
Abstract
Nowadays, coatings need to fulfill a variety of requirements such as having excellent mechanical, chemical, and optical properties at low baking temperatures. On a large scale, polyisocyanates, amines or melamines are used as crosslinking agents in the coatings industry. In this work, a new self-crosslinking agent based on a hydroxy functional 6-membered carbonate with high ring tension and thus presumably lower baking temperature was synthesized and the behavior as self-crosslinking agent was compared to the crosslinking agent derived from the commercially available 5-membered glycerol carbonate. The hydroxy functional 6-membered carbonate monomer was synthesized enzymatically under mild reaction conditions from commercially available substances, linked to a hexamethylene diisocyanate trimer and self-polymerized afterward. NMR- and IR-spectroscopy and GC-MS analysis were found to be suitable techniques to characterize monomers and crosslinking agents. DSC measurements were performed to evaluate appropriate reaction parameters for the attachment reaction of the 6-membered cyclic carbonate to the polyisocyanate without ring opening. The progress of self-crosslinking has been followed by characteristic changes in IR spectra as well as time and temperature-dependent changes of storage and loss modulus while oscillating rheological crosslinking. Furthermore, glass transition temperatures of the resulting coating films are determined, and sol gel analysis was performed to estimate the degree of crosslinking. After application on steel, aluminum and glass plates application tests were performed. In addition to excellent mechanical and chemical properties, the coating film showed good adhesion to the surface and was colorless. Combining these properties with relatively low baking temperatures, 6-membered cyclic carbonate crosslinking agents could represent a new technology for the coatings industry.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
For coatings showing high resistance and enhanced mechanical properties, reactive systems based on a crosslinking component in combination with a binder are usually applied rather than systems based on physically drying binders. These systems often contain melamine, polyisocyanates or acrylate monomers or adducts, which have toxicologically questionable properties, when applied by end consumers, before the network is formed.1
In order to develop more sustainable systems, the coatings industry has been researching new crosslinking technologies such as non-isocyanate polyurethanes (NIPU) for a number of years.2 Promising results have already been reported by Wunschik et al.3,4 by means of 5-membered cyclic carbonates as crosslinking agents producing polyester networks including good mechanical properties without using isocyanates, melamines or amines as crosslinking agents. The lower ring tension of 5-membered cyclic carbonates leads to higher activation energies for ring opening polymerization and thus to higher baking temperatures of around 160°C, which limits the areas of application.4,5 For this reason, thermodynamically less stable 6-membered cyclic carbonates are preferred toward 5-membered cyclic carbonates.5
Typically, organic carbonates are synthesized by reaction of diols with phosgene, which requires the utilization of toxic chemicals and results in the formation of chloride salts as side products. Therefore, the compliance to high hygienic and safety standards are required when using or working with these substances. Regarding to the production process based on phosgene, the usage of carbonate monomers as self-crosslinking agents is not beneficial in terms of sustainability and working safety.6 Different 6-membered cyclic carbonates with different functionalities except hydroxyl were synthesized by thermal treatment of different diols with diphenyl dimethyl carbonate without any catalyst.7 The synthesis of a hydroxyl-functionalized cyclic carbonate from trimethylolpropane and dimethyl carbonate following a two-step synthesis based on enzymatic catalysis was described previously.8,9 First, trimethylolpropane was transesterified with dimethyl carbonate by lipase catalysis, which leads to three types of carbonates. In the second reaction step, thermal cyclization was performed. However, long reaction times are disadvantageous during the second step (Fig. 1).

The hydroxyl functionalized 6-membered cyclic carbonate could also be formed at long reaction times by thermal treatment of trimethylolpropane with diphenyl carbonate.11
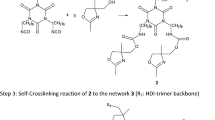
In this paper, a hydroxy functional 6-membered cyclic carbonate (5-Ethyl-5-(hydroxymethyl)-1,3-dioxan-2-on) was synthesized via the enzymatic reaction of trimethylolpropane and dimethyl carbonate and subsequent thermal cyclization (Fig. 1). Performing the second synthesis step, we obtained a composition of 41.8% trimethylolpropane, 9.1% 2,2-bis(hydroxymethyl)butyl methyl carbonate, 3.2% 2-(hydroxymethyl)-2-(((methoxycarbonyl)oxy)methyl)butyl methyl carbonate and 45.9% (5-Ethyl-5(hydroxymethyl)1,3-dioxan-2-on) according to gas chromatography mass spectroscopy (Table 3). After column chromatography to extract the 6-membered carbonate, DSC measurements with and without catalyst were performed to gain reliable parameters for the last reaction step. The hydroxy functional 6-membered cyclic carbonate has been linked to a hexamethylene diisocyanate trimer (HDI-trimer) without ring opening polymerization as shown in Fig. 2, so that a polyisocyanate-carbonate adduct based on urethane moieties was formed.

Attaching 5-Ethyl-5-(hydroxymethyl)-1,3-dioxan-2-on to HDI-trimer without ring opening polymerization
The polymeric resin was then able to undergo a ring opening reaction upon curing as pictured in Fig. 3 to form a polymeric network based on urethane and carbonate groups without the need of the second hardener component.

Ring opening polymerization of trimeric 6-membered cyclic carbonate-trimer forming a polymeric network with urethane and carbonate groups
Network building reaction was followed by changes in IR spectra and time and temperature-dependent oscillating rheometer measurements. After baking, mechanical properties of resulting crosslinked polymer films were determined.
Furthermore, a 5-membered crosslinking agent from glycerol 1,2-carbonate and HDI-trimer has been synthesized in analogy to Fig. 2, ring opening starting temperatures were determined and mechanical and chemical properties of resulting polymer films were compared to the 6-membered cyclic carbonate-polyisocyanate adduct (6-membered cyclic carbonate trimer).
Raw materials
Novozyme-435 was a kind gift from Novozymes Denmark and HDI-Trimer Desmodur® ultra N 3600 was kindly provided by Covestro Deutschland AG. Acetone (≥ 99.0%), acetonitrile (HPLC grade), dichloromethane (≥ 99.5%), dimethyl carbonate (≥ 99.8%), ethyl acetate (≥ 99.5%), dimethyl sulfoxide (≥ 99.5%) molecular sieves (4Å) and silica gel 60 0.02–0.045 mm were purchased from Carl Roth. Trimethylolpropane (98%) and glycerol 1,2-carbonate (90%) were purchased from abcr, and molecular sieves (4Å) (for cyclization) were purchased from Merck. All other compounds were purchased from Aldrich Chemicals and were used without further purification.
All coatings were applied by a squeegee on steel panels (DX51D+z), aluminum samples (type Al99,5) or glass plates.
Analytical methods
1H-NMR spectra were measured on a Bruker Avance Neo 400 spectrometer at 400 MHz. Deuterated dimethyl sulfoxide was used as solvent and internal standard.
IR spectra were measured with the Bruker Vertex 70 FTIR-spectrometer (4000–375 cm−1) with a resolution of 4 cm−1, and they were baseline corrected and normalized on the CH2-deformation signal at 1462 cm−1 as this signal does not change during urethane formation or crosslinking reaction.
Gas chromatography mass spectroscopy analysis was performed on a system from Agilent Technologies with a GC-System “7890 A” equipped with an Agilent J&W VF-5ms column (30 m × 0.25 mm, 0.5 μm) and a MS-System “5975C” with a triplet axis detector. The samples were diluted to a concentration of 0.1–0.5 mg/mL in acetonitrile or ethyl acetate and injected in split injection (30:1) at 50°C. The initial oven temperature was increased by the following temperature program. The temperature was held for 3 min at 50°C. The initial oven temperature was increased from 50 to 150°C at a rate of 15°C/min and increased from 150 to 320°C at a rate of 35°C/min. The final temperature was held for 10 min.
Differential scanning calorimetry (DSC) was performed on a DSC Q200 from TA Instruments using Tzero aluminum pans with lids, and sample weights were between 13 and 18 mg. Heating or cooling rates were carried out with a value of 5°C/min. Catalyzed samples include 1% DABCO.
The rheological experiments to achieve starting polymerization temperatures, values for storage and loss modulus and glass transition temperatures were performed on a Modular Compact Rheometer MCR 102 from Anton Paar using plate-plate single use systems with a diameter of 25 mm and a measuring gap of 1 mm. All carbonate trimer samples applied on the rheometer plate were containing 50% solvents. A solvent matrix is advantageous for the application, since on the one hand the resulting higher mobility of the chains ensures that the glass state is reached later and thus a higher network density can be achieved, and on the other hand it promotes the flow of the coating film. However, a high solvent content has a negative effect on the accuracy of the measurement data for the rheometer measurements, so the samples are dried overnight at 50°C to reduce the solvent content. The samples still may contain up to twenty percent solvent residues. Values for G’ and G’’ are always lower in comparison to DMA measurements of a free film because the plate-plate single use system influences measuring data by the pliability of the lower aluminum plate dependent on the diameter of the upper plate. The diameter of 25 mm is a compromise to gain accurate data in all steps during the curing reaction. All measurements were performed in the linear viscoelastic range (LVE-range) of the samples, determined by changing shear deformation γ = 0.01–4.0% and constant frequency ω = 10 rad/s and temperature (stress sweep). Parameters for crosslinking measurements were: γ = 0.1% and ω = 10 rad/s, heating rate = 1°C/min.
The pendulum damping values were obtained according to DIN EN ISO 1522 using a Königspendel from Erichsen. Three measurements for each panel were performed 4 h after baking. Average values are given.
The adhesion of the coating films was determined by performing the cross-cut test according to DIN EN ISO 2409 after application on steel panels. Furthermore, the resistance of the samples in the event of sudden deformation based on DIN EN ISO 6272-1:2011 has been determined. For the evaluation, a drop height of 1 m was chosen and it was judged if the film was cracked and if it still adhered to the steel surface after the indirect impact of the one kilogram weight.
Synthesis of the compounds
6-membered cyclic carbonate monomer (5-Ethyl-5-(hydroxymethyl)-1,3-dioxan-2-on)
The synthesis of the cyclic carbonate (5-Ethyl-5-(hydroxymethyl)-1,3-dioxan-2-on) was performed with small variations according to Pyo et al.9 and Bornadel et al.10 First, 5.0 g trimethylolpropane (0.04 mol) was dissolved in 150 mL dimethyl carbonate (1.8 mol) at 60°C and 10% (w/v) molecular sieves (15.0 g) and 18% Novozyme-435 (0.9 g) were added. After 6 h the reaction was stopped by filtering off the molecular sieves and Novozyme-435. Dimethyl carbonate was evaporated under vacuum. The remaining liquid was dissolved in acetonitrile (2.4 mol, 130 mL). The cyclization reaction of 10 mL of the dissolved product was performed with 15% (w/v) molecular sieves (4Å, Merck) at 110°C for 24 h in vials, which were closed with a septum. After removal of the molecular sieves and the solvent the composition according to gas chromatography mass spectroscopy was 41.8% trimethylolpropane, 9.1% 2,2-bis(hydroxymethyl)butyl methyl carbonate, 3.2% 2-(hydroxymethyl)-2-(((methoxycarbonyl)oxy)methyl)butyl methyl carbonate and 45.9% (5-Ethyl-5(hydroxymethyl)1,3-dioxan-2-on) (Table 3). The solvent was removed. Afterward the cyclic carbonate was separated from the reaction mixture by silica column chromatography with dichloromethane: ethyl acetate: acetone (2:1:1) as solvent mixture. The solvents were removed under vacuum, and the composition was quantified by gas chromatography mass spectroscopy. The product was characterized by 1H-NMR and IR spectroscopy.
1H NMR (400 MHz, DMSO, δ in ppm): 0.82 (t, J = 7.6 Hz, 3H, –CH3); 1.37 (q, J = 7.6 Hz, 2H, CH2–CH3); 3.38 (d, J = 5.3 Hz, 2H, –CH2–OH); 4.20 (dt, J = 12.1, 10.5 Hz, 4H, 2x –CH2–); 4.99 (t, J = 5.3 Hz, 1H, –OH).
IR (resonance in [cm−1] and type): 3400 (OH valence), 2920 (CH2 stretch), 1755 (cycl. carbonate), 1462 (CH2 deformation), 1730 (C=O), 1405 (CH2 wagging), 1175, 1115 (=C–O–C valence), 1047 (C–OH, C–O valence), 770 (carbonate bending in the plane), compare Socrates12, Hesse et al.13
6-membered carbonate-trimer
For the mixture consisting of 89.7% 6-membered carbonate (5-Ethyl-5-(hydroxymethyl)-1,3-dioxan-2-on), 2.7% 2,2-bis(hydroxymethyl)butyl methyl carbonate and 7.6% ethyl acetate an OH-equivalent mass of 167.31 g/mol OH has been calculated from GC-MS data. Isocyanate equivalent weight of HDI-Trimer Desmodur® ultra N 3600 is 182.6 g/mol NCO, so that reaction ratio was 1.09 g Desmodur® ultra N 3600 and 1.00 g 6-membered carbonate mixture. Next, 6.27 g ethyl acetate or dried dimethyl sulfoxide was added in a 50 mL two-necked flask with magnetic stirrer to 3.27 g Desmodur® ultra N 3600 under nitrogen atmosphere. After homogenization 3.00 g of the 6-membered cyclic carbonate mixture was added while stirring. Then, 800 ppm dibutyltin dilaurate (DBTDL) was added for catalysis. The clear solution was heated to 45°C. The reaction time was 6–10 h, and the yield was controlled by IR-spectroscopy.
IR (resonance in [cm−1] and type): 3350 (NH–COO and NH stretch), 2963 (CH3 stretch), 2937 (CH2 stretch), 2904 (CHx stretch), 2862 (CH3 stretch), 1748 (cyclic carbonate), 1712 amide I (C=O, urethane), 1677 amide II (C=O, urethane, isocyanurate), 1525 (NH–COO and NH bending), 1462 (CH2. deformation), 1242 (C–O–C, urethane), 1180, 1115 (=C–O–C valence), 765 (C–N stretch, isocyanurate), 729 (CH2 rocking), compare Ming et al.14 and Defeyt et al.15
5-membered carbonate trimer
First, 27.8 mmol glycerol 1,2-carbonate and 8.07 g ethyl acetate or dimethyl sulfoxide were added under nitrogen atmosphere to a 50 mL two-necked flask with magnetic stirrer. Then, 9.2 mmol Desmodur® ultra N 3600 was added while stirring. At last, 1100 ppm dibutyltin dilaurate (DBTDL) were added for catalysis. After that the temperature was increased to 50°C. The reaction course was controlled using IR spectroscopy. Within 5–8 h, the reaction was completed.
IR (resonance in [cm−1] and type): 3300 (NH–COO and NH stretch), 2963 (CH3 stretch), 2937 (CH2 stretch), 2904 (CHx stretch), 2862 (CH3 stretch), 1790 (cyclic carbonate), 1715 amide I (C=O, urethane), 1677 amide II (C=O, urethane, isocyanurate), 1540 (NH–COO and NH bending), 1462 (CH2 deformation), 1242 (C–O–C, urethane), 1160, 1087 (=C–O–C valence), 765 (C–N stretch, isocyanurate), 729 (CH2 rocking).12,14,15
Both 5- and 6-membered carbonate-trimer products contained dimethyl sulfoxide as a solvent and were used without purification for one-compound crosslinking reactions and IR evaluation before and after baking. For DSC and rheometer measurements ethyl acetate was used for trimer building reaction and distilled off at 40°C, 180 mbar.
Application testing
For application testing, a formulation consisting of 5- or 6-membered cyclic carbonate- trimer, diluted 50% in dimethyl sulfoxide was prepared with 1.0% DABCO catalyst and 1.0% 1-butanol. The formulation was applied by drawing down a bar on aluminum sheets and curing at 90–160°C for 30 min to achieve a dry film thickness of 40 µm.
Results and discussion
In this work, the self-polymerization of 6-membered cyclic carbonate derivatives was studied and compared to a 5-membered cyclic carbonate crosslinking agent. A novel polyisocyanate-carbonate adduct was prepared by reaction of HDI-trimer and OH-functionalized 6-membered cyclic carbonate upon urethane formation without ring opening polymerization. The crosslinking ability of the obtained adduct is based on a ring opening polymerization of the carbonate moiety and occurs without carbon dioxide elimination.
OH-functionalized 6-membered cyclic carbonate was obtained by enzymatic transesterification between trimethylolpropane and dimethyl carbonate and subsequent thermal cyclization for 24 h at 110°C. After column chromatography the product was characterized by NMR- and IR-spectroscopy and GC-MS analysis. Product spectra are shown in supporting information, Figs. 19, 20, and 27. To enhance the carbonate-functionality of the later polyisocyanate carbonate adduct, several column chromatography fractions were combined including 2.7% dihydroxy functional 2,2-bis(hydroxymethyl)butyl methyl carbonate and 7.6% of residual solvent EtAc. Gas chromatography mass spectrogram of the mixture is pictured in Fig. 4

Sample diluted to a concentration of 0.1–0.5 mg/mL in acetonitrile or ethyl acetate, 50–150°C (15°C/min) and 150–320°C (35°C/min)
To gain reliable reaction parameters for urethane formation of the 6-membered cyclic carbonate trimer, differential scanning calorimetry (DSC) measurements of catalyzed and uncatalyzed 6-membered carbonate monomers were performed and are pictured in Fig. 5

DSC-measurements of uncatalyzed and DABCO-catalyzed 6-membered cyclic carbonate, 1. heating run
The onset temperature of DABCO-catalyzed sample has been decreased by around 20°C to approximately 72°C compared to 92°C for uncatalyzed sample. Endothermic peaks around 90°C could be traced back to solvent residues of ethyl acetate in both samples. Decomposition temperatures were also dependent on whether the sample was catalyzed or not. For DABCO-catalyzed samples decomposition began at 150°C, whereas decomposition temperatures for uncatalyzed samples were 225°C. The higher ring tension of 6-membered cyclic carbonates resulted in small values for reaction enthalpy, because ring opening process is slightly endothermic and almost simultaneously ongoing polymerization is slightly exothermic. Considering solvent residues ∆Hreact.-values around ∆Hreact. = −1.2 ± 0.4 kcal/mol were measured for 6-membered cyclic carbonate monomers. Furthermore, the glass transition temperature of the polymerized carbonate was determined in the second heating run: TG = −6°C, Fig. 24, supplementary material. Thus, DSC-measurements allow the conclusion to perform catalyzed urethane formation reaction at 45°C without ring opening polymerization.
IR-spectroscopy turned out to be suitable to verify successful urethane formation of polyisocyanate-carbonate adduct as shown in Fig. 6. As depicted in Fig. 7, the amide I vibration of the urethane/isocyanurate group can be detected at 1712 and 1680 cm−1 in the IR spectrum with the corresponding amide II signal at 1525 cm−1 caused by the N–H group of the emerging urethane groups. An amide II signal is visible, if at least one proton is bound at the nitrogen of the amide group, so that this signal proves the formation or the existence of an amide with an N–H group. The isocyanate peak at 2270 cm−1 decreased after reaction, whereas urethane peak amide I at 1712 cm−1, the amide II at 1525 cm−1 (NH–COO, N–H bending) and 1242 cm−1 (=C–O–C urethane) arised. The OH-valence peak at 3400 cm−1 decreased and was shifted to 3300 cm−1 (NH–COO and NH-bending) during urethane formation. Furthermore, peaks, which would indicate ring-opening polymerization such as 1748 cm−1 (cyclic carbonate) and 1115, 1180 cm−1 (=C–O–C valence carbonate) remained unaffected during urethane formation. Spectra of successful urethane reaction of the 5-membered carbonate with HDI-trimer are shown in supplementary material, Figs. 25 and 26.

ATR-spectra of 6-membered cyclic carbonate before and after urethane formation by reaction with HDI-trimer, diluted 50% in DMSO-D6

Enlargement of Fig. 6 from 1500 to 1800 cm−1
DSC-measurements of DABCO and BuOH-catalyzed self-polymerization of the 6-membered cyclic carbonate trimer were performed to determine onset temperature, reaction enthalpy ∆Hreact. and glass transition temperature TG of resulting polymer network. Figure 8 shows changes of heat flow in the first and second heating run.

DSC measurement of catalyzed self-polymerization of 6-membered cyclic carbonate trimer
Onset temperature of the sample was around 85°C followed by an endothermic peak of ethyl acetate solvent residues. After that, endothermic ring opening and exothermic polymerization took place simultaneously. Thus, a reaction enthalpy excluding solvent residues of ∆Hreact. = −1.78 kcal/mol was measured. Reaction was stopped at 140°C to ensure there was no beginning decomposition. In the second heating run a glass transition temperature of TG = 16°C for the crosslinked polymer was measured.
To verify that enthalpy values are measured in the correct order of magnitude, a combined measurement of urethane formation between 6-membered cyclic carbonate monomer and HDI-trimer and subsequent self-polymerization of resulting carbonate-polyisocyanate adduct was carried out. For urethane formation ∆Hreact. = −46.3 kJ/mol was measured, which can be compared to the results of Lovering and Laidler,16 for polymerization ∆Hreact. = −3.6 kJ/mol was determined.
To further study the crosslinking mechanism leading to a polycarbonate urethane network, mechanical and spectroscopic changes while curing were investigated.
A direct comparison of IR spectra of uncured and cured polyisocyanate-carbonate adduct is suitable to describe successful ongoing ring opening polymerization at a temperature of 130°C for the 6-membered cyclic carbonate. Additionally, to prove successful crosslinking, storage and loss modulus in the context of temperature and time-dependent oscillating rheological measurements are discussed, too.
IR spectra are baseline corrected and normalized on the CH2-deformation signal at 1462 cm−1 as this signal does not change during crosslinking reaction (Figs. 9 and 10).

ATR spectra of 6-membered cyclic carbonate trimer (1.0% DABCO, 1.0% butanol, 50% in DMSO) and the cured coating after 30 min at 130°C applied on aluminum panels

Enlargement of Fig. 9 from 1100 to 1300 cm−1
Successful ring opening polymerization can be detected by decreased cyclic carbonate peak at 1748 cm−1 and decreased cyclic carbonate characteristic =C–O–C-valence vibrations at 1115 and 1180 cm−1. During the polymerization, it could be assumed that many short-chained polymer chains were formed due to the steric hindrance of the trimeric carbonate-polyisocyanate adduct. According to polymerization mechanism, these have an OH-bond at the start and the end of each chain, which can be detected by means of ATR. Spectra show a significant increase in OH-valence peak at 3400 cm−1 during curing reaction. Urethane peaks at 1712, 1525 and 1240 cm−1 are slightly shifted, but do not change during ring opening polymerization.
Due to the lower ring tension of the 5-membered cyclic carbonate-polyisocyanate adduct, higher baking temperatures of around 160°C were necessary for crosslinking reaction. IR spectra before and after curing are shown in Figs. 11 and 12.

ATR spectra of 5-membered cyclic carbonate-polyisocyanate adduct (1.0% DABCO, 1.0% butanol, 50% in DMSO) and the cured coating after 30 min at 160°C applied on aluminum panels

Enlargement of Fig. 11 from 1075 to 1900 cm−1
In contrast to the complete curing of the 6-membered cyclic carbonate-trimer the ring opening of the 5-membered crosslinking agent was different: The cyclic carbonate peak at 1790 cm−1 decreased approximately by 60%. Furthermore, signals of =C–O–C-carbonate valence vibrations at 1087 and 1160 cm−1 do not disappear after curing reaction. Thus, it can be assumed that ring opening polymerization of the 5-membered carbonate-polyisocyanate adduct was incomplete. This assumption was confirmed by the lower gel part of 65.4%, which was measured for the 160°C sample of the 5-membered carbonate adduct. It is possible that at the increased temperatures there was no longer sufficient OH chain starter available, so that the start of the polymerization was restricted. Furthermore, at elevated temperatures around 160°C, huge parts of solvents are already evaporated, and the mobility of the chains is lower, such that the glassy state was reached at lower crosslinking degrees.
Gel parts were measured by the extraction of polymer films for 16 h using MEK after curing with a Soxhlet extractor to draw conclusions on the crosslinking degree at different curing temperatures for 5- and 6-membered carbonate-trimer.
The curing temperature for successful ring opening polymerization of the 5-membered carbonate-trimer was at least 160°C for the studied system. After Soxhlet extraction of a 120°C cured film, there was no gel part remaining, and thus no ring opening polymerization, that would lead to a crosslinked polymer and a measurable gel part. For the 6-membered cyclic carbonate-trimer even at 110°C a gel part of 72.0% can be determined, and at 130°C the gel part is 91.4%, which indicates a high degree of crosslinking at lower temperatures compared to the 5-membered carbonate-trimer.
The crosslinking ability of the carbonate-functional crosslinkers in the presence of DABCO catalyst and butanol was further studied using rheological experiments to determine gel points, values for storage and loss modulus and glass transition temperatures. The TG was obtained in a temperature dependent measurement of storage and loss modulus at the maximum of G’’ in the second heating run. All rheological measurements must be performed in the LVE-range.17 Figures 21 and 22 show DMTA measurement results of 6-membered carbonate trimer (Fig. 13).

DMTA (amplitude 0.1%, frequency 10 rad/s, temperature ramp: 1°C/min. to 160°C) experiment with 6-membered cyclic carbonate trimer (1.0% DABCO, 1.0% butanol, EtAc residues)
The network formation is visible by an increase in the values for storage and loss modulus after reaching approximately 100°C. Starting temperatures for the reaction, which were measured by DMTA, are higher compared to DSC, because of higher film thickness and delayed heat conduction of the plate-plate single measurement system and the Peltier-element. The determined glass transition temperature was TG = 53°C (Fig. 14).

2. heating run of 6-membered cyclic carbonate trimer (1.0% DABCO, 1.0% butanol, amplitude 0.1%, frequency 10 rad/s, temperature ramp: 1°C/min. to 100°C)
The TG values of physically dried acrylate coatings were in the range of 32–48°C, whereas with the same resins cured with crosslinking agents, the resulting coating show TG in the range of 61–72°C.18 HDI-trimer, which was also used in this work, formed a network with a branched polyester with a TG of 54°C.19 Therefore, the new polyurethane-carbonate network is in the standard range of the glass transition temperature of crosslinked coatings with a TG of 53°C.
Upon curing of the 5-membered cyclic carbonate-polyisocyanate adduct, the gel point, i.e., G’ = G’’ was achieved some minutes after reaching 160°C. Below 160°C, no increase in storage and loss modulus resulting from ring opening polymerization was observed (Figs. 15 and 16).

DMTA (amplitude 0.1%, frequency 10 rad/s, temperature ramp: 1°C/min. to 160°C), 5-membered cyclic carbonate-polyisocyanate adduct (1.0% DABCO, 1.0% butanol, EtAc residues)

2. heating run of 5-membered cyclic carbonate trimer (1.0% DABCO, 1.0% butanol, amplitude 0.1%, frequency 10 rad/s, temperature ramp: 1°C/min. to 100°C)
Compared to the 6-membered cyclic carbonate trimer, glass transition temperature of the polymer crosslinked by the 5-membered carbonate was TG = 38°C. The lower value for TG is probably caused by the lower crosslinking degree of this sample.
In order to be able to finally assess a new crosslinking reaction, the resulting properties of the final coating surface are very important. With the focus of the coating properties of polymer network, the impact test and pendulum damping were performed. Further-more, the adhesion on steel was tested. To ensure comparability, all samples were adjusted to 50% solid content and were catalyzed with 1% DABCO and 1% butanol.
Table 1 summarizes the coatings properties of the 5- and 6-membered cyclic carbonate trimers cured at different temperatures. As shown before, the 5-membered cyclic carbonate crosslinked at 160°C, at lower temperatures films were sticky or very soft and did not resist to the effects of tested chemicals. At a curing temperature of 160°C, a high film hardness with 120 pendulum swings in combination with a good adhesion (GT = 1) was determined. The 6-membered cyclic carbonate trimer cured at 90°C did not crosslink, because of low film hardness (15 pendulum swings) in combination with brittleness and thus no adhesion to the surface (GT = 5). Samples baked at 110 and 130°C showed elasticity (GT = 2 and GT = 0) and high film hardness (104 and 124 pendulum swings). Furthermore, the resistance to tested chemicals with the exception of MEK-impact was very good. All coating films were colorless and thus meet the high optical requirements, which is pictured in Fig. 17 (Table 2).

Coating of 6-membered cyclic carbonate-trimer (1.0% DABCO, 1.0% butanol, cured at 130°C for 30 min)
The gloss of the coating was only visually evaluated as high and more or less independent of the curing temperature, since no films applied on contrast cards were available for a measurement according to DIN.
Finally, the resulting coatings properties are very promising using 6-membered carbonate trimer as self-polymerizable crosslinking agents for coating applications.
Conclusions
This work shows that 6-membered cyclic carbonates are advantageous compared to cyclic 5-membered carbonates as crosslinking agents due to the lower ring tension and thus also lower baking temperatures.
The trimeric 6-membered carbonate could undergo a ring opening polymerization that led to colorless urethane-carbonate networks, which had high crosslinking degrees and excellent mechanical properties.
Furthermore, the synthesis of the 6-membered cyclic carbonate could be done enzymatically and did not require the use of phosgene. With that, enzymatically synthesized cyclic 6-membered carbonates are promising educts for crosslinking agents and of high interest for the coatings industry as they combine the need for sustainable products with excellent properties of the resulting polymer network.
References
Mischke, P, Film Formation, Vincentz Network, Hanover (2010)
Guan, J, Song, Y, Lin, Y, Yin, X, Zuo, M, Zhao, Y, et al. “Progress in Study of Non-Isocyanate Polyurethane.” Ind. Eng. Chem. Res., 50 (11) 6517–6527. https://doi.org/10.1021/ie101995j (2011)
Wunschik, DS, Ingenbosch, KN, Zähres, M, Horst, J, Mayer, C, Jäger, M, Strehmel, V, Dornbusch, M, Hoffmann-Jacobsen, K, “Biocatalytic and Solvent-Free Synthesis of a Bio-Based Biscyclocarbonate.” Green Chem., 20 4738–4745 (2018)
Wunschik, D, Hoffmann-Jacobsen, K, Dornbusch, M, “Lipase Catalyzed Modification of Functionalized Polymers.” Prog. Org. Coat., 20 (5) 9358–9379 (2019)
Tomita, H, Sanda, F, Endo, T, “Reactivity Comparison of Five- and Six-Membered Cyclic Carbonates with Amines: Basic Evaluation for Synthesis of Poly(Hydroxyurethane).” J. Polym. Sci. A Polym. Chem., 39 162–168. https://doi.org/10.1002/1099-0518(20010101)39:1%3c162::AID-POLA180%3e3.0.CO;2-O (2001)
Shaikh, A-AG, Sivaram, S, “Organic Carbonates.” Chem. Rev., 96 951–976. https://doi.org/10.1021/cr950067i (1996).
Pyo, S-H, Hatti-Kaul, R, “Chlorine-Free Synthesis of Organic Alkyl Carbonates and Five- and Six-Membered Cyclic Carbonates.” Adv. Synth. Catal., 358 834–839. https://doi.org/10.1002/adsc.201500654 (2016)
Bornadel, A, Ismail, M, Sayed, M, Hatti-Kaul, R, et al. “Six-Membered Cyclic Carbonates from Trimethylolpropane: Lipase-Mediated Synthesis in a Flow Reactor and In Silico Evaluation of the Reaction.” Biotechnol. Prog., 33 375–382. https://doi.org/10.1002/btpr.2406 (2017)
Pyo, S-H, Persson, P, Lundmark, S, Hatti-Kaul, R, “Solvent-Free Lipase-Mediated Synthesis of Six-Membered Cyclic Carbonates from Trimethylolpropane and Dialkyl Carbonates.” Green Chem., 13 976. https://doi.org/10.1039/c0gc00783h (2011)
Bornadel, A, Hatti-Kaul, R, Sörensen, K, Lundmark, S, Pyo, SH, “Optimization of a Two-Step Process Comprising Lipase Catalysis and Thermal Cyclization Improves the Efficiency of Synthesis of Six-Membered Cyclic Carbonate from Trimethylolpropane and Dimethylcarbonate.” Biotechnol. Prog., 29 (1) 66–73. https://doi.org/10.1002/btpr.1662 (2013)
Matsukizono, H, Endo, T, “Synthesis and Hydrolytic Properties of Water-Soluble Poly(Carbonate–Hydroxyurethane)s from Trimethylolpropane.” Polym. Chem., 7 958–969. https://doi.org/10.1039/c5py01733e (2016)
Socrates, GB, Infrared and Raman Characteristic Group Frequencies, 3rd edn. Wiley, England (2004)
Hesse, M, Meier, H, Zeeh, B, Spektroskopische Methoden in der organischen Chemie, Thieme, 8th edn. Georg Thieme Verlag, Stuttgart, New York (2011)
Ming, W, Tian, M, van de Grampel, RD, Melis, F, Jia, X, Loos, J, van der Linde, R, “Low Surface Energy Polymeric Films from Solventless Liquid Oligoesters and Partially Fluorinated Isocyanates.” Macromolecules, 35 6920–6929 (2002)
Defeyt, C, Langenbacher, J, Rivenc, R, “Polyurethane Coatings Used in Twentieth Century Outdoor Painted Sculptures. Part I: Comparative Study of Various Systems by Means of ATR-FTIR Spectroscopy.” Herit. Sci., 5 11 (2017)
Lovering, E, Laidler, K, “Kinetic Studies of Some Alcohol-Isocyanate Reactions.” Can. J. Chem., 40 31–36. https://doi.org/10.1139/v62-006 (2011)
Mezger, TG, Angewandte Rheologie. Anton Paar GmbH, Österreich (2014)
Ma, X, Qiao, Z, Huang, Z, Jing, X, “The Dependence of Pendulum Hardness on the Thickness of Acrylic Coating.” J. Coat. Technol. Res., 10 (3) 433–439 (2013)
Matner, M, Casselmann, H, Zhuang, W, Achten, D, Ehlers, M, “Transparent Polyurethanes with High Glass Transition Temperature Tg.” US020150246998A1
Acknowledgments
This work was supported by the German “Bundesministerium für Bildung und Forschung (BMBF)” in the framework of the program “FHProfUnt,” grant 13FH125PX6, and Covestro Deutschland AG. We thank Dr. T. Schaller, University of Duisburg-Essen, for acquisition of NMR spectra and Dipl.-Ing., Joachim Horst, Niederrhein University of Applied Sciences, for supporting GC-MS measurements.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Seithümmer, J., Knospe, P., Reichmann, R. et al. Comparison of 5- and 6-membered cyclic carbonate-polyisocyanate adducts for high performance coatings. J Coat Technol Res 20, 173–186 (2023). https://doi.org/10.1007/s11998-022-00665-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11998-022-00665-3