
Abstract
Background
Giant cell lesions are locally aggressive intraosseous neoplasms with capacity to metastasize. The role of immune surveillance in the pathophysiology of giant cell lesions is poorly understood, and understanding what role the immune system plays in giant cell lesions may lead to the development of more effective treatment. The aim of this study was to explore the role of immune surveillance in giant cell lesions by examining the expression of the HLA class I and class II antigens and tumor infiltrating lymphocytes. In addition, we examined the role of the immune modulating surface antigen B7-H3, which belongs to the B7 superfamily, a group of molecules that modulates T-cell responses.
Questions/Purposes
(1) Is an immune response elicited by giant cell lesions? (2) Do clinically relevant human leukocyte antigen (HLA) defects exist in giant cell lesions? (3) Is B7-H3 a clinically relevant immune modulator?
Methods
The study sample was derived from the population of patients presenting to the Massachusetts General Hospital for evaluation and management of giant cell lesions from 1993 to 2008. We included patients with histologically confirmed giant cell lesions with a minimum followup of 6 months. Patients with systemic diseases (n = 4 [3%]), syndromes associated with giant cell lesions (n = 4 [3%]), and those without sufficient followup (n = 26 [19%]), inadequate records (n = 7 [5%]), or inadequate tissue available (n = 2 [1%]) were excluded. Tissue microarray, containing 288 tissue cores for 93 patients, was carefully constructed. This contained tissue from 45 patients with maxillofacial lesions, 38 with aggressive and seven with nonaggressive lesions, and 48 patients with axial and appendicular lesions, 30 with aggressive lesions and 18 with nonaggressive lesions. The population mean age was 28 ± 12 years and the duration of followup was 4 ± 3 years. The tissue microarray was immunohistochemically stained with monoclonal antibodies specific for HLA classes I and II and B7-H3 antigens and analyzed for tumor infiltrating lymphocytes. Antigen expression was examined in multinucleated giant cells and mononuclear stromal cells. The results were correlated with local invasion and tumor aggressiveness, which is based on accepted staging criteria.
Results
Tumor infiltrating lymphocytes were detected in all the tumors. The mean number of CD8+ T cell infiltration was lower in aggressive tumors (median, 4.8; interquartile range [IQR], 0.4–13.4), when compared with nonaggressive tumors (median, 15.8; IQR, 4.3–46.3; p = 0.007). HLA class I antigens were highly expressed by multinucleated giant cells in all tumors, but were lightly expressed on mononuclear stromal cells in 53% (45 of 84) to 73% (56 of 77) of tumors. HLA class I antigen low expression in mononuclear stromal cells was associated with tumor aggressiveness (odds ratio [OR], 4.3; p = 0.005). Low HLA class I expression combined with low CD8+ T cell infiltration was most highly associated with tumor aggressiveness (OR, 7.81; p = 0.011). B7-H3 antigen was expressed in 36.9% mononuclear stroma cells and also was associated with local tumor invasion (OR, 1.36; p < 0.001). Similarly, giant cell lesions with high B7-H3 expression and low CD8+ tumor infiltrating lymphocytes were associated with increased tumor aggressiveness (OR, 8.89; p = 0.0491).
Conclusions
Locally aggressive giant cell lesions are associated with low HLA class 1 antigen expression, low CD8+T cell infiltration, and high expression of the immune modulator B7-H3.
Clinical Relevance
Failure of immune surveillance implies that there may be an opportunity to target aspects of the immune surveillance machinery to treat giant cell lesions.
Similar content being viewed by others

Introduction
Giant cell lesions of bone are relatively rare tumors with low metastatic potential but they can be locally aggressive [2, 3, 29]. Giant cell lesions include central giant cell lesions of the maxillofacial skeleton and of the axial and appendicular skeleton [42]. Clinical and radiographic criteria are used to stratify giant cell lesions into aggressive and nonaggressive tumors as histologic criteria have not been correlated with tumor behavior [2, 18, 38]. Surgery remains the dominant approach but systemic therapy targeting receptor activator of nuclear factor kappa-B ligand (RANKL) with a monoclonal antibody (denosumab) has been shown to be an effective adjuvant [6]. However, the long-term effect of systemically targeting RANKL is poorly understood and it is not curative as giant cell lesions recur once the therapy is stopped [22]. This has prompted further investigation in the pathophysiology of giant cell lesions with the hopes of identifying other targets. Histologically, there are mainly two cell populations in giant cell lesions that can act as potential targets for novel therapies. These are multinucleated osteoclastlike giant cells and mononuclear, spindle-shaped fibroblastlike, stromal cells [20, 23, 27]. Mononuclear stromal cells are thought to be the neoplastic cell as the multinucleated giant cells disappear with denosumab treatment yet the mononuclear stromal cells persist, and mononuclear stromal cells can be indefinitely propagated in culture [57]. For these reasons, it is useful to consider mononuclear stromal cells separately from multinucleated giant cells.
The role of immune surveillance in giant cell tumors of bone was suggested in an in vitro analysis of lymphocytic activation where radiologically more-aggressive tumors had decreased lymphocytic activation [35]. The role of immune surveillance has regained importance as tumor infiltrating lymphocytes have been correlated with outcomes in several cancers. Unfortunately, many cancer cells have evolved tumor escape mechanisms to avoid immune surveillance such as defects in human leukocyte antigen (HLA) class I antigen processing machinery components leading to ineffective presentation of tumor antigen-derived peptide complexes to their cognate T cell receptor [9], and blocking activation of cytotoxic lymphocytes via the interaction of immune modulators such as B7-H3 [41].The HLA class I molecule is a heterodimer that consists of two polypeptide heavy α chains and a β2-microglobulin light chain. However, multiple intracellular molecules are needed to effectively present antigens to their cognate T cells, and defects in the intracellular processing of antigens and other stages of antigen processing machinery also have been correlated with poor survival in several malignancies [32, 47]. Despite having functional HLA class I antigen, some tumors can still escape the immune system by expressing coinhibitory molecules that deactivate cytotoxic lymphocytes cells. B7-H3 is a member of the B7 family of molecules involved in immune regulation and is overexpressed on a wide variety of solid tumor types [41]. It has been shown that B7-H3 exerts an immunosuppressive action in various cancers [11, 55, 56]. The aims of our study were to assess the frequency of HLA class I antigen defects in a large collection of well-annotated giant cell lesions, and to correlate these results with lymphocyte infiltration and the clinical course of the disease. In addition, we explored the immunologic role of B7-H3 and its association with local invasion in giant cell lesions.
Therefore we asked: (1) Was an immune response elicited by giant cell lesions? (2) Did clinically relevant HLA defects exist in giant cell lesions? (3) Was B7-H3 a clinically relevant immune modulator?
Methods
Study Design and Setting
This was a longitudinal study of a cohort of patients with giant cell lesions of bone treated at Massachusetts General Hospital during a 15-year period (January 1993 to June 2008).
Participants and Study Subjects
Subjects were identified through the Massachusetts General Hospital giant cell patient registry [38, 39]. Patients were included if they had histologically confirmed central giant cell lesions of the maxillofacial skeleton that were treated by the Department of Oral and Maxillofacial Surgery or axial and appendicular giant cell tumors that were treated by the Department of Orthopaedic Surgery, with a minimum followup of 6 months. Histologic examination results were confirmed as reported by the official interpretation of our musculoskeletal pathologist (WCF) during clinical care of the patient. Patients with followup less than 6 months (n = 26) were excluded. Patients with systemic diseases (n = 4), such as hyperparathyroidism, and giant cell lesions associated syndromes (n = 4), such as cherubism, and Noonan syndrome, also were excluded as these conditions are known to be associated with giant cell lesions and may represent a separate pathogenesis from a solitary giant cell lesion. Patients also were excluded if insufficient tumor tissue was available (n = 2), or if insufficient documentation was encountered (n = 7). A total of 43 patients with mean age of 36 ±14 years (52% were female patients) were excluded. Ninety-three patients with a mean age of 28 ± 12 years (56% were female patients) who underwent surgical resection of their giant cell lesions were included in this study (Table 1). Any bias inherent in this process was unintentional. It seems unlikely that bias inherent in this retrospective study altered the outcome sufficiently. The project was approved by the institutional review board (protocol 2008-P-000563).
Description of Experiment, Treatment, or Surgery
Selected tissue blocks and accompanying hematoxylin and eosin-stained slides were reviewed by a senior musculoskeletal pathologist (WCF) to assess for adequate tumor tissue. A representative region of each tumor block was identified and extracted to build the tissue microarray. One-millimeter cores were extracted with a maximum of three cores per block. Cores from human cartilage, human spleen, human liver, human lymph node, human melanoma metastasis, melanoma xenograft, and mouse liver also were included in the tissue microarray. Monoclonal antibodies (mAb) hydroxycarboxylic acid receptors (HCA2), which recognize β2-microglobulin-free HLA-A (excluding-A24), -B7301, and -G heavy chains [48, 49]; mAb HC-10, which recognizes β2-microglobulin-free HLA-A3, -A10, -A28, -A29, -A30, -A31, -A32, -A33, and all β2-microglobulin-free-HLA-B (excluding -B5702, -B5804, and -B73) and HLA-C heavy chains [40, 48, 49]; and mAb NAMB-1, which recognize HLA class I heavy chain- associated and free β2 microglobulin [7]; and mAb LGII-612.14 which recognizes a monomorphic epitope expressed on the β chain of HLA-DR, -DQ, and -DP antigens [51] were developed and characterized as described. mAbs were purified from ascitic fluid by affinity chromatography on a Protein G column (GE Healthcare Life Sciences, Pittsburgh, PA, USA). The purity and activity of mAb preparations was monitored by sodium dodecyl sulfate polyacrylamide gel electrophoresis and by binding assays with the cognate antigen, respectively. The B7-H3-specific mAb 1027 was obtained from R&D Systems (Minneapolis, MN, USA) [10, 15, 53]. A binding assay using purified recombinant B7-H3 was used to validate the affinity of the B7-H3-specific mAb 1027 to B7-H3. The human CD8 (clone 4B12) and CD4-specific (EPR6844) mAbs were obtained from DAKO (Carpinteria, CA, USA) and Abcam (Cambridge, MA, USA), respectively.
Secondary Antibody System
The DAKO EnVisionTM+System horseradish peroxidase-labeled polymer antimouse (K4001) kit was obtained from DAKO.
Immunohistochemical Staining
Four micrometers of formalin-fixed, paraffin-embedded tissue sections from the giant cell lesions tissue microarray block were used as substrates in immunohistochemical reactions. Tissue microarrays were stained with HLA class I-, HLA class II- and B7-H3-specific mAb as previously described [37]. Staining intensity and percentage of stained tumor cells in each lesion were reviewed and enumerated by two investigators (CH, LC) who had no knowledge of the patients’ characteristics or clinical outcomes. Results were graded as positive, heterogeneous, or negative when the total score in an entire lesion was greater than 75, 75 to 25, and less than 25, respectively [54]. Staining with CD8- and CD4-specific mAbs was performed according to the manufacturer’s (DAKO) instructions, and the results of staining were calculated by counting the number of stained infiltrating cells in four high-powered fields (×40) of the giant cell lesion cores. Giant cell lesions are formed principally by two cell types which are multinucleated osteoclastlike giant cells and mononuclear, spindle-shaped fibroblastlike, stromal cells [57]. Immunohistochemical staining was scored individually for each of the two cellular subtypes.
Clinical Data Stratification of Aggressive Versus Nonaggressive Lesions
We used Enneking’s staging system for benign bone tumors [18]. This system focuses on the clinical behavior of the tumors. Inactive lesions are designated Stage I, whereas tumors that are active (show progressive growth, radiologic deformation of the bony cortex) are Stage II, and aggressive lesions (destructive lesions with a soft tissue mass) are Stage III. For the purpose of our study, we classified the tumor lesions as either aggressive or nonaggressive, as described previously [42]. Briefly, for axial and appendicular lesions all Enneking Stage III tumors and any Stage II tumors with pathologic fracture and/or recurrence were considered aggressive. All other axial and appendicular tumors were categorized as nonaggressive [42]. Central giant cell lesions of the maxillofacial skeleton were characterized as aggressive or nonaggressive according to the clinical and radiographic criteria defined by Chuong et al. [13] in 1986 and modified by Kaban et al. [28] in 2007.
Research Questions
The first question we asked is whether the immune system is recognizing and responding to the giant cell lesions. One way to assess whether the host is responding to tumor cells is to assess whether CD8+ lymphocytes are infiltrating the tumors. Many of our normal cells express HLA class I antigens on their cell surface and it is here that they present antigens to CD8+ lymphocytes. If the antigen presented by the HLA expressing cell is abnormal, then the CD8+ lymphocyte will destroy the tumor cell. This is part of the normal immunosurveillance function of our immune system [16]. This leads to our second question, which is whether HLA class I antigen expression was low in our aggressive giant cell lesions. If HLA class I antigen expression is low in our tumor cells, CD8+ lymphocytes will not be able to detect the tumor cells, and therefore they will not be activated nor will they mount an immune response and the tumor cells will be able to evade this aspect of the immune system leading to a more aggressive phenotype [9]. The last question we asked pertains to the B7-H3 molecule which is an immune-modulator [35]. Immune-modulators are an important mechanism by which our host prevents an overzealous response by lymphocytes, which, if left unchecked, could lead to autoimmune disease. These immune-modulators are essentially check points that can suppress or stimulate the host’s response. Unfortunately, some cancers have adopted the immune suppressive effects of inhibitory immune-modulators, such as B7-H3, which allows them to escape the immune response and invade surrounding tissues [34]. We examined the association between B7-H3 and tumor aggressiveness and local tumor invasion.
Statistical Analysis and Study Size
Statistical analyses were performed using IBM SPSS statistical software Version 20.0 (IBM Corporation, Armonk, NY, USA). A chi-square test was used to assess association between the different parameters, and the Mann-Whitney U test was used to compare means of lymphocyte cell infiltration. Spearman’s rank correlation coefficient was used to assess correlations. A probability less than 0.05 was considered statistically significant.
Results
Was an Immune Response Elicited by Giant Cell Lesions?
Tumor infiltrating lymphocytes in primary giant cell lesions CD4+ and CD8+ cell infiltration were found in 90% (76 of 85) and 84% (71 of 85) of giant cell lesions, respectively. The number of CD4+ cells ranged between 0 and 93 (mean number, 14.3), whereas that of CD8+ cells ranged between 1 and 184 (mean number, 24.2). Staining patterns of giant cell lesions with CD4+ and CD8+ tumor infiltrating lymphocytes are shown (Fig. 1). There was a weak positive correlation between CD4+ and CD8+ cell infiltration (correlation coefficient, 0.224; p = 0.039). Interestingly, the number of CD8+ cells was lower in aggressive tumors (median, 4.8; interquartile range [IQR], 0.4–13.4) when compared with nonaggressive tumors (median, 15.8; IQR, 4.3–46.3; p = 0.007) (Fig. 2). We then investigated the prognostic value of HLA class I antigen expression in combination with CD8+ T cell infiltration level. Low expression of HLA-B and -C antigens in combination with low CD8+ T cell infiltration was associated with highest risk of tumor aggressiveness (odds ratio [OR],7.81; 95% CI, 1.4–43.5; p = 0.011) when compared with high HLA-B and -C antigen expression and high CD8+ T cell infiltration (Fig. 3).

Representative chromogenic immunohistochemistry staining results of the giant cell lesion tissue microarray are shown for CD4+ and CD8+ tumor infiltrating lymphocytes. The staining was performed using CD8+ and CD4+ specific mAbs according to the manufacturer’s instructions (DAKO, Carpinteria, CA, USA). The results of the staining were calculated by counting the number of stained infiltrating cells in four high-powered fields (×40) in each tissue microarray core. Representative high T cell infiltration and low T cell infiltration are shown. The red arrows point to the stained T cells.

The boxplot shows a comparison of CD8+ tumor infiltrating cell count between aggressive and nonaggressive giant cell lesions of the bone. Aggressive tumors show a significantly (p = 0.007) low number of CD8+ tumor infiltrating cells (median, 4.8; IQR, 0.4–13.4) compared with nonaggressive tumors (median, 15.8; IQR, 4.3–46.3).

The bar graph shows the relative number of giant cell lesions with high HLA-B and -C expression and high CD8+ tumor infiltrating cells (TILs) versus low HLA-B and -C expression and low CD8+ TILs stratified by tumor aggressiveness. HLA-B and -C antigen low expression in combination with low CD8+ T cell infiltration was associated with highest risk of tumor aggressiveness (OR, 7.81; 95% CI, 1.4–43.5; p = 0.011) when compared with high HLA-B and -C antigen expression and high CD8+ T cell infiltration.
Did Clinically Relevant HLA Defects Exist in Giant Cell Lesions?
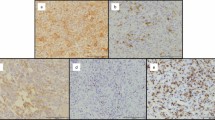
Expression of HLA-A antigens and HLA-B and -C antigens in mononuclear stromal cells were low and heterogeneous in 68% (56 of 83) and 53% (45 of 84) of the tumors, respectively. Similarly, expression of HLA-A antigens and HLA-B and -C antigens in multinucleated giant cells were low and heterogeneous in 65% (49 of 75) and 73% (56 of 77) of the tumors, respectively. There was no difference in the percentage of HLA-A and HLA-B and -C expression in mononuclear stromal cells (percentage difference, 1%; 95% CI, 12%–14%; p = 0.85) and multinucleated giant cells (percentage difference, 2%; 95% CI, −17% to 20%; p = 0.83). Representative staining patterns of giant cell lesions with HLA-A-specific mAb HCA2 and with HLA-B and -C specific mAb HC-10 are shown (Fig. 4). Beta 2-microglobulin expression was low and heterogeneous in 89% (75 of 84) of the mononuclear stromal cells and 51% (39 of 76) of the multinucleated giant cells. Low β2-microglobulin expression level in mononuclear stromal cells was associated with low HLA-A heavy chain (OR, 22.05; 95% CI, 4.00–121.58; p < 0.001) and low HLA-B and -C heavy chain (OR, 13.00; 95% CI, 2.79–60.49; p < 0. 001). Similarly, low β2-microglobulin expression level in multinucleated giant cells of β2-microglobulin was associated with low HLA-A heavy chain (OR, 7.81; 95% CI, 1.4–43.5; p = 0.011) and low HLA-B and -C heavy chain expression (OR, 7.81; 95% CI, 1.4–43.5; p = 0.011). Low expression of HLA-B and -C heavy chain, but not HLA-A heavy chain was associated with aggressive tumors (OR, 4.30; 95% CI, 1.50–12.35; p = 0.005) (Table 2). Similarly, low and heterogeneous β2-microglobulin expression was associated with aggressive tumors (OR, 4.67; 95% CI, 1.29–12.35; p = 0.031). However, when adjusting the data for HLA-B and -C heavy chain expression, we observed no association between low and heterogeneous β2-microglobulin and aggressive tumors (OR, 0.25; 95% CI, 0.03– 2.35; p = 0.383). HLA class II antigens were expressed by mononuclear stromal cells in 69% (58 of 84) of the giant cell lesions; the expression was scored as positive in 26% (22 of 84) lesions and heterogeneous in 43% (36 of 84). HLA class II antigen expression in mononuclear stromal cells was not associated with HLA class I antigen expression in mononuclear stromal cells. However, high expression of HLA class II in mononuclear stromal cells was associated with local tumor invasion to surrounding soft tissue (OR, 6.56; 95% CI, 1.40–30.56; Fisher’s exact test, p = 0.019). HLA class II antigen expression was low or negative in 75 of the 77 samples tested. Two samples were heterogenous and none of them had high expression. HLA class II antigen expression was not associated with HLA class I, or with either CD4+ or CD8+ T cell infiltration.

Representative chromogenic immunohistochemistry staining results of the giant cell lesions tissue microarray are shown for the five immunologic markers HLA class I HLA-A heavy chain (stained using hydroxycarboxylic acid receptors (HCA2) mAb), HLA class I HLA-B and -C (stained using HC-10 mAb), HLA class I β-2 using microglobulin (β2m) light chain (stained using namb-1 mAb), HLA class II (stained using lgii-612.14 mAb), and B7-H3 (stained using B7-H3-specific mAb 1027). Each tissue microarray core was scored as low, heterogeneous, or high if the percentage of the stained area lesion was less than 25%, 25% to 75%, and greater than 75%, respectively.
Was B7-H3 a Clinically and Immunologically Relevant Marker?
B7-H3 was expressed in all giant cell lesions (100%; n = 83). The expression was low and heterogeneous in 63% (52 of 83), heterogeneous in 60% (50 of 83), and low in 3% (two of 83); and in multinucleated giant cells in 51% (39 of 76), heterogeneous in 41% (31 of 76), and low in 10% (eight of 76) of tumors. High B7-H3 expression in mononuclear stromal cells was associated with high HLA-B and -C heavy chain expression (OR, 2.9; 95% CI, 1.16–7.33; p = 0.022), but not with HLA-A heavy chain expression. High B7-H3 expression in mononuclear stromal cells was associated with high CD8+ cell infiltration (OR, 3.64; 95% CI, 1.4–9.38; p = 0.006), but not with CD4+ cell infiltration. High B7-H3 expression in mononuclear stromal cells was associated with high HLA class II antigen expression (OR, 6.32; 95% CI, 1.70–23.50; p = 0.003). B7-H3 expression, by itself, was not associated with tumor aggressiveness as defined in Methods (OR, 1.42; 95% CI, 0.51–3.97; p = 0.50). However, high B7-H3 expression was associated with local tumor invasion (soft tissue mass) (relative risk [RR], 3.12; 95% CI, 2.13–4.54; p < 0.001). When stratifying the data according to CD8+ cell infiltration (high versus low), the association between high B7-H3 and local tumor invasion was seen only in the high CD8 T cell infiltration group (RR, 1.97; 95% CI, 1.2–4.27; p = 0.028). When stratifying the data according to the expression of B7-H3 and CD8+ tumor infiltrating lymphocytes in mononuclear stromal cells, the high B7-H3 expression combined with low CD8+ tumor infiltrating cells stratum was associated with increased risk of having an aggressive tumor when compared with low B7-H3 expression and high CD8+ tumor infiltrating cells stratum (OR, 8.89; p = 0.0491) (Fig. 5).

The bar graph shows the relative number of giant cell lesions and their respective expression of B7-H3 and CD8+ tumor infiltrating cells stratified by tumor aggressiveness. The giant cell lesions with low B7-H3 and low CD8+ tumor infiltrating cells were statistically more likely to be aggressive tumors (p < 0.001).
Discussion
The role of the immune system in giant cell lesions is unknown. However, in general, there has been renewed interest in the role of the immune system in how cancers progress with the hopes of improving our understanding and possibly identifying targets for therapy [16]. Clinical trials have shown encouraging results in a subset of patients with various types of malignancies [17, 21, 25, 43, 50]. Specifically, the administration of mAbs which blocks interaction of immune regulatory checkpoint molecules such as programmed cell death protein 1 (PD-1) with their ligands programmed cell death ligand 1 (PD-L1), have shown a major effect on the clinical course of various types of malignancies [1, 5, 12, 25, 26, 52]. However the efficacy of this novel immunotherapeutic strategy is limited to less than 30% of patients [45]. These findings have stimulated an interest in exploring other immune regulatory checkpoints such as B7-H3 and characterizing the host’s immune response. Currently there are three ongoing Phase 1 clinical trials (NCT02381314, NCT02475213, NCT02982941) that are evaluating enoblituzumab, which is a monoclonal antibody that targets B7-H3 that is overly expressed by tumor cells and its associated vasculature to mount antibody-dependent cell-mediated cytotoxicity [33]. We sought to identify the role of the immune system in giant cell lesions by studying the expression of components of HLA classes I and II and the immune modulator B7-H3. Our study provides, for the first time to our knowledge, evidence that giant cell lesions may escape immune surveillance secondary to defects in HLA class I expression and the inhibitory effect of B7-H3.
Limitations
This study has several limitations worth noting. First, we chose to combine central giant cell lesions of the maxillofacial skeleton with giant cell tumors of the axial and appendicular skeleton. This has been a controversial choice and there have been reports that suggest that these tumors are molecularly distinct. One study failed to show a histone mutation in central giant cell lesions (n = 9) [24] that was reported in 92% of giant cell tumors (49 of 53) [4]. However, these two studies were not methodologically equivalent, as one study separated the giant cells from stromal cells [4] and the other did not [24]. Furthermore, it is possible that other mutations in histones would be seen if whole genome sequencing had been performed. Furthermore, it is unclear what the functional importance of these histone mutations is as neither group assessed the corresponding protein expression. We believe that there are enough similarities between these groups that combining them makes clinical sense. This is particularly true for aggressive giant cell lesions.
It was reported that giant cell lesions in the axial and appendicular skeleton are more aggressive than those seen in the jaw [19]. However, in the jaw, there are many, very small nonaggressive lesions that, if lumped together with aggressive lesions, lead to an underestimation of the aggressiveness of the latter group. By excluding the indolent groups from the jaw, the two groups become comparable, which is what we have seen [38].
In a previous study, Resnick et al. [42] showed that if you look at aggressive giant cell lesions of the jaws and aggressive giant cell lesions of the axial and appendicular skeleton, they are similarly aggressive—except that a low percentage of the axial and appendicular lesions metastasize. In that study, the classification of Chuong et al. [13] was used which categorizes giant cell lesions in two categories as aggressive and nonaggressive. Using this model, clinical and radiologic features of the Enneking classification [18] were converted to a binary system (aggressive and nonaggressive). The theory was that when you categorize giant cell lesions based on their behavior (ie, aggressive versus nonaggressive), they are found to behave similarly in both groups. Resnick et al. [42] hypothesized that “maxillofacial and axial and appendicular giant cell lesions represent the same entity when assessed by comparable phenotypic parameters, comparing the clinical behavior of lesions from each group might reconcile some of differences noted in previous studies, and differences noted between maxillofacial and axial and appendicular lesions are a function of their anatomic locations, which results in the earlier diagnosis of maxillofacial lesions because of exposure of the face and the prevalence of screening dental examinations and radiographs.” They found that the lesions were similar when grouped in aggressive and nonaggressive categories.
In the study by Peacock et al. [39] on gene expression and tissue microarray analysis, the same argument was made by looking at gene expression from laser capture microdissection experiments confirmed with tissue microarray. Again, the lesions were found to be similar. In addition, in another study, Peacock et al. [38] asked pathologists to differentiate between aggressive and nonaggressive giant cell lesions based on histology and on location. The pathologists were not able to differentiate the phenotypic behavior of the tumors nor their site of origin based on histology alone [38]. In another study [46], the aggressive nature of giant cell lesions of the jaw was described in a 20-year followup study of the adjuvant interferon protocol. This study [46] corroborates the aggressive nature of giant cell lesions occurring in the jaw.
A second limitation is that our study proves an association but it does not definitely show causation. However, we feel that our findings are compelling when read in the context of the cited studies. Third, while we have a large series of tumors, there is inherently a selection bias that we cannot control given that not all of our patients were included in the study. The reasons why some patients were not included were many but they were unique to the practice of each surgeon whose patients were eligible for the study. It is does not seem likely that this selection bias inherently flaws the data we have collected.
Was an Immune Response Elicited by Giant Cell Lesions?
Muscolo and Ayerza [35] identified the potential role of a T cell immune response in giant cell lesions when they were able to show a graded activation of T lymphocytes when cocultured with cells taken from giant cell tumors. The Enneking system [18] was correlated with activation of the lymphocytes where more aggressive Stage III tumors did not elicit a robust T cell activation when cocultured with giant cell tumor cells, whereas Stage I tumors did elicit T cell activation. Their study provided evidence that behavior of giant cell lesions may be inversely related to T cell activation [35]. Our study provides a mechanistic framework, which helps to explain the lack of T cell activation seen in the study by Muscolo and Ayerza [35]. Our study showed that a T cell immune response is present in giant cell lesions as lymphocyte infiltrates were identified in all tumors. Low CD8+ T cell infiltration was associated with aggressive tumors, further suggesting that the immune system plays an active role in determining disease status.
Did Clinically Relevant HLA Defects Exist in Giant Cell Lesions?
Lymphocytes appear to impose selective pressure on tumor cell populations, as HLA class I antigen defects have been found in more than 50% of the mononuclear stromal cells analyzed. These defects reflect the outgrowth of tumor cells which escape from recognition and destruction by the host immune system because of abnormalities in HLA class I antigens. The latter may cause a defective expression of HLA class I antigen-tumor antigen derived peptide complexes which mediate the interaction of tumor cells with cognate cytotoxic T lymphocytes. While defects in β2-microglobulin were detected in almost 90% of mononuclear stromal cells, they did not show any correlation with tumor aggressiveness when stratified by HLA class I expression. Similarly, there was no association between β2-microglobulin expression and CD8+ infiltration. This suggests that a defect in β-2 microglobulin light chain is unlikely to play a role in tumor immune evasion in giant cell lesions. HLA class II antigens have classically been thought to be expressed on antigen processing cells with limited expression in other normal cells, although there is considerable evidence to suggest that HLA class II antigens are expressed on many normal cells other than those of the hematopoietic lineage [9]. In addition, HLA class II antigens are thought to be induced in some cancer cells. These “nonprofessional” antigen-presenting cells can present antigens to CD4+ T lymphocytes. However, there is evidence that this antigen-presenting cell and CD4 interaction leads to immunosuppression rather than activation of the immune response [8]. Our data show that HLA class II antigens are present in the majority of mononuclear stromal cells and that HLA class II expression is associated with local tumor invasion. The same was not true with HLA class II expression in multinucleated giant cells where 97% of the samples were negative for HLA class II expression. This is interesting in a historical context where osteoclasts were once thought to be the giant cells in giant cell tumors [14]. Mature osteoclasts express HLA class II antigens yet the giant cells in our giant cell lesions did not express HLA class II antigens. HLA class II antigen expression was not associated with HLA class I, or with either CD4+ or CD8+ T cell infiltration.
Was B7-H3 a Clinically Relevant Immune Modulator?
Defects in HLA class I antigen expression are not the only escape mechanism used by giant cell lesion cells to avoid destruction by host immune cells. High expression of B7-H3 expression, which we found on mononuclear stromal cells in 37% of the giant cell lesions, is associated with local tumor invasion. This is similar to what has been shown in glioblastoma where B7-H3 expression by tumor cells was associated with greater local tumor invasiveness [31]. Similarly, a previous study showed silencing of B7-H3 reduced migration and invasion of melanoma and breast cancer cell lines [36]. B7-H3 has been associated with immune suppression and worse prognosis in multiple cancers including lung, glioblastoma, renal cell carcinoma, prostate, and pancreatic ductal adenocarcinoma cancer cells [11, 15, 31, 44, 58]. B7-H3 exerts an inhibitory immune effect by preventing activation of CD8+ T lymphocytes [30]. Our data indicate that high B7-H3 expression in the presence of high CD8+ tumor infiltrating cells is associated with local tumor invasiveness suggesting an inhibitory immune function for B7-H3 in giant cell lesions.
Conclusions
Locally aggressive giant cell lesions are associated with lower HLA class I expression, lower CD8+T cell infiltration, and higher expression of the immune modulator B7-H3 when compared with nonaggressive lesions. Giant cell lesions may become aggressive because they evade immune surveillance. The data presented here provide a mechanistic view into how some giant cell lesions may progress and become infiltrative as they overcome the host’s immune system. Exploring antigen processing machinery could shed some light on how tumors can regulate their HLA class I. It also offers potential sites one could target with immune therapy such as blockage of B7-H3.
References
Armand P, Nagler A, Weller EA, Devine SM, Avigan DE, Chen YB, Kaminski MS, Kent Holland H, Winter JN, Mason JR, Fay JW, Rizzieri DA, Hosing CM, Ball ED, Uberti JP, Lazarus HM, Mapara MY, Gregory SA, Timmerman JM, Andorsky D, Or R, Waller EK, Rotem-Yehudar R, Gordon LI. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol. 2013;31:4199–4206.
Balke M, Schremper L, Gebert C, Ahrens H, Streitbuerger A, Koehler G, Hardes J, Gosheger G. Giant cell tumor of bone: treatment and outcome of 214 cases. J Cancer Res Clin Oncol. 2008;134:969–978.
Beebe-Dimmer JL, Cetin K, Fryzek JP, Schuetze SM, Schwartz K. The epidemiology of malignant giant cell tumors of bone: an analysis of data from the Surveillance, Epidemiology and End Results Program (1975-2004). Rare Tumors. 2009;1:e52.
Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, Nik-Zainal S, Martin S, McLaren S, Goody V, Robinson B, Butler A, Teague JW, Halai D, Khatri B, Myklebost O, Baumhoer D, Jundt G, Hamoudi R, Tirabosco R, Amary MF, Futreal PA, Stratton MR, Campbell PJ, Flanagan AM. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013;45:1479–1482.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465.
Branstetter DG, Nelson SD, Manivel JC, Blay JY, Chawla S, Thomas DM, Jun S, Jacobs I. Denosumab induces tumor reduction and bone formation in patients with giant-cell tumor of bone. Clin Cancer Res. 2012;18:4415–4424.
Brodsky FM, Bodmer WF, Parham P. Characterization of a monoclonal anti-beta 2-microglobulin antibody and its use in the genetic and biochemical analysis of major histocompatibility antigens. Eur J Immunol. 1979;9:536–545.
Byrne SN, Halliday GM. High levels of Fas ligand and MHC class II in the absence of CD80 or CD86 expression and a decreased CD4+ T cell Infiltration, enables murine skin tumours to progress. Cancer Immunol Immunother. 2003;52:396–402.
Campoli M, Ferrone S. HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene. 2008;27:5869–5885.
Chavin G, Sheinin Y, Crispen PL, Boorjian SA, Roth TJ, Rangel L, Blute ML, Sebo TJ, Tindall DJ, Kwon ED, Karnes RJ. Expression of immunosuppresive B7-H3 ligand by hormone-treated prostate cancer tumors and metastases. Clin Cancer Res. 2009;15:2174–2180.
Chen C, Shen Y, Qu QX, Chen XQ, Zhang XG, Huang JA. Induced expression of B7-H3 on the lung cancer cells and macrophages suppresses T-cell mediating anti-tumor immune response. Exp Cell Res. 2013;319:96–102.
Chouaib S. [At the crossroads of cancer][in French]. Bull Cancer. 2013;100:569–574.
Chuong R, Kaban LB, Kozakewich H, Perez-Atayde A. Central giant cell lesions of the jaws: a clinicopathologic study. J Oral Maxillofac Surg. 1986;44:708–713.
Coley BL. Giant Cell Tumor (Osteoclastoma). Neoplasms of Bone and Related Conditions: Their Etiology, Pathogenesis, Diagnosis and Treatment. New York, NY: Paul B. Hoeber Inc; 1949:162–205.
Crispen PL, Sheinin Y, Roth TJ, Lohse CM, Kuntz SM, Frigola X, Thompson RH, Boorjian SA, Dong H, Leibovich BC, Blute ML, Kwon ED. Tumor cell and tumor vasculature expression of B7-H3 predict survival in clear cell renal cell carcinoma. Clin Cancer Res. 2008;14:5150–5157.
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998.
Emens LA, Butterfield LH, Hodi FS Jr, Marincola FM, Kaufman HL. Cancer immunotherapy trials: leading a paradigm shift in drug development. J Immunother Cancer. 2016;4:42.
Enneking WF. A system of staging musculoskeletal neoplasms. Clin Orthop Relat Res. 1986;204:9–24.
Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
Fellenberg J, Saehr H, Lehner B, Depeweg D. A microRNA signature differentiates between giant cell tumor derived neoplastic stromal cells and mesenchymal stem cells. Cancer Lett. 2012;321:162–168.
Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, Lee JS, Hellmann MD, Hamid O, Goldman JW, Soria JC, Dolled-Filhart M, Rutledge RZ, Zhang J, Lunceford JK, Rangwala R, Lubiniecki GM, Roach C, Emancipator K, Gandhi L; KEYNOTE-001 Investigators. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–2028.
Gaston CL, Grimer RJ, Parry M, Stacchiotti S, Dei Tos AP, Gelderblom H, Ferrari S, Baldi GG, Jones RL, Chawla S, Casali P, LeCesne A, Blay JY, Dijkstra SP, Thomas DM, Rutkowski P. Current status and unanswered questions on the use of Denosumab in giant cell tumor of bone. Clin Sarcoma Res. 2016;6:15.
Ghert M, Simunovic N, Cowan RW, Colterjohn N, Singh G. Properties of the stromal cell in giant cell tumor of bone. Clin Orthop Relat Res. 2007;459:8–13.
Gomes CC, Diniz MG, Amaral FR, Antonini Guimaraes BV, Gomez RS. The highly prevalent H3F3A mutation in giant cell tumours of bone is not shared by sporadic central giant cell lesion of the jaws. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:583–585.
Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Mateus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723.
Huang L, Teng XY, Cheng YY, Lee KM, Kumta SM. Expression of preosteoblast markers and Cbfa-1 and Osterix gene transcripts in stromal tumour cells of giant cell tumour of bone. Bone. 2004;34:393–401.
Kaban LB, Troulis MJ, Wilkinson MS, Ebb D, Dodson TB. Adjuvant antiangiogenic therapy for giant cell tumors of the jaws. J Oral Maxillofac Surg. 2007;65:2018–2024; discussion 2024.
Larsson SE, Lorentzon R, Boquist L. Giant-cell tumor of bone: a demographic, clinical, and histopathological study of all cases recorded in the Swedish Cancer Registry for the years 1958 through 1968. J Bone Joint Surg Am. 1975;57:167–173.
Leitner J, Klauser C, Pickl WF, Stockl J, Majdic O, Bardet AF, Kreil DP, Dong C, Yamazaki T, Zlabinger G, Pfistershammer K, Steinberger P. B7-H3 is a potent inhibitor of human T-cell activation: no evidence for B7-H3 and TREML2 interaction. Eur J Immunol. 2009;39:1754–1764.
Lemke D, Pfenning PN, Sahm F, Klein AC, Kempf T, Warnken U, Schnolzer M, Tudoran R, Weller M, Platten M, Wick W. Costimulatory protein 4IgB7H3 drives the malignant phenotype of glioblastoma by mediating immune escape and invasiveness. Clin Cancer Res. 2012;18:105–117.
Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V. MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst. 2013;105:1172–1187.
Loo D, Alderson RF, Chen FZ, Huang L, Zhang W, Gorlatov S, Burke S, Ciccarone V, Li H, Yang Y, Son T, Chen Y, Easton AN, Li JC, Rillema JR, Licea M, Fieger C, Liang TW, Mather JP, Koenig S, Stewart SJ, Johnson S, Bonvini E, Moore PA. Development of an Fc-enhanced anti-B7-H3 monoclonal antibody with potent antitumor activity. Clin Cancer Res. 2012;18:3834–3845.
Loos M, Hedderich DM, Friess H, Kleeff J. B7-h3 and its role in antitumor immunity. Clin Dev Immunol. 2010;2010:683875.
Muscolo DL, Ayerza MA. Lymphocyte in vitro response to human giant cell tumors. Clin Orthop Relat Res. 1996;326:55–62.
Nygren MK, Tekle C, Ingebrigtsen VA, Fodstad O. B7-H3 and its relevance in cancer; immunological and non-immunological perspectives. Front Biosci (Elite Ed). 2011;3:989–993.
Ogino T, Bandoh N, Hayashi T, Miyokawa N, Harabuchi Y, Ferrone S. Association of tapasin and HLA class I antigen down-regulation in primary maxillary sinus squamous cell carcinoma lesions with reduced survival of patients. Clin Cancer Res. 2003;9:4043–4051.
Peacock ZS, Resnick CM, Susarla SM, Faquin WC, Rosenberg AE, Nielsen GP, Schwab JH, Hornicek F, Ebb DH, Dodson TB, Kaban LB. Do histologic criteria predict biologic behavior of giant cell lesions? J Oral Maxillofac Surg. 2012;70:2573–2580.
Peacock ZS, Schwab JH, Faquin WC, Hornicek FJ, Benita Y, Ebb DH, Kaban LB. Genetic analysis of giant cell lesions of the maxillofacial and axial/appendicular skeletons. J Oral Maxillofac Surg. 2017;75:298–308.
Perosa F, Luccarelli G, Prete M, Favoino E, Ferrone S, Dammacco F. Beta 2-microglobulin-free HLA class I heavy chain epitope mimicry by monoclonal antibody HC-10-specific peptide. J Immunol. 2003;171:1918–1926.
Picarda E, Ohaegbulam KC, Zang X. Molecular pathways: targeting B7-H3 (CD276) for human cancer immunotherapy. Clin Cancer Res. 2016;22:3425–3431.
Resnick CM, Margolis J, Susarla SM, Schwab JH, Hornicek FJ, Dodson TB, Kaban LB. Maxillofacial and axial/appendicular giant cell lesions: unique tumors or variants of the same disease? A comparison of phenotypic, clinical, and radiographic characteristics. J Oral Maxillofac Surg. 2010;68:130–137.
Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, Weber JS, Joshua AM, Hwu WJ, Gangadhar TC, Patnaik A, Dronca R, Zarour H, Joseph RW, Boasberg P, Chmielowski B, Mateus C, Postow MA, Gergich K, Elassaiss-Schaap J, Li XN, Iannone R, Ebbinghaus SW, Kang SP, Daud A. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109–1117.
Roth TJ, Sheinin Y, Lohse CM, Kuntz SM, Frigola X, Inman BA, Krambeck AE, McKenney ME, Karnes RJ, Blute ML, Cheville JC, Sebo TJ, Kwon ED. B7-H3 ligand expression by prostate cancer: a novel marker of prognosis and potential target for therapy. Cancer Res. 2007;67:7893–7900.
Sathyanarayanan V, Neelapu SS. Cancer immunotherapy: strategies for personalization and combinatorial approaches. Mol Oncol. 2015;9:2043–2053.
Schreuder WH, Peacock ZS, Ebb D, Chuang SK, Kaban LB. Adjuvant antiangiogenic treatment for aggressive giant cell lesions of the jaw: a 20-year experience at Massachusetts General Hospital. J Oral Maxillofac Surg. 2017;75:105–118.
Seliger B, Maeurer MJ, Ferrone S. Antigen-processing machinery breakdown and tumor growth. Immunol Today. 2000;21:455–464.
Sernee MF, Ploegh HL, Schust DJ. Why certain antibodies cross-react with HLA-A and HLA-G: epitope mapping of two common MHC class I reagents. Mol Immunol. 1998;35:177–188.
Stam NJ, Spits H, Ploegh HL. Monoclonal antibodies raised against denatured HLA-B locus heavy chains permit biochemical characterization of certain HLA-C locus products. J Immunol. 1986;137:2299–2306.
Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–1028.
Temponi M, Kekish U, Hamby CV, Nielsen H, Marboe CC, Ferrone S. Characterization of anti-HLA class II monoclonal antibody LGII-612.14 reacting with formalin fixed tissues. J Immunol Methods. 1993;161:239–256.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454.
Tran CN, Thacker SG, Louie DM, Oliver J, White PT, Endres JL, Urquhart AG, Chung KC, Fox DA. Interactions of T cells with fibroblast-like synoviocytes: role of the B7 family costimulatory ligand B7-H3. J Immunol. 2008;180:2989–2998.
12th International Histocompatibility Conference. Genetic diversity of HLA: functional and medical implications. Paris, France, June 9-12, 1996. Abstracts. Hum Immunol. 1996;47:1–184.
Wang L, Kang FB, Shan BE. B7-H3-mediated tumor immunology: friend or foe? Int J Cancer. 2014;134:2764–2771.
Wang L, Zhang Q, Chen W, Shan B, Ding Y, Zhang G, Cao N, Liu L, Zhang Y. B7-H3 is overexpressed in patients suffering osteosarcoma and associated with tumor aggressiveness and metastasis. PLoS One. 2013;8:e70689.
Wulling M, Delling G, Kaiser E. The origin of the neoplastic stromal cell in giant cell tumor of bone. Hum Pathol. 2003;34:983–993.
Yamato I, Sho M, Nomi T, Akahori T, Shimada K, Hotta K, Kanehiro H, Konishi N, Yagita H, Nakajima Y. Clinical importance of B7-H3 expression in human pancreatic cancer. Br J Cancer. 2009;101:1709–1716.
Acknowledgment
We thank William C. Faquin MD, PhD (Department of Pathology, Massachusetts, General Hospital, Harvard Medical School) for help in reviewing the tissue microarray to assess for adequate tumor tissue; Clair Hart BSc, MPhil (Institute of Cancer Sciences, University of Manchester, Manchester, UK) for help in scoring the stained tissue microarray; and Lei Cai MD, PhD (Department of Surgery, Massachusetts General Hospital, Harvard Medical School), for help in staining and scoring the tissue microarray for tumor infiltrating cells.
Author information
Authors and Affiliations
Corresponding author
Additional information
One author (AA-S) received funding from the Pathological Society and British Medical and Dental Students’ Trust.
Each author certifies that he has no commercial associations (eg consultancies, stock ownership, equity interest, patent/licensing arrangements, etc) that might pose a conflict of interest in connection with the submitted article.
All ICMJE Conflict of Interest Forms for authors and Clinical Orthopaedics and Related Research ® editors and board members are on file with the publication and can be viewed on request.
Each author certifies that his institution approved the approval for the human protocol for this investigation and that all investigations were conducted in conformity with ethical principles of research.
This work was done at Massachusetts General Hospital, Boston, MA, USA.
About this article

Cite this article
Al-Sukaini, A., Hornicek, F.J., Peacock, Z.S. et al. Immune Surveillance Plays a Role in Locally Aggressive Giant Cell Lesions of Bone. Clin Orthop Relat Res 475, 3071–3081 (2017). https://doi.org/10.1007/s11999-017-5451-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11999-017-5451-1