Pociot F (2005) CBLB variants in type 1 diabetes and their genetic interaction with CTLA4. J Leukoc Biol 77(4):579–585. doi:10.1189/jlb.0904524
Gomez-Martin D, Ibarra-Sanchez M, Romo-Tena J, Cruz-Ruiz J, Esparza-Lopez J, Galindo-Campos M, Diaz-Zamudio M, Alcocer-Varela J (2013) Casitas B lineage lymphoma b is a key regulator of peripheral tolerance in systemic lupus erythematosus. Arthritis Rheum 65(4):1032–1042. doi:10.1002/art.3783
Recher M, Burns SO, de la Fuente MA, Volpi S, Dahlberg C, Walter JE, Moffitt K, Mathew D, Honke N, Lang PA et al (2012) B cell-intrinsic deficiency of the Wiskott-Aldrich syndrome protein (WASp) causes severe abnormalities of the peripheral B-cell compartment in mice. Blood 119(12):2819–2828
Schurman SH, Candotti F (2003) Autoimmunity in Wiskott-Aldrich syndrome. Curr Opin Rheumatol 15(4):446–453
Rieck M, Arechiga A, Onengut-Gumuscu S, Greenbaum C, Concannon P, Buckner JH (2007) Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J Immunol 179(7):4704–4710
Jacob CO, Eisenstein M, Dinauer MC, Ming W, Liu Q, John S, Quismorio FP Jr, Reiff A, Myones BL, Kaufman KM et al (2012) Lupus-associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proc Natl Acad Sci U S A 109(2):E59–E67. doi:10.1073/pnas.1113251108
Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, Ortmann W, Kosoy R, Ferreira RC, Nordmark G et al (2009) A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet 41(11):1228–1233. doi:10.1038/ng.468
Morris DL, Sheng Y, Zhang Y, Wang YF, Zhu Z, Tombleson P, Chen L, Cunninghame Graham DS, Bentham J, Roberts AL et al (2016) Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat Genet 48(8):940–946. doi:10.1038/ng.3603
Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD, Nath SK et al (2008) Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 40(2):204–210. doi:10.1038/ng.81
Raychaudhuri S, Thomson BP, Remmers EF, Eyre S, Hinks A, Guiducci C, Catanese JJ, Xie G, Stahl EA, Chen R et al (2009) Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat Genet 41(12):1313–1318. doi:10.1038/ng.479
Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, Xu JH, Cai ZM, Huang W, Zhao GP et al (2009) Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet 41(11):1234–1237. doi:10.1038/ng.472
Kozyrev SV, Abelson AK, Wojcik J, Zaghlool A, Linga Reddy MV, Sanchez E, Gunnarsson I, Svenungsson E, Sturfelt G, Jonsen A et al (2008) Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet 2008 40(2):211–216. doi:10.1038/ng.79
Sawalha AH, Kaufman KM, Kelly JA, Adler AJ, Aberle T, Kilpatrick J, Wakeland EK, Li QZ, Wandstrat AE, Karp DR et al (2008) Genetic association of interleukin-21 polymorphisms with systemic lupus erythematosus. Ann Rheum Dis 67(4):458–461. doi:10.1136/ard.2007.075424
Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, Lee AT, Chung SA, Ferreira RC, Pant PV et al (2008) Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med 358(9):900–909. doi:10.1056/NEJMoa0707865
Castillejo-Lopez C, Delgado-Vega AM, Wojcik J, Kozyrev SV, Thavathiru E, Wu YY, Sanchez E, Pollmann D, Lopez-Egido JR, Fineschi S et al (2012) Genetic and physical interaction of the B-cell systemic lupus erythematosus-associated genes BANK1 and BLK. Ann Rheum Dis 71(1):136–142. doi:10.1136/annrheumdis-2011-200085
Raychaudhuri S, Remmers EF, Lee AT, Hackett R, Guiducci C, Burtt NP, Gianniny L, Korman BD, Padyukov L, Kurreeman FA et al (2008) Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat Genet 40(10):1216–1223. doi:10.1038/ng.233
Yang W, Shen N, Ye DQ, Liu Q, Zhang Y, Qian XX, Hirankarn N, Ying D, Pan HF, Mok CC et al (2010) Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet 6(2):e1000841. doi:10.1371/journal.pgen.1000841
Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM, Kelly JA, Dozmorov MG, Miceli-Richard C, Bowman S et al (2013) Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjogren’s syndrome. Nat Genet 45(11):1284–1292. doi:10.1038/ng.2792
Kobayashi T, Shimabukuro-Demoto S, Yoshida-Sugitani R, Furuyama-Tanaka K, Karyu H, Sugiura Y, Shimizu Y, Hosaka T, Goto M, Kato N et al (2014) The histidine transporter SLC15A4 coordinates mTOR-dependent inflammatory responses and pathogenic antibody production. Immunity 41(3):375–388. doi:10.1016/j.immuni.2014.08.011
Yang J, Yang W, Hirankarn N, Ye DQ, Zhang Y, Pan HF, Mok CC, Chan TM, Wong RW, Mok MY et al (2011) ELF1 is associated with systemic lupus erythematosus in Asian populations. Hum Mol Genet 20(3):601–607. doi:10.1093/hmg/ddq474
Manjarrez-Orduno N, Marasco E, Chung SA, Katz MS, Kiridly JF, Simpfendorfer KR, Freudenberg J, Ballard DH, Nashi E, Hopkins TJ et al (2012) CSK regulatory polymorphism is associated with systemic lupus erythematosus and influences B-cell signaling and activation. Nat Genet 44(11):1227–1230. doi:10.1038/ng.2439
Ding C, Ma Y, Chen X, Liu M, Cai Y, Hu X, Xiang D, Nath S, Zhang HG, Ye H et al (2013) Integrin CD11b negatively regulates BCR signalling to maintain autoreactive B cell tolerance. Nat Commun 4:2813. doi:10.1038/ncomms3813
Lessard CJ, Adrianto I, Ice JA, Wiley GB, Kelly JA, Glenn SB, Adler AJ, Li H, Rasmussen A, Williams AH et al (2012) Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as susceptibility loci for systemic lupus erythematosus in a large-scale multiracial replication study. Am J Hum Genet 90(4):648–660. doi:10.1016/j.ajhg.2012.02.023
Sheng YJ, Gao JP, Li J, Han JW, Xu Q, Hu WL, Pan TM, Cheng YL, Yu ZY, Ni C et al (2011) Follow-up study identifies two novel susceptibility loci PRKCB and 8p11.21 for systemic lupus erythematosus. Rheumatology (Oxford) 50(4):682–688. doi:10.1093/rheumatology/keq313
Lewis MJ, Vyse S, Shields AM, Boeltz S, Gordon PA, Spector TD, Lehner PJ, Walczak H, Vyse TJ (2015) UBE2L3 polymorphism amplifies NF-kappaB activation and promotes plasma cell development, linking linear ubiquitination to multiple autoimmune diseases. Am J Hum Genet 96(2):221–234. doi:10.1016/j.ajhg.2014.12.024
Kaufman KM, Zhao J, Kelly JA, Hughes T, Adler A, Sanchez E, Ojwang JO, Langefeld CD, Ziegler JT, Williams AH et al (2013) Fine mapping of Xq28: both MECP2 and IRAK1 contribute to risk for systemic lupus erythematosus in multiple ancestral groups. Ann Rheum Dis 72(3):437–444. doi:10.1136/annrheumdis-2012-201851
He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, Shan M, Xiong H, Bussel JB, Chiu A et al (2010) The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol 11(9):836–845. doi:10.1038/ni.1914
Gregersen PK, Amos CI, Lee AT, Lu Y, Remmers EF, Kastner DL, Seldin MF, Criswell LA, Plenge RM, Holers VM et al (2009) REL, encoding a member of the NF-kappaB family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat Genet 41(7):820–823. doi:10.1038/ng.395
Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T (2004) Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med 350(25):2572–2581. doi:10.1056/NEJMoa032534
Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, McGee PF, Moran AM et al (2009) Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med 361(22):2143–2152. doi:10.1056/NEJMoa0904452
Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, Kallenberg CG, St Clair EW, Turkiewicz A, Tchao NK (2010) Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 363(3):221–232. doi:10.1056/NEJMoa0909905
Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, HERMES Trial Group et al (2008) B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 358(7):676–688. doi:10.1056/NEJMoa0706383
Daoussis D, Andonopoulos AP (2011) Rituximab in the treatment of systemic sclerosis-associated interstitial lung disease: comment on the article by Yoo. Rheumatol Int 31(6):841–842. doi:10.1007/s00296-010-1485-3
Jordan S, Distler JH, Maurer B, Huscher D, van Laar JM, Allanore Y, Distler O, group ERs (2015) Effects and safety of rituximab in systemic sclerosis: an analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann Rheum Dis 74(6):1188–1194. doi:10.1136/annrheumdis-2013-204522
Devauchelle-Pensec V, Morvan J, Rat AC, Jousse-Joulin S, Pennec Y, Pers JO, Jamin C, Renaudineau Y, Quintin-Roué I, Cochener B et al (2011) Effects of rituximab therapy on quality of life in patients with primary Sjögren’s syndrome. Clin Exp Rheumatol 29(1):6–12
Jousse-Joulin S, Devauchelle-Pensec V, Morvan J, Guias B, Pennec Y, Pers JO, Daridon C, Jamin C, Renaudineau Y, Roué IQ et al (2007) Ultrasound assessment of salivary glands in patients with primary Sjögren’s syndrome treated with rituximab: quantitative and Doppler waveform analysis. Biologics 1(3):311–319
Pers JO, Devauchelle V, Daridon C, Bendaoud B, Le Berre R, Bordron A, Hutin P, Renaudineau Y, Dueymes M, Loisel S et al (2007) BAFF-modulated repopulation of B lymphocytes in the blood and salivary glands of rituximab-treated patients with Sjögren’s syndrome. Arthritis Rheum 56(5):1464–1477. doi:10.1002/art.22603
Devauchelle-Pensec V, Pennec Y, Morvan J, Pers JO, Daridon C, Jousse-Joulin S, Roudaut A, Jamin C, Renaudineau Y, Roué IQ et al (2007) Improvement of Sjögren’s syndrome after two infusions of rituximab (anti-CD20). Arthritis Rheum 57(2):310–317. doi:10.1002/art.22536
Taher TE, Muhammad HA, Bariller E, Flores-Borja F, Renaudineau Y, Isenberg DA, Mageed RA (2013) B-lymphocyte signalling abnormalities and lupus immunopathology. Int Rev Immunol 32(4):428–444. doi:10.3109/08830185.2013.788648
Isenberg DA (2012) Rituximab—it was the best of times, it was the worst of times. Autoimmun Rev 11(11):790–791. doi:10.1016/j.autrev.2012.02.005
Seret G, Le Meur Y, Renaudineau Y, Youinou P (2012) Mesangial cell-specific antibodies are central to the pathogenesis of lupus nephritis. Clin Dev Immunol 2012:579670. doi:10.1155/2012/579670
Cancro MP (2009) Signalling crosstalk in B cells: managing worth and need. Nat Rev Immunol 9(9):657–661. doi:10.1038/nri2621
Cambier JC, Gauld SB, Merrell KT, Vilen BJ (2007) B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nat Rev Immunol 7(8):633–643. doi:10.1038/nri2133
Hartley SB, Crosbie J, Brink R, Kantor AB, Basten A, Goodnow CC (1991) Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature 353(6346):765–769. doi:10.1038/353765a0
Nemazee D (2006) Receptor editing in lymphocyte development and central tolerance. Nat Rev Immunol 6(10):728–740. doi:10.1038/nri1939
Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, Kutok JL, Kearney JF, Otipoby KL, Rajewsky K (2009) PI3 kinase signals BCR-dependent mature B cell survival. Cell 139(3):573–586. doi:10.1016/j.cell.2009.08.041
Wang LD, Clark MR (2003) B-cell antigen-receptor signalling in lymphocyte development. Immunol 110(4):411–420
Mocsai A, Ruland J, Tybulewicz VL (2010) The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol 10(6):387–402. doi:10.1038/nri2765
Koretzky GA, Abtahian F, Silverman MA (2006) SLP76 and SLP65: complex regulation of signalling in lymphocytes and beyond. Nat Rev Immunol 6(1):67–78. doi:10.1038/nri1750
Kurosaki T, Hikida M (2009) Tyrosine kinases and their substrates in B lymphocytes. Immunol Rev 228(1):132–148. doi:10.1111/j.1600-065X.2008.00748.x
Oellerich T, Bremes V, Neumann K, Bohnenberger H, Dittmann K, Hsiao HH, Engelke M, Schnyder T, Batista FD, Urlaub H et al (2011) The B-cell antigen receptor signals through a preformed transducer module of SLP65 and CIN85. EMBO J 30(17):3620–3634. doi:10.1038/emboj.2011.251
Oh-hora M, Rao A (2008) Calcium signaling in lymphocytes. Curr Opin Immunol 20(3):250–258. doi:10.1016/j.coi.2008.04.004
Gross AJ, Lyandres JR, Panigrahi AK, Prak ET, DeFranco AL (2009) Developmental acquisition of the Lyn-CD22-SHP-1 inhibitory pathway promotes B cell tolerance. J Immunol 182(9):5382–5392. doi:10.4049/jimmunol.0803941
Cornall RJ, Cyster JG, Hibbs ML, Dunn AR, Otipoby KL, Clark EA, Goodnow CC (1998) Polygenic autoimmune traits: Lyn, CD22, and SHP-1 are limiting elements of a biochemical pathway regulating BCR signaling and selection. Immunity 8(4):497–508
Lamagna C, Hu Y, DeFranco AL, Lowell CA (2014) B cell-specific loss of Lyn kinase leads to autoimmunity. J Immunol 192(3):919–928. doi:10.4049/jimmunol.1301979
Hua Z, Gross AJ, Lamagna C, Ramos-Hernandez N, Scapini P, Ji M, Shao H, Lowell CA, Hou B, DeFranco AL (2014) Requirement for MyD88 signaling in B cells and dendritic cells for germinal center anti-nuclear antibody production in Lyn-deficient mice. J Immunol 192(3):875–885. doi:10.4049/jimmunol.1300683
Lu R, Vidal GS, Kelly JA, Delgado-Vega AM, Howard XK, Macwana SR, Dominguez N, Klein W, Burrell C, Harley IT et al (2009) Genetic associations of LYN with systemic lupus erythematosus. Genes Immun 10(5):397–403. doi:10.1038/gene.2009.19
Hermiston ML, Xu Z, Weiss A (2003) CD45: a critical regulator of signaling thresholds in immune cells. Ann Rev Immunol 21:107–137. doi:10.1146/annurev.immunol.21.120601.140946
Irie-Sasaki J, Sasaki T, Matsumoto W, Opavsky A, Cheng M, Welstead G, Griffiths E, Krawczyk C, Richardson CD, Aitken K et al (2001) CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature 409(6818):349–354. doi:10.1038/35053086
Roach T, Slater S, Koval M, White L, Cahir McFarland ED, Okumura M, Thomas M, Brown E (1997) CD45 regulates Src family member kinase activity associated with macrophage integrin-mediated adhesion. Curr Biol 7(6):408–417
Piercy J, Petrova S, Tchilian EZ, Beverley PC (2006) CD45 negatively regulates tumour necrosis factor and interleukin-6 production in dendritic cells. Immunology 118(2):250–256. doi:10.1111/j.1365-2567.2006.02363.x
Gregori S, Mangia P, Bacchetta R, Tresoldi E, Kolbinger F, Traversari C, Carballido JM, de Vries JE, Korthauer U, Roncarolo MG (2005) An anti-CD45RO/RB monoclonal antibody modulates T cell responses via induction of apoptosis and generation of regulatory T cells. J Exp Med 201(8):1293–1305. doi:10.1084/jem.20040912
Brooks WP, Lynes MA (2001) Effects of hemizygous CD45 expression in the autoimmune Fasl(gld/gld) syndrome. Cell Immunol 212(1):24–34. doi:10.1006/cimm.2001.1845
Greer SF, Justement LB (1999) CD45 regulates tyrosine phosphorylation of CD22 and its association with the protein tyrosine phosphatase SHP-1. J Immunol 162(9):5278–5286
Benatar T, Carsetti R, Furlonger C, Kamalia N, Mak T, Paige CJ (1996) Immunoglobulin-mediated signal transduction in B cells from CD45-deficient mice. J Exp Med 183(1):329–334
Surolia I, Pirnie SP, Chellappa V, Taylor KN, Cariappa A, Moya J, Liu H, Bell DW, Driscoll DR, Diederichs S et al (2010) Functionally defective germline variants of sialic acid acetylesterase in autoimmunity. Nature 466(7303):243–247. doi:10.1038/nature09115
Cyster JG, Healy JI, Kishihara K, Mak TW, Thomas ML, Goodnow CC (1996) Regulation of B-lymphocyte negative and positive selection by tyrosine phosphatase CD45. Nature 381(6580):325–328. doi:10.1038/381325a0
Aiba Y, Yamazaki T, Okada T, Gotoh K, Sanjo H, Ogata M, Kurosaki T (2006) BANK negatively regulates Akt activation and subsequent B cell responses. Immunity 24(3):259–268. doi:10.1016/j.immuni.2006.01.002
Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM et al (2004) A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet 75(2):330–337. doi:10.1086/422827
Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M et al (2004) A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet 36(4):337–338. doi:10.1038/ng1323
Kyogoku C, Langefeld CD, Ortmann WA, Lee A, Selby S, Carlton VE, Chang M, Ramos P, Baechler EC, Batliwalla FM et al (2004) Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet 75(3):504–507. doi:10.1086/423790
Bootman MD (2012) Calcium signaling. Cold Spring Harb Perspect Biol 4(7):a011171. doi:10.1101/cshperspect.a011171
Matthews SA, Cantrell DA (2009) New insights into the regulation and function of serine/threonine kinases in T lymphocytes. Immunol Rev 228(1):241–252. doi:10.1111/j.1600-065X.2008.00759.x
Im SH, Rao A (2004) Activation and deactivation of gene expression by Ca2+/calcineurin-NFAT-mediated signaling. Mol Cells 18(1):1–9
Muller MR, Rao A (2010) NFAT, immunity and cancer: a transcription factor comes of age. Nat Rev Immunol 10(9):645–656. doi:10.1038/nri2818
Garaud S, Morva A, Lemoine S, Hillion S, Bordron A, Pers JO, Berthou C, Mageed RA, Renaudineau Y, Youinou P (2011) CD5 promotes IL-10 production in chronic lymphocytic leukemia B cells through STAT3 and NFAT2 activation. J Immunol 186(8):4835–4844. doi:10.4049/jimmunol.1003050
Lyubchenko T, dal Porto J, Cambier JC, Holers VM (2005) Coligation of the B cell receptor with complement receptor type 2 (CR2/CD21) using its natural ligand C3dg: activation without engagement of an inhibitory signaling pathway. J Immunol 174(6):3264–3272
Yarkoni Y, Getahun A, Cambier JC (2010) Molecular underpinning of B-cell anergy. Immunol Rev 237(1):249–263. doi:10.1111/j.1600-065X.2010.00936.x
Gauld SB, Benschop RJ, Merrell KT, Cambier JC (2005) Maintenance of B cell anergy requires constant antigen receptor occupancy and signaling. Nat Immunol 6(11):1160–1167. doi:10.1038/ni1256
Bednarski JJ, Lyssiotis CA, Roush R, Boitano AE, Glick GD, Opipari AW Jr (2004) A novel benzodiazepine increases the sensitivity of B cells to receptor stimulation with synergistic effects on calcium signaling and apoptosis. J Biol Chem 279(28):29615–29621. doi:10.1074/jbc.M403507200
Dhand R, Hara K, Hiles I, Bax B, Gout I, Panayotou G, Fry MJ, Yonezawa K, Kasuga M, Waterfield MD (1994) PI 3-kinase: structural and functional analysis of intersubunit interactions. EMBO J 13(3):511–521
Lehmann K, Muller JP, Schlott B, Skroblin P, Barz D, Norgauer J, Wetzker R (2009) PI3Kgamma controls oxidative bursts in neutrophils via interactions with PKCalpha and p47phox. Biochem J 419(3):603–610. doi:10.1042/BJ20081268
Geering B, Cutillas PR, Nock G, Gharbi SI, Vanhaesebroeck B (2007) Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc Natl Acad Sci U S A 104(19):7809–7814. doi:10.1073/pnas.0700373104
Pirola L, Zvelebil MJ, Bulgarelli-Leva G, Van Obberghen E, Waterfield MD, Wymann MP (2001) Activation loop sequences confer substrate specificity to phosphoinositide 3-kinase alpha (PI3Kalpha ). Functions of lipid kinase-deficient PI3Kalpha in signaling. J Biol Chem 276(24):21544–21554. doi:10.1074/jbc.M011330200
Dyson JM, Fedele CG, Davies EM, Becanovic J, Mitchell CA (2012) Phosphoinositide phosphatases: just as important as the kinases. Subcell Biochem 58:215–279. doi:10.1007/978-94-007-3012-0_7
Finlay D, Cantrell D (2011) The coordination of T-cell function by serine/threonine kinases. Cold Spring Harb Perspect Biol 3(1):a002261. doi:10.1101/cshperspect.a002261
Laplante M, Sabatini DM (2012) mTOR signaling. Cold Spring Harb Perspect Biol 4(2). doi:10.1101/cshperspect.a011593
Kuo TC, Schlissel MS (2009) Mechanisms controlling expression of the RAG locus during lymphocyte development. Curr Opin Immunol 21(2):173–178. doi:10.1016/j.coi.2009.03.008
Hart GT, Wang X, Hogquist KA, Jameson SC (2011) Kruppel-like factor 2 (KLF2) regulates B-cell reactivity, subset differentiation, and trafficking molecule expression. Proc Natl Acad Sci U S A 108(2):716–721. doi:10.1073/pnas.1013168108
Bi L, Okabe I, Bernard DJ, Nussbaum RL (2002) Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm Genome 13(3):169–172. doi:10.1007/s00335-001-2123-x
Bilancio A, Okkenhaug K, Camps M, Emery JL, Ruckle T, Rommel C, Vanhaesebroeck B (2006) Key role of the p110delta isoform of PI3K in B-cell antigen and IL-4 receptor signaling: comparative analysis of genetic and pharmacologic interference with p110delta function in B cells. Blood 107(2):642–650. doi:10.1182/blood-2005-07-3041
Clayton E, Bardi G, Bell SE, Chantry D, Downes CP, Gray A, Humphries LA, Rawlings D, Reynolds H, Vigorito E et al (2002) A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J Exp Med 196(6):753–763
Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP (2000) Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 287(5455):1049–1053
Jou ST, Carpino N, Takahashi Y, Piekorz R, Chao JR, Carpino N, Wang D, Ihle JN (2002) Essential, nonredundant role for the phosphoinositide 3-kinase p110delta in signaling by the B-cell receptor complex. Mol Cell Biol 22(24):8580–8591
Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I et al (2000) Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 287(5455):1040–1046
Barber DF, Bartolome A, Hernandez C, Flores JM, Fernandez-Arias C, Rodriguez-Borlado L, Hirsch E, Wymann M, Balomenos D, Carrera AC (2006) Class IB-phosphatidylinositol 3-kinase (PI3K) deficiency ameliorates IA-PI3K-induced systemic lupus but not T cell invasion. J Immunol 176(1):589–593
Barber DF, Bartolome A, Hernandez C, Flores JM, Redondo C, Fernandez-Arias C, Camps M, Ruckle T, Schwarz MK, Rodriguez S et al (2005) PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med 11(9):933–935. doi:10.1038/nm1291
Maxwell MJ, Tsantikos E, Kong AM, Vanhaesebroeck B, Tarlinton DM, Hibbs ML (2012) Attenuation of phosphoinositide 3-kinase delta signaling restrains autoimmune disease. J Autoimmun 38(4):381–391. doi:10.1016/j.jaut.2012.04.001
Suarez-Fueyo A, Barber DF, Martinez-Ara J, Zea-Mendoza AC, Carrera AC (2011) Enhanced phosphoinositide 3-kinase delta activity is a frequent event in systemic lupus erythematosus that confers resistance to activation-induced T cell death. J Immunol 187(5):2376–2385. doi:10.4049/jimmunol.1101602
Omori SA, Cato MH, Anzelon-Mills A, Puri KD, Shapiro-Shelef M, Calame K, Rickert RC (2006) Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity 25(4):545–557. doi:10.1016/j.immuni.2006.08.015
Omori SA, Rickert RC (2007) Phosphatidylinositol 3-kinase (PI3K) signaling and regulation of the antibody response. Cell Cycle 6(4):397–402. doi:10.4161/cc.6.4.3837
Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, Pearce W, Meek SE, Salpekar A, Waterfield MD et al (2002) Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 297(5583):1031–1034. doi:10.1126/science.1073560
Durand CA, Hartvigsen K, Fogelstrand L, Kim S, Iritani S, Vanhaesebroeck B, Witztum JL, Puri KD, Gold MR (2009) Phosphoinositide 3-kinase p110 delta regulates natural antibody production, marginal zone and B-1 B cell function, and autoantibody responses. J Immunol 183(9):5673–5684. doi:10.4049/jimmunol.0900432
Beer-Hammer S, Zebedin E, von Holleben M, Alferink J, Reis B, Dresing P, Degrandi D, Scheu S, Hirsch E, Sexl V et al (2010) The catalytic PI3K isoforms p110gamma and p110delta contribute to B cell development and maintenance, transformation, and proliferation. J Leukoc Biol 87(6):1083–1095. doi:10.1189/jlb.0809585
Dil N, Marshall AJ (2009) Role of phosphoinositide 3-kinase p110 delta in TLR4- and TLR9-mediated B cell cytokine production and differentiation. Mol Immunol 46(10):1970–1978. doi:10.1016/j.molimm.2009.03.010
Marone R, Cmiljanovic V, Giese B, Wymann MP (2008) Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta 1784(1):159–185. doi:10.1016/j.bbapap.2007.10.003
Rommel C, Camps M, Ji H (2007) PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol 7(3):191–201. doi:10.1038/nri2036
Camps M, Ruckle T, Ji H, Ardissone V, Rintelen F, Shaw J, Ferrandi C, Chabert C, Gillieron C, Francon B et al (2005) Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med 11(9):936–943. doi:10.1038/nm1284
Gonzalez-Martin A, Adams BD, Lai M, Shepherd J, Salvador-Bernaldez M, Salvador JM, Lu J, Nemazee D, Xiao C (2016) The microRNA miR-148a functions as a critical regulator of B cell tolerance and autoimmunity. Nat Immunol 17(4):433–440. doi:10.1038/ni.3385
Getahun A, Beavers NA, Larson SR, Shlomchik MJ, Cambier JC (2016) Continuous inhibitory signaling by both SHP-1 and SHIP-1 pathways is required to maintain unresponsiveness of anergic B cells. J Exp Med 213(5):751–769. doi:10.1084/jem.20150537
Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67:425–479. doi:10.1146/annurev.biochem.67.1.425
Chen J, Chen ZJ (2013) Regulation of NF-kappaB by ubiquitination. Curr Opin Immunol 25(1):4–12. doi:10.1016/j.coi.2012.12.005
Haglund K, Di Fiore PP, Dikic I (2003) Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem Sci 28(11):598–603. doi:10.1016/j.tibs.2003.09.005
Hitotsumatsu O, Ahmad RC, Tavares R, Wang M, Philpott D, Turer EE, Lee BL, Shiffin N, Advincula R, Malynn BA et al (2008) The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity 28(3):381–390. doi:10.1016/j.immuni.2008.02.002
Duan L, Reddi AL, Ghosh A, Dimri M, Band H (2004) The Cbl family and other ubiquitin ligases: destructive forces in control of antigen receptor signaling. Immunity 21(1):7–17. doi:10.1016/j.immuni.2004.06.012
Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, Oliveira-dos-Santos A, Mariathasan S, Bouchard D, Wakeham A, Itie A et al (2000) Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature 403(6766):211–216. doi:10.1038/35003228
Keane MM, Rivero-Lezcano OM, Mitchell JA, Robbins KC, Lipkowitz S (1995) Cloning and characterization of cbl-b: a SH3 binding protein with homology to the c-cbl proto-oncogene. Oncogene 10(12):2367–2377
Lupher ML Jr, Rao N, Eck MJ, Band H (1999) The Cbl protooncoprotein: a negative regulator of immune receptor signal transduction. Immunol Today 20(8):375–382
Ota Y, Beitz LO, Scharenberg AM, Donovan JA, Kinet JP, Samelson LE (1996) Characterization of Cbl tyrosine phosphorylation and a Cbl-Syk complex in RBL-2H3 cells. J Exp Med 184(5):1713–1723
Yasuda T, Maeda A, Kurosaki M, Tezuka T, Hironaka K, Yamamoto T, Kurosaki T (2000) Cbl suppresses B cell receptor-mediated phospholipase C (PLC)-gamma2 activation by regulating B cell linker protein-PLC-gamma2 binding. J Exp Med 191(4):641–650
Yasuda T, Tezuka T, Maeda A, Inazu T, Yamanashi Y, Gu H, Kurosaki T, Yamamoto T (2002) Cbl-b positively regulates Btk-mediated activation of phospholipase C-gamma2 in B cells. J Exp Med 196(1):51–63
Rao N, Dodge I, Band H (2002) The Cbl family of ubiquitin ligases: critical negative regulators of tyrosine kinase signaling in the immune system. J Leukoc Biol 71(5):753–763
Joazeiro CA, Wing SS, Huang H, Leverson JD, Hunter T, Liu YC (1999) The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science 286(5438):309–312
Drake L, McGovern-Brindisi EM, Drake JR (2006) BCR ubiquitination controls BCR-mediated antigen processing and presentation. Blood 108(13):4086–4093. doi:10.1182/blood-2006-05-025338
Katkere B, Rosa S, Caballero A, Repasky EA, Drake JR (2010) Physiological-range temperature changes modulate cognate antigen processing and presentation mediated by lipid raft-restricted ubiquitinated B cell receptor molecules. J Immunol 185(9):5032–5039. doi:10.4049/jimmunol.1001653
Katkere B, Rosa S, Drake JR (2012) The Syk-binding ubiquitin ligase c-Cbl mediates signaling-dependent B cell receptor ubiquitination and B cell receptor-mediated antigen processing and presentation. J Biol Chem 287(20):16636–16644. doi:10.1074/jbc.M112.357640
Zhang M, Veselits M, O’Neill S, Hou P, Reddi AL, Berlin I, Ikeda M, Nash PD, Longnecker R, Band H et al (2007) Ubiquitinylation of Ig beta dictates the endocytic fate of the B cell antigen receptor. J Immunol 179(7):4435–4443
Veselits M, Tanaka A, Lipkowitz S, O’Neill S, Sciammas R, Finnegan A, Zhang J, Clark MR (2014) Recruitment of Cbl-b to B cell antigen receptor couples antigen recognition to Toll-like receptor 9 activation in late endosomes. PLoS One 9(3):e89792. doi:10.1371/journal.pone.0089792
Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O et al (2004) The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 5(10):1052–1060. doi:10.1038/ni1110
Heyninck K, De Valck D, Vanden Berghe W, Van Criekinge W, Contreras R, Fiers W, Haegeman G, Beyaert R (1999) The zinc finger protein A20 inhibits TNF-induced NF-kappaB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-kappaB-inhibiting protein ABIN. J Cell Biol 145(7):1471–1482
Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A (2000) Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 289(5488):2350–2354
Shembade N, Ma A, Harhaj EW (2010) Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 327(5969):1135–1139. doi:10.1126/science.1182364
Beyaert R, Heyninck K, Van Huffel S (2000) A20 and A20-binding proteins as cellular inhibitors of nuclear factor-kappa B-dependent gene expression and apoptosis. Biochem Pharmacol 60(8):1143–1151
Bosanac I, Wertz IE, Pan B, Yu C, Kusam S, Lam C, Phu L, Phung Q, Maurer B, Arnott D, Kirkpatrick DS et al (2010) Ubiquitin binding to A20 ZnF4 is required for modulation of NF-kappaB signaling. Mol Cell 40(4):548–557. doi:10.1016/j.molcel.2010.10.009
Lu TT, Onizawa M, Hammer GE, Turer EE, Yin Q, Damko E, Agelidis A, Shifrin N, Advincula R, Barrera J et al (2013) Dimerization and ubiquitin mediated recruitment of A20, a complex deubiquitinating enzyme. Immunity 38(5):896–905. doi:10.1016/j.immuni.2013.03.008
Skaug B, Chen J, Du F, He J, Ma A, Chen ZJ (2011) Direct, noncatalytic mechanism of IKK inhibition by A20. Mol Cell 44(4):559–571. doi:10.1016/j.molcel.2011.09.015
Tokunaga F, Nishimasu H, Ishitani R, Goto E, Noguchi T, Mio K, Kamei K, Ma A, Iwai K, Nureki O (2012) Specific recognition of linear polyubiquitin by A20 zinc finger 7 is involved in NF-kappaB regulation. EMBO J 31(19):3856–3870. doi:10.1038/emboj.2012.241
Verhelst K, Carpentier I, Kreike M, Meloni L, Verstrepen L, Kensche T, Dikic I, Beyaert R (2012) A20 inhibits LUBAC-mediated NF-kappaB activation by binding linear polyubiquitin chains via its zinc finger 7. EMBO J 31(19):3845–3855. doi:10.1038/emboj.2012.240
Lin SC, Chung JY, Lamothe B, Rajashankar K, Lu M, Lo YC, Lam AY, Darnay BG, Wu H (2008) Molecular basis for the unique deubiquitinating activity of the NF-kappaB inhibitor A20. J Mol Biol 376(2):526–540. doi:10.1016/j.jmb.2007.11.092
Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL et al (2004) De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430(7000):694–699. doi:10.1038/nature02794
Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, Jang IK, Gutkind JS, Shevach E, Gu H (2000) Cbl-b regulates the CD28 dependence of T-cell activation. Nature 403:216(6766)–216(67220. doi:10.1038/35003235
Rudd CE, Schneider H (2000) Lymphocyte signaling: Cbl sets the threshold for autoimmunity. Curr Biol 10(9):R344–R347
Vereecke L, Beyaert R, van Loo G (2009) The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol 30(8):383–391. doi:10.1016/j.it.2009.05.007
Graham RR, Cotsapas C, Davies L, Hackett R, Lessard CJ, Leon JM, Burtt NP, Guiducci C, Parkin M, Gates C et al (2008) Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet 40(9):1059–1061. doi:10.1038/ng.200
Hughes LB, Reynolds RJ, Brown EE, Kelley JM, Thomson B, Conn DL, Jonas BL, Westfall AO, Padilla MA, Callahan LF et al (2010) Most common single-nucleotide polymorphisms associated with rheumatoid arthritis in persons of European ancestry confer risk of rheumatoid arthritis in African Americans. Arthritis Rheum 62(12):3547–3553. doi:10.1002/art.27732
Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, Gudjonsson JE, Li Y, Tejasvi T, Feng BJ et al (2009) Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet 41(2):199–204. doi:10.1038/ng.311
Eyre S, Hinks A, Bowes J, Flynn E, Martin P, Wilson AG, Morgan AW, Emery P, Steer S, Hocking LJ, Early Arthritis Consortium et al (2010) Overlapping genetic susceptibility variants between three autoimmune disorders: rheumatoid arthritis, type 1 diabetes and coeliac disease. Arthritis Res Ther 12(5):R175. doi:10.1186/ar3139
Fung EY, Smyth DJ, Howson JM, Cooper JD, Walker NM, Stevens H, Wicker LS, Todd JA (2009) Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locus. Genes Immun 10(2):188–191. doi:10.1038/gene.2008.99
Dieude P, Guedj M, Wipff J, Ruiz B, Riemekasten G, Matucci-Cerinic M, Melchers I, Hachulla E, Airo P, Diot E et al (2010) Association of the TNFAIP3 rs5029939 variant with systemic sclerosis in the European Caucasian population. Ann Rheum Dis 69(11):1958–1964. doi:10.1136/ard.2009.127928
Koumakis E, Giraud M, Dieude P, Cohignac V, Cuomo G, Airo P, Hachulla E, Matucci-Cerinic M, Diot E, Caramaschi P et al (2012) Brief report: Candidate gene study in systemic sclerosis identifies a rare and functional variant of the TNFAIP3 locus as a risk factor for polyautoimmunity. Arthritis Rheum 64(8):2746–2752. doi:10.1002/art.34490
Adrianto I, Wen F, Templeton A, Wiley G, King JB, Lessard CJ, Bates JS, Hu Y, Kelly JA, Kaufman KM et al (2011) Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet 43(3):253–258. doi:10.1038/ng.766
Boonyasrisawat W, Eberle D, Bacci S, Zhang YY, Nolan D, Gervino EV, Johnstone MT, Trischitta V, Shoelson SE, Doria A (2007) Tag polymorphisms at the A20 (TNFAIP3) locus are associated with lower gene expression and increased risk of coronary artery disease in type 2 diabetes. Diabetes 56(2):499–505. doi:10.2337/db06-0946
Elsby LM, Orozco G, Denton J, Worthington J, Ray DW, Donn RP (2010) Functional evaluation of TNFAIP3 (A20) in rheumatoid arthritis. Clin Exp Rheumatol 28(5):708–714
Koczan D, Drynda S, Hecker M, Drynda A, Guthke R, Kekow J, Thiesen HJ (2008) Molecular discrimination of responders and nonresponders to anti-TNF alpha therapy in rheumatoid arthritis by etanercept. Arthritis Res Ther 10(3):R50. doi:10.1186/ar2419
Nocturne G, Tarn J, Boudaoud S, Locke J, Miceli-Richard C, Hachulla E, Dubost JJ, Bowman S, Gottenberg JE, Criswell LA et al (2016) Germline variation of TNFAIP3 in primary Sjögren’s syndrome-associated lymphoma. Ann Rheum Dis 75(4):780–783. doi:10.1136/annrheumdis-2015-207731
Orozco G, Eyre S, Hinks A, Bowes J, Morgan AW, Wilson AG, Wordsworth P, Steer S, Hocking L, Consortium U et al (2011) Study of the common genetic background for rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis 70(3):463–468. doi:10.1136/ard.2010.137174
Wang S, Adrianto I, Wiley GB, Lessard CJ, Kelly JA, Adler AJ, Glenn SB, Williams AH, Ziegler JT, Comeau ME et al (2012) A functional haplotype of UBE2L3 confers risk for systemic lupus erythematosus. Genes Immun 13(5):380–387. doi:10.1038/gene.2012.6
Lewis M, Vyse S, Shields A, Boeltz S, Gordon P, Spector T, Lehner P, Walczak H, Vyse T (2015) Effect of UBE2L3 genotype on regulation of the linear ubiquitin chain assembly complex in systemic lupus erythematosus. Lancet 385(Suppl 1):S9. doi:10.1016/S0140-6736(15)60324-5
Browne EP (2012) Regulation of B-cell responses by Toll-like receptors. Immunology 136(4):370–379. doi:10.1111/j.1365-2567.2012.03587.x
Kawagoe T, Sato S, Matsushita K, Kato H, Matsui K, Kumagai Y, Saitoh T, Kawai T, Takeuchi O, Akira S (2008) Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat Immunol 9(6):684–691. doi:10.1038/ni.160
Negishi H, Fujita Y, Yanai H, Sakaguchi S, Ouyang X, Shinohara M, Takayanagi H, Ohba Y, Taniguchi T, Honda K (2006) Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc Natl Acad Sci U S A 103(41):15136–15141. doi:10.1073/pnas.0607181103
Schoenemeyer A, Barnes BJ, Mancl ME, Latz E, Goutagny N, Pitha PM, Fitzgerald KA, Golenbock DT (2005) The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J Biol Chem 280(17):17005–17012. doi:10.1074/jbc.M412584200
Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, Kano S, Honda K, Ohba Y, Mak TW et al (2005) Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 434(7030):243–249. doi:10.1038/nature03308
Jackson SW, Kolhatkar NS, Rawlings DJ (2015) B cells take the front seat: dysregulated B cell signals orchestrate loss of tolerance and autoantibody production. Curr Opin Immunol 33:70–77. doi:10.1016/j.coi.2015.01.018
Suurmond J, Diamond B (2015) Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest 125(6):2194–2202. doi:10.1172/JCI78084
Chaturvedi A, Dorward D, Pierce SK (2008) The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity 28(6):799–809. doi:10.1016/j.immuni.2008.03.019
Chaturvedi A, Pierce SK (2009) How location governs toll-like receptor signaling. Traffic 10(6):621–628. doi:10.1111/j.1600-0854.2009.00899.x
Berland R, Fernandez L, Kari E, Han JH, Lomakin I, Akira S, Wortis HH, Kearney JF, Ucci AA, Imanishi-Kari T (2006) Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity 25(3):429–440. doi:10.1016/j.immuni.2006.07.014
Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ (2005) Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med 202(2):321–331. doi:10.1084/jem.20050338
Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ (2006) Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 25(3):417–428. doi:10.1016/j.immuni.2006.07.013
Lartigue A, Courville P, Auquit I, Francois A, Arnoult C, Tron F, Gilbert D, Musette P (2006) Role of TLR9 in anti-nucleosome and anti-DNA antibody production in lpr mutation-induced murine lupus. J Immunol 177(2):1349–1354
Jackson SW, Scharping NE, Kolhatkar NS, Khim S, Schwartz MA, Li QZ, Hudkins KL, Alpers CE, Liggitt D, Rawlings DJ (2014) Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol 192(10):4525–4532. doi:10.4049/jimmunol.1400098
Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA (1994) A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr 125(6 Pt 1):876–885
Becker-Herman S, Meyer-Bahlburg A, Schwartz MA, Jackson SW, Hudkins KL, Liu C, Sather BD, Khim S, Liggitt D, Song W et al (2011) WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity. J Exp Med 208(10):2033–2042. doi:10.1084/jem.20110200
Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV (2006) TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med 203(3):553–561
Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, Smyth MJ, Mackay CR, Mackay F (2007) BAFF and MyD88 signals promote a lupus like disease independent of T cells. J Exp Med 204(8):1959–1971. doi:10.1084/jem.20062567
Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ (2013) Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity 38(3):528–540. doi:10.1016/j.immuni.2012.11.017
Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S (2007) Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 27(5):801–810. doi:10.1016/j.immuni.2007.09.009
Walsh ER, Pisitkun P, Voynova E, Deane JA, Scott BL, Caspi RR, Bolland S (2012) Dual signaling by innate and adaptive immune receptors is required for TLR7-induced B-cell-mediated autoimmunity. Proc Natl Acad Sci U S A 109(40):16276–16281. doi:10.1073/pnas.1209372109
Sun X, Wiedeman A, Agrawal N, Teal TH, Tanaka L, Hudkins KL, Alpers CE, Bolland S, Buechler MB, Hamerman JA et al (2013) Increased ribonuclease expression reduces inflammation and prolongs survival in TLR7 transgenic mice. J Immunol 190(6):2536–2543. doi:10.4049/jimmunol.1202689
Garcia-Ortiz H, Velazquez-Cruz R, Espinosa-Rosales F, Jimenez-Morales S, Baca V, Orozco L (2010) Association of TLR7 copy number variation with susceptibility to childhood-onset systemic lupus erythematosus in Mexican population. Ann Rheum Dis 69(10):1861–1865. doi:10.1136/ard.2009.124313
Kawasaki A, Furukawa H, Kondo Y, Ito S, Hayashi T, Kusaoi M, Matsumoto I, Tohma S, Takasaki Y, Hashimoto H et al (2011) TLR7 single-nucleotide polymorphisms in the 3′ untranslated region and intron 2 independently contribute to systemic lupus erythematosus in Japanese women: a case-control association study. Arthritis Res Ther 13(2):R41. doi:10.1186/ar3277
Shen N, Fu Q, Deng Y, Qian X, Zhao J, Kaufman KM, Wu YL, Yu CY, Tang Y, Chen JY et al (2010) Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci U S A 107(36):15838–15843. doi:10.1073/pnas.1001337107
Konsta OD, Le Dantec C, Charras A, Brooks WH, Arleevskaya MI, Bordron A, Renaudineau Y (2015) An in silico approach reveals associations between genetic and epigenetic factors within regulatory elements in B cells from primary Sjögren’s syndrome patients. Front Immunol 6:437. doi:10.3389/fimmu.2015.00437
Desnues B, Macedo AB, Roussel-Queval A, Bonnardel J, Henri S, Demaria O, Alexopoulou L (2014) TLR8 on dendritic cells and TLR9 on B cells restrain TLR7-mediated spontaneous autoimmunity in C57BL/6 mice. Proc Natl Acad Sci U S A 111(4):1497–1502. doi:10.1073/pnas.1314121111
Isnardi I, Ng YS, Srdanovic I, Motaghedi R, Rudchenko S, von Bernuth H, Zhang SY, Puel A, Jouanguy E, Picard C et al (2008) IRAK-4- and MyD88-dependent pathways are essential for the removal of developing autoreactive B cells in humans. Immunity 29(5):746–757. doi:10.1016/j.immuni.2008.09.015
Kurosaki T, Shinohara H, Baba Y (2010) B cell signaling and fate decision. Annu Rev Immunol 28:21–55. doi:10.1146/annurev.immunol.021908.132541
Mageed RA, Garaud S, Taher TE, Parikh K, Pers JO, Jamin C, Renaudineau Y, Youinou P (2012) CD5 expression promotes multiple intracellular signaling pathways in B lymphocyte. Autoimmun Rev 11(11):795–798. doi:10.1016/j.autrev.2012.02.007
Renaudineau Y, Hillion S, Saraux A, Mageed RA, Youinou P (2005) An alternative exon 1 of the CD5 gene regulates CD5 expression in human B lymphocytes. Blood 106(8):2781–2789. doi:10.1182/blood-2005-02-0597
Youinou P, Renaudineau Y (2007) The paradox of CD5-expressing B cells in systemic lupus erythematosus. Autoimmun Rev 7(2):149–154. doi:10.1016/j.autrev.2007.02.016
Daeron M, Lesourne R (2006) Negative signaling in Fc receptor complexes. Adv Immunol 89:39–86. doi:10.1016/S0065-2776(05)89002-9
Parry RV, Harris SJ, Ward SG (2010) Fine tuning T lymphocytes: a role for the lipid phosphatase SHIP-1. Biochim Biophys Acta 1804(3):592–597. doi:10.1016/j.bbapap.2009.09.019
Séïté JF, Cornec D, Renaudineau Y, Youinou P, Mageed RA, Hillion S (2010) IVIg modulates BCR signaling through CD22 and promotes apoptosis in mature human B lymphocytes. Blood 116(10):1698–1704. doi:10.1182/blood-2009-12-261461
Kumanogoh A, Watanabe C, Lee I, Wang X, Shi W, Araki H, Hirata H, Iwahori K, Uchida J, Yasui T et al (2000) Identification of CD72 as a lymphocyte receptor for the class IV semaphorin CD100: a novel mechanism for regulating B cell signaling. Immunity 13(5):621–631
Wu Y, Nadler MJ, Brennan LA, Gish GD, Timms JF, Fusaki N, Jongstra-Bilen J, Tada N, Pawson T, Wither J et al (1998) The B-cell transmembrane protein CD72 binds to and is an in vivo substrate of the protein tyrosine phosphatase SHP-1. Curr Biol 8(18):1009–1017
Li DH, Tung JW, Tarner IH, Snow AL, Yukinari T, Ngernmaneepothong R, Martinez OM, Parnes JR (2006) CD72 down-modulates BCR-induced signal transduction and diminishes survival in primary mature B lymphocytes. J Immunol 176(9):5321–5328
Shi W, Kumanogoh A, Watanabe C, Uchida J, Wang X, Yasui T, Yukawa K, Ikawa M, Okabe M, Parnes JR et al (2000) The class IV semaphorin CD100 plays nonredundant roles in the immune system: defective B and T cell activation in CD100-deficient mice. Immunity 13(5):633–642
Early GS, Zhao W, Burns CM (1996) Anti-CD40 ligand antibody treatment prevents the development of lupus-like nephritis in a subset of New Zealand black x New Zealand white mice. Response correlates with the absence of an anti-antibody response. J Immunol 157(7):3159–3164
Ma J, Xu J, Madaio MP, Peng Q, Zhang J, Grewal IS, Flavell RA, Craft J (1996) Autoimmune lpr/lpr mice deficient in CD40 ligand: spontaneous Ig class switching with dichotomy of autoantibody responses. J Immunol 157(2):417–426
Wang X, Huang W, Schiffer LE, Mihara M, Akkerman A, Hiromatsu K, Davidson A (2003) Effects of anti-CD154 treatment on B cells in murine systemic lupus erythematosus. Arthritis Rheum 48(2):495–506. doi:10.1002/art.10929
Kyburz D, Corr M, Brinson DC, Von Damm A, Tighe H, Carson DA (1999) Human rheumatoid factor production is dependent on CD40 signaling and autoantigen. J Immunol 163(6):3116–3122
Tellander AC, Michaelsson E, Brunmark C, Andersson M (2000) Potent adjuvant effect by anti-CD40 in collagen-induced arthritis. Enhanced disease is accompanied by increased production of collagen type-II reactive IgG2a and IFN-gamma. J Autoimmun 14(4):295–302. doi:10.1006/jaut.2000.0374
Baker RL, Wagner DH Jr, Haskins K (2008) CD40 on NOD CD4 T cells contributes to their activation and pathogenicity. J Autoimmun 31(4):385–392. doi:10.1016/j.jaut.2008.09.001
Balasa B, Krahl T, Patstone G, Lee J, Tisch R, McDevitt HO, Sarvetnick N (1997) CD40 ligand-CD40 interactions are necessary for the initiation of insulitis and diabetes in nonobese diabetic mice. J Immunol 159(9):4620–4627
Bour-Jordan H, Salomon BL, Thompson HL, Szot GL, Bernhard MR, Bluestone JA (2004) Costimulation controls diabetes by altering the balance of pathogenic and regulatory T cells. J Clin Invest 114(7):979–987. doi:10.1172/JCI20483
Green EA, Wong FS, Eshima K, Mora C, Flavell RA (2000) Neonatal tumor necrosis factor alpha promotes diabetes in nonobese diabetic mice by CD154-independent antigen presentation to CD8(+) T cells. J Exp Med 191(2):225–238
Gerritse K, Laman JD, Noelle RJ, Aruffo A, Ledbetter JA, Boersma WJ, Claassen E (1996) CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci U S A 93(6):2499–2504
Howard LM, Miga AJ, Vanderlugt CL, Dal Canto MC, Laman JD, Noelle RJ, Miller SD (1999) Mechanisms of immunotherapeutic intervention by anti-CD40L (CD154) antibody in an animal model of multiple sclerosis. J Clin Invest 103(2):281–290. doi:10.1172/JCI5388
Boon L, Brok HP, Bauer J, Ortiz-Buijsse A, Schellekens MM, Ramdien-Murli S, Blezer E, van Meurs M, Ceuppens J, de Boer M et al (2001) Prevention of experimental autoimmune encephalomyelitis in the common marmoset (Callithrix jacchus) using a chimeric antagonist monoclonal antibody against human CD40 is associated with altered B cell responses. J Immunol 167(5):2942–2949
Boon L, Laman JD, Ortiz-Buijsse A, den Hartog MT, Hoffenberg S, Liu P, Shiau F, de Boer M (2002) Preclinical assessment of anti-CD40 Mab 5D12 in cynomolgus monkeys. Toxicology 174(1):53–65
Laman JD, t Hart BA, Brok H, Meurs M, Schellekens MM, Kasran A, Boon L, Bauer J, Boer M, Ceuppens J (2002) Protection of marmoset monkeys against EAE by treatment with a murine antibody blocking CD40 (mu5D12). Eur J Immunol 32(8):2218–2228. doi:10.1002/1521-4141(200208)32:8<2218::AID-IMMU2218>3.0.CO;2-0
Boumpas DT, Furie R, Manzi S, Illei GG, Wallace DJ, Balow JE, Vaishnaw A, Group BGLNT (2003) A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum 48(3):719–727. doi:10.1002/art.10856
Ford ML, Adams AB, Pearson TC (2004) Targeting co-stimulatory pathways: transplantation and autoimmunity. Nat Rev Nephrol 10(1):14–24. doi:10.1038/nrneph.2013.183
Kalunian KC, Davis JC Jr, Merrill JT, Totoritis MC, Wofsy D, Group I-LS (2002) Treatment of systemic lupus erythematosus by inhibition of T cell costimulation with anti-CD154: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 46(12):3251–3258. doi:10.1002/art.10681
Zouali M, Sarmay G (2004) B lymphocyte signaling pathways in systemic autoimmunity: implications for pathogenesis and treatment. Arthritis Rheum 50(9):2730–2741. doi:10.1002/art.20487
Jiang Y, Hirose S, Sanokawa-Akakura R, Abe M, Mi X, Li N, Miura Y, Shirai J, Zhang D, Hamano Y, Shirai T (1999) Genetically determined aberrant down-regulation of FcgammaRIIB1 in germinal center B cells associated with hyper-IgG and IgG autoantibodies in murine systemic lupus erythematosus. Int Immunol 11(10):1685–1691
Li DH, Winslow MM, Cao TM, Chen AH, Davis CR, Mellins ED, Utz PJ, Crabtree GR, Parnes JR (2008) Modulation of peripheral B cell tolerance by CD72 in a murine model. Arthritis Rheum 58(10):3192–3204. doi:10.1002/art.23812
Healy JI, Dolmetsch RE, Timmerman LA, Cyster JG, Thomas ML, Crabtree GR, Lewis RS, Goodnow CC (1997) Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity 6(4):419–428
Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI (1997) Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386(6627):855–858. doi:10.1038/386855a0
Chiles TC (2004) Regulation and function of cyclin D2 in B lymphocyte subsets. J Immunol 173(5):2901–2907
Winslow MM, Gallo EM, Neilson JR, Crabtree GR (2006) The calcineurin phosphatase complex modulates immunogenic B cell responses. Immunity 24(2):141–152. doi:10.1016/j.immuni.2005.12.013
Ghoreschi K, Laurence A, O’Shea JJ (2009) Janus kinases in immune cell signaling. Immunol Rev 228(1):273–287. doi:10.1111/j.1600-065X.2008.00754.x
Harrison DA (2012) The Jak/STAT pathway. Cold Spring Harb Perspect Biol 4(3). doi:10.1101/cshperspect.a011205
Cantrell DA (2003) GTPases and T cell activation. Immunol Rev 192:122–130
Okkenhaug K, Fruman DA (2010) PI3Ks in lymphocyte signaling and development. Curr Top Microbiol Immunol 346:57–85. doi:10.1007/82_2010_45
O’Shea JJ, Paul WE (2010) Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 327(5969):1098–1102. doi:10.1126/science.1178334
Batten M, Groom J, Cachero TG, Qian F, Schneider P, Tschopp J, Browning JL, Mackay F (2000) BAFF mediates survival of peripheral immature B lymphocytes. J Exp Med 192(10):1453–1466
Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, Frew E, Scott ML (2001) An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science 293(5537):2111–2114. doi:10.1126/science.1061964
Marsters SA, Yan M, Pitti RM, Haas PE, Dixit VM, Ashkenazi A (2000) Interaction of the TNF homologues BLyS and APRIL with the TNF receptor homologues BCMA and TACI. Curr Biol 10(13):785–788
Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, Hession C, Schneider P, Sizing ID, Mullen C et al (2001) BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science 293(5537):2108–2111. doi:10.1126/science.1061965
Fairfax KA, Tsantikos E, Figgett WA, Vincent FB, Quah PS, LePage M, Hibbs ML, Mackay F (2015) BAFF-driven autoimmunity requires CD19 expression. J Autoimmun 62:1–10. doi:10.1016/j.jaut.2015.06.001
Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ (2009) Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev 229(1):152–172. doi:10.1111/j.1600-065X.2009.00782.x
Rickert RC, Jellusova J, Miletic AV (2011) Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunol Rev 244(1):115–133. doi:10.1111/j.1600-065X.2011.01067.x
Brightbill HD, Jackman JK, Suto E, Kennedy H, Jones C 3rd, Chalasani S, Lin Z, Tam L, Roose-Girma M, Balazs M et al (2015) Conditional deletion of NF-kappaB-inducing kinase (NIK) in adult mice disrupts mature B cell survival and activation. J Immunol 195(3):953–964. doi:10.4049/jimmunol.1401514
Mackay F, Schneider P (2009) Cracking the BAFF code. Nat Rev Immunol 9(7):491–502. doi:10.1038/nri2572
Otipoby KL, Sasaki Y, Schmidt-Supprian M, Patke A, Gareus R, Pasparakis M, Tarakhovsky A, Rajewsky K (2008) BAFF activates Akt and Erk through BAFF-R in an IKK1-dependent manner in primary mouse B cells. Proc Natl Acad Sci U S A 105(34):12435–12438. doi:10.1073/pnas.0805460105
Hobeika E, Levit-Zerdoun E, Anastasopoulou V, Pohlmeyer R, Altmeier S, Alsadeq A, Dobenecker MW, Pelanda R, Reth M (2015) CD19 and BAFF-R can signal to promote B-cell survival in the absence of Syk. EMBO J 34(7):925–939. doi:10.15252/embj.201489732
Schweighoffer E, Vanes L, Nys J, Cantrell D, McCleary S, Smithers N, Tybulewicz VL (2013) The BAFF receptor transduces survival signals by co-opting the B cell receptor signaling pathway. Immunity 38(3):475–488. doi:10.1016/j.immuni.2012.11.015
Lund FE (2008) Cytokine-producing B lymphocytes—key regulators of immunity. Curr Opin Immunol 20(3):332–338. doi:10.1016/j.coi.2008.03.003
Dang VD, Hilgenberg E, Ries S, Shen P, Fillatreau S (2014) From the regulatory functions of B cells to the identification of cytokine-producing plasma cell subsets. Curr Opin Immunol 28:77–83. doi:10.1016/j.coi.2014.02.009
Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, Ries S, Dang VD, Jaimes Y, Daridon C et al (2014) IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 507(7492):366–370. doi:10.1038/nature12979
Sweet RA, Lee SK, Vinuesa CG (2012) Developing connections amongst key cytokines and dysregulated germinal centers in autoimmunity. Curr Opin Immunol 24(6):658–664. doi:10.1016/j.coi.2012.10.003
Blenman KR, Duan B, Xu Z, Wan S, Atkinson MA, Flotte TR, Croker BP, Morel L (2006) IL-10 regulation of lupus in the NZM2410 murine model. Lab Investig 86(11):1136–1148. doi:10.1038/labinvest.3700468
Llorente L, Richaud-Patin Y, Garcia-Padilla C, Claret E, Jakez-Ocampo J, Cardiel MH, Alcocer-Varela J, Grangeot-Keros L, Alarcon-Segovia D, Wijdenes J et al (2000) Clinical and biologic effects of anti-interleukin-10 monoclonal antibody administration in systemic lupus erythematosus. Arthritis Rheum 43(8):1790–1800. doi:10.1002/1529-0131(200008)43:8<1790::AID-ANR15>3.0.CO;2-2
Watanabe R, Ishiura N, Nakashima H, Kuwano Y, Okochi H, Tamaki K, Sato S, Tedder TF, Fujimoto M (2010) Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. J Immunol 184(9):4801–4809. doi:10.4049/jimmunol.0902385
Capper ER, Maskill JK, Gordon C, Blakemore AI (2004) Interleukin (IL)-10, IL-1ra and IL-12 profiles in active and quiescent systemic lupus erythematosus: could longitudinal studies reveal patient subgroups of differing pathology? Clin Exp Immunol 138(2):348–356. doi:10.1111/j.1365-2249.2004.02607.x
Braun D, Caramalho I, Demengeot J (2002) IFN-alpha/beta enhances BCR-dependent B cell responses. Int Immunol 14(4):411–419
Graham RR, Kozyrev SV, Baechler EC, Reddy MV, Plenge RM, Bauer JW, Ortmann WA, Koeuth T, Gonzalez Escribano MF et al (2006) A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet 38(5):550–555. doi:10.1038/ng1782
Ronnblom L, Eloranta ML, Alm GV (2006) The type I interferon system in systemic lupus erythematosus. Arthritis Rheum 54(2):408–420. doi:10.1002/art.21571
Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, Morinobu A, Gadina M, O’Shea JJ, Biron CA (2002) Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science 297(5589):2063–2066. doi:10.1126/science.1074900
Graninger WB, Hassfeld W, Pesau BB, Machold KP, Zielinski CC, Smolen JS (1991) Induction of systemic lupus erythematosus by interferon-gamma in a patient with rheumatoid arthritis. J Rheumatol 18(10):1621–1622
Jackson SW, Jacobs HM, Arkatkar T, Dam EM, Scharping NE, Kolhatkar NS, Hou B, Buckner JH, Rawlings DJ (2016) B cell IFN-gamma receptor signaling promotes autoimmune germinal centers via cell-intrinsic induction of BCL-6. J Exp Med 213(5):733–750. doi:10.1084/jem.20151724
Ettinger R, Kuchen S, Lipsky PE (2008) Interleukin 21 as a target of intervention in autoimmune disease. Ann Rheum Dis 67(Suppl 3):iii83–iii86. doi:10.1136/ard.2008.098400
Moisini I, Davidson A (2009) BAFF: a local and systemic target in autoimmune diseases. Clin Exp Immunol 158(2):155–163. doi:10.1111/j.1365-2249.2009.04007.x
Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, Tschopp J, Browning JL (1999) Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med 190(11):1697–1710
Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu HB, Cyster JG (2004) Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity 20(4):441–453
Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, Brink R (2004) Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity 20(6):785–798. doi:10.1016/j.immuni.2004.05.010
Haapaniemi EM, Kaustio M, Rajala HL, van Adrichem AJ, Kainulainen L, Glumoff V, Doffinger R, Kuusanmaki H, Heiskanen-Kosma T, Trotta L et al (2015) Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT3. Blood 125(4):639–648. doi:10.1182/blood-2014-04-570101
Kotkowska A, Sewerynek E, Domańska D, Pastuszak-Lewandoska D, Brzeziańska E (2015) Single nucleotide polymorphisms in the STAT3 gene influence AITD susceptibility, thyroid autoantibody levels, and IL6 and IL17 secretion. Cell Mol Biol Lett 20(1):88–101. doi:10.1515/cmble-2015-0004
Nakayamada S, Kubo S, Iwata S, Tanaka Y (2016) Chemical JAK inhibitors for the treatment of rheumatoid arthritis. Expert Opin Pharmacother. doi:10.1080/14656566.2016.1241237
Deng GM, Kyttaris VC, Tsokos GC (2016) Targeting Syk in autoimmune rheumatic diseases. Front Immunol 7(7):78–82. doi:10.3389/fimmu.2016.00078
Schwartz DM, Bonelli M, Gadina M, O’Shea JJ (2016) Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol 12(1):25–36. doi:10.1038/nrrheum.2015.167
Lazaro E, Scherlinger M, Truchetet ME, Chiche L, Schaeverbeke T, Blanco P, Richez C (2016) Biotherapies in systemic lupus erythematosus: new targets. Joint Bone Spine 20(16):30123–30133. doi:10.1016/j.jbspin.2016.07.004
Lazzerini PE, Capecchi PL, Guidelli GM, Selvi E, Acampa M, Laghi-Pasini F (2016) Spotlight on sirukumab for the treatment of rheumatoid arthritis: the evidence to date. Drug Des Devel Ther 26(10):3083–3098
Burmester GR, Rigby WF, van Vollenhoven RF, Kay J, Rubbert-Roth A, Kelman A, Dimonaco S, Mitchell N (2016) Tocilizumab in early progressive rheumatoid arthritis: FUNCTION, a randomised controlled trial. Ann Rheum Dis 75(6):1081–1091. doi:10.1136/annrheumdis-2015-207628
Oon S, Wilson NJ, Wicks I (2016) Targeted therapeutics in SLE: emerging strategies to modulate the interferon pathway. Clin Transl Immunology 5(5):e79. doi:10.1038/cti.2016.26
Achek A, Yesudhas D, Choi S (2016) Toll-like receptors: promising therapeutic targets for inflammatory diseases. Ach Pharm Res 39(8):1032–1049. doi:10.1007/s12272-016-0806-9
Chalmers SA, Doerner J, Bosanac T, Khalil S, Smith D, Harcken C, Dimock J, Der E, Herlitz L, Webb D et al (2016) Therapeutic blockade of immune complex-mediated glomerulonephritis by highly selective inhibition of Bruton’s tyrosine kinase. Sci Rep 6:26164. doi:10.1038/srep26164
Park JK, Byun JY, Park JA, Kim YY, Lee YJ, Oh JI, Jang SY, Kim YH, Song YW, Son J et al (2015) HM71224, a novel Bruton’s tyrosine kinase inhibitor, suppresses B cell and monocyte activation and ameliorates arthritis in a mouse model: a potential drug for rheumatoid arthritis. Arthritis Res Ther 18:91. doi:10.1186/s13075-016-0988-z
Acknowledgments
Grant UC5 from the Raynaud’s and Scleroderma Association (new name: Scleroderma research-UK) awarded to Rizgar Mageed and David Abraham.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Funding
Dr. Taher E Taher was funded by grant UC5 from Scleroderma and Raynaud’s-UK (SR-UK) awarded to Professors Rizgar Mageed and David Abraham.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Taher, T.E., Bystrom, J., Ong, V.H. et al. Intracellular B Lymphocyte Signalling and the Regulation of Humoral Immunity and Autoimmunity. Clinic Rev Allerg Immunol 53, 237–264 (2017). https://doi.org/10.1007/s12016-017-8609-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-017-8609-4