
Abstract
Purpose of review
Autoimmune encephalitis (AE) is increasingly recognized as a treatable cause of encephalitis in children. While prior observational studies demonstrate improved motor outcomes with early immunotherapy, less is known about long-term management and treatment for relapsing disease. In this review, we present current treatment approaches to pediatric AE, in particular relapse risk and treatment for relapsing AE in children.
Recent findings
A recent meta-analysis of anti-NMDAR encephalitis demonstrated that disease onset in adolescence was associated with an increased odds of relapse whereas treatment with rituximab and IVIG for 6 months or longer were associated with a non-relapsing course. However, no specific pediatric sub-analyses were reported. A single-center study on adult and pediatric AE showed that rituximab use was associated with a reduction in time to relapse and recurring relapses although the data for the pediatric cohort did not achieve statistical significance.
Summary
The use of second-line immunotherapy during the initial attack may reduce the risk for relapsing disease in pediatric AE. Larger studies are needed to investigate relapse risk and treatment in both anti-NMDAR and non-NMDAR encephalitis in children.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Autoimmune encephalitis (AE) is an inflammatory brain disease that affects both adults and children. Antibody-associated encephalitis encompasses a broad spectrum of diseases affecting the central nervous system (CNS). In this review, the term “autoimmune encephalitis” refers to those AE associated with auto-antibodies directed at the cell surface or synaptic proteins such as ion channels or synaptic receptors, or directed at intracellular antigens in the central nervous system. Each autoantibody associates with a specific clinical syndrome. In general, antibodies that target the cell surface or synaptic proteins have been shown to be pathogenic and may be paraneoplastic or non-paraneoplastic in origin [1]. Patients with these antibodies often have good treatment response to immunotherapy [2]. Antibodies targeting intracellular antigens are not typically directly pathogenic and are almost always a paraneoplastic phenomenon. Disorders associated with intracellular antigens generally have poorer immunotherapy response. However, these disorders rarely occur in children.
While the overall prevalence for AE is 13.7/100,000, similar to that of infectious or viral encephalitis, when subcategorizing for definite antibody positive AE (including intracellular, synaptic receptors, ion channels, and cell surface antibodies), the prevalence is lower and closer to 6.5/100,000 [3]. A study on pediatric antibody-mediated AE from the Netherlands showed a prevalence of 1.18–1.54/1,000,000 between 2015 and 2018 that included anti-N-methyl-D-aspartate receptor (NMDAR), anti-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA), and anti-leucine-rich glioma-inactivated protein 1 (LGI1) encephalitis [4]. NMDAR encephalitis (NMDARE) is the most common AE subtype in children, typically presenting in early to mid-adolescence [5, 6••]. Approximately 20% of anti-NMDARE cases are associated with ovarian teratomas, but this frequency is less in children [6••, 7]. In anti-NMDAR encephalitis, the auto-antibodies crosslink the NMDA receptor causing receptor internalization and subsequent changes in synaptic transmission [8]. Glutamic acid decarboxylase 65 (GAD65) is another common antibody in pediatric AE [1]. However, it should be noted that GAD65 antibodies may be mildly elevated non-specifically in the serum [1]. In such cases, the presence of a specific clinical syndrome (limbic encephalitis, cerebellitis, or stiff person syndrome) along with an elevated GAD antibody titer in CSF is helpful to determine the relevance for the antibody positivity. Other antibodies reported in children are more rare, including metabotropic glutamate receptor 5 (GluR5), γ-aminobutyric acid A receptor (GABAAR) and AMPA receptor, GFAP, and Ma2 [1, 2, 9,10,11,12,13,14,15]. Symptoms for AE vary depending on the antibody subtype, but most commonly, patients present with acute to subacute onset of neuropsychiatric symptoms, altered mental status, sleep disturbances, seizures, and/or movement disorders. Unlike adult onset AE, pediatric AE typically do not present with discrete clinical syndromes such as limbic encephalitis or cerebellar ataxia but rather with symptoms related to neuropsychiatric complaints or developmental regression [1].
Diagnostic evaluation
Pediatric AE differs from adult onset AE in clinical findings, comorbidities, prognosis, and treatment response [1]. Current diagnostic evaluation for pediatric AE is based on a consensus statement from an expert panel and includes symptom time course (acute vs subacute), clinical evidence of neurologic dysfunction, and paraclinical evidence of neuroinflammation (cerebrospinal fluid (CSF) analysis, magnetic resonance imaging (MRI), positive serology in serum and/or CSF, and exclusion of alternative causes) [1]. Other diagnostic evaluations including brain biopsy can be considered if the diagnosis remains uncertain or concerning for an alternative neoplastic or autoimmune process, as the diagnosis for AE does not require brain biopsy [1]. Cellucci et al. proposed a diagnostic classification for pediatric AE which includes possible AE, probable antibody-negative AE, or definite antibody-positive AE [1]. This criteria [1] includes myelin oligodendrocyte glycoprotein (MOG) antibody associated with AE phenotype. However, since these guidelines were published, separate criteria for MOG antibody-associated disease (MOGAD) was published by Banwell et al. in 2023 which defined MOGAD as a distinct entity for which cortical cerebral encephalitis is a rare subtype [16].
Diagnosing AE in pediatric patients necessitates a broad differential consideration for potential mimickers. The timing of symptom onset and a thorough review of systems along with salient features on diagnostic testing can help narrow the diagnosis. Differential diagnoses include infectious meningitis/encephalitis, CNS demyelinating disorders (acute disseminated encephalomyelitis, MOGAD, and multiple sclerosis), rheumatologic diseases (systemic lupus erythematous, neurosarcoidosis, primary CNS vasculitis, and systemic vasculitis), malignancy (gliomas, lymphoma), metabolic or mitochondrial disease, and primary psychiatric disease [5]. Work up should include blood tests, CSF evaluation, neuroimaging (MRI brain and spine), and EEG if clinically relevant. Laboratory testing should be broad to screen for infection (meningoencephalitis panel including enterovirus, herpes simplex virus, varicella zoster virus, West Nile virus), CSF cell counts and differential, oligoclonal bands, inflammatory markers (C-reactive protein, sedimentation rate), C3 and C4 complement, anti-nuclear antibody, metabolic screening (vitamin B12 level, vitamin D level, lactate, thyroid function, and copper/ceruloplasmin), and autoimmune encephalopathy antibody panels from serum and CSF [1, 5]. A malignancy screening should be pursued, depending on the antibody subtype. If anti-NMDAR antibody is found, an abdominal and pelvic MRI or pelvic/transvaginal/testicular ultrasound should be performed to evaluate for ovarian teratoma in female patients and testicular tumors in male patients [5]. Other than anti-NMDAR antibody AE, paraneoplastic syndromes are less frequent than in adults.
Relapsing autoimmune encephalitis
Clinical relapsing AE is defined as a return of previously resolved symptoms or new or acute worsening neuropsychiatric symptoms, seizures, or other neurologic symptoms after at least 1 month of clinical stability [6••, 7]. Relapses in pediatric AE are mostly reported as frequencies. In children with AE, relapses occur in approximately 10–30% of patients, which varies depending on antibody subtype [6••, 7, 17,18,19,20,21,22,23, 24••, 25, 26]. A retrospective cohort study on pediatric and adult AE found that among 30 pediatric patients with AE, frequency of at least one relapse in antibody positive AE was 31% [6••]. For anti-NMDARE (adult and children), relapse frequency range from 8.3 to 32.6% [17,18,19]. In a study of pediatric anti-NMDARE only, 21% had a relapse, and the median time to first relapse was 31.5 months [10]. A meta-analysis found that adolescent age at disease onset was associated with relapsing disease [27]. While most relapse data are from anti-NMDARE cohorts, there are published cases of relapses in other types of AE. Combining pediatric and adult cases, relapse frequencies are 15.4–40% for LGI-1, 25% for CASPR2, and 9% for GABAR, and while the focus of this chapter is antibody positive AE, relapses have also been reported for antibody negative AE at frequency of 11.8–35.1% [6••, 19, 24••, 28, 29]. Relapse frequencies appear to decrease in more recent years likely due to earlier disease recognition and diagnosis as well as more aggressive early immunotherapy [19, 30].
Current treatment strategies
Immunotherapy should be started as soon as the diagnosis is suspected with reasonable exclusion of other etiologies (Fig. 1). First-line immunotherapies include intravenous (IV) steroids, intravenous immunoglobulins (IVIG), and plasma exchange (PLEX) [2, 28, 31,32,33]. Up to half of patients may not respond to first-line treatment within the first month. Therefore, second-line treatment should be considered if there is no improvement within 10–14 days of initiating first-line therapy [34].

Proposed treatment algorithm
Second-line immunotherapies include disease-modifying therapies (DMT) such a rituximab and cyclophosphamide. Steroid-sparing oral DMTs such as mycophenolate mofetil or azathioprine are also used in the outpatient setting and should be considered for patients where risk of relapse is high [2, 28, 32,33,34, 38].
One of the most common DMTs is rituximab, an anti-CD20 monoclonal antibody that binds to CD20 expressing B cells causing complement and cell-mediated destruction of B cells [34, 35]. Side effect includes infusion reactions, hypogammaglobulinemia, and liver enzyme elevation [34, 36, 37]. Induction dosing is typically 500–750 mg/m2 (max 1000 mg) for two doses spaced 14 days apart or 375 mg/m2 once weekly for 4 doses [5, 7]. Maintenance immunotherapy is not usually required. If repeat dosing is indicated, subsequent dosing is generally 350–750 mg/m2 (max 1000 mg) every 6 months or based on CD19 counts [34]. Another B cell depleting agent is inebilizumab, an anti-CD19 monoclonal antibody. The ExTINGUISH Trial of Inebilizumab in NMDAR Encephalitis (ExTINGUISH) is underway (clinicaltrials.gov, NCT04372615) to investigate the safety and efficacy of inebilizumabin adult patients with anti-NMDARE. Cyclophosphamide, an alkylating agent that inhibits DNA synthesis affecting both B and T cell populations [34], is another second-line DMT used in AE. Although rituximab is generally preferred over cyclophosphamide due to its safety profile, cyclophosphamide is used for cases that are refractory to rituximab. Side effects of cyclophosphamide include infections, malignancy and fertility risks [34]. For cyclophosphamide, the intravenous formulation is preferred over oral due to fewer side effects. Induction dosing is 500–1000 mg/m2 (max 1500 mg) monthly for 3–6 months depending on severity and therapy response [7, 34].
Safety monitoring is recommended prior to starting all DMTs. Prior to starting B-cell depleting agents, patients should be screened for tuberculosis, hepatitis B and C infections, varicella IgG, liver enzymes, complete blood count, and baseline immunoglobulins levels (IgA, IgM, and IgG) and B and T cell counts. If re-dosing is warranted, CD19 counts should be obtained 3 months after initial dose and repeated every 3–6 months to determine timing of the next dose. Infusion reactions are often mitigated with pre-hydration, anti-pyretic therapy, anti-histamines, and IV steroids. Monitoring for cyclophosphamide include complete blood counts and urinalysis. All DMTs are immunosuppressive; thus, patients should take extra precaution with infection exposures and stay to date on routine and seasonal vaccinations except live vaccines during the treatment period.
Treating refractory cases
Several hypotheses exist on the pathophysiology of refractory AE. Long-lived plasma cells that do not express CD20 and produce autoantibodies even after pre-B cell and mature B-cells depletion are thought to be involved [31]. Drug penetration is another consideration. Rituximab does not cross the blood brain barrier and may not target immune cells in the CNS compartment [31]. AE subtypes with intracellular-targeted antibodies often lead to T cell mediated cytotoxicity. This subtype of AE is typically not responsive to immunotherapy with the exception of anti-Ma2 [2]. In these cases, treatment with cyclophosphamide or oral agents should be considered earlier. If the AE is paraneoplastic, treatment of the primary tumor (surgical removal and/or chemotherapy) should be initiated as soon as possible. However, even if there is tumor resection, acute immunotherapy is still recommended.
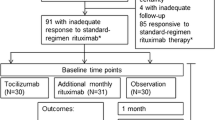
Third-line immunotherapy includes tocilizumab and bortezomib. Toclizumab is a monoclonal antibody directed at interleukin(IL)-6 involved in both T and B cell signal pathways [39], though it should be noted that data is limited for tocilizumab use in pediatric AE. In children, most published literature regarding tocilizumab use is in patients with anti-NMDARE [7, 40]. Lee et al. published an institutional cohort study of adult AE refractory to rituximab treatment comparing next steps in treatment with tocilizumab, additional rituximab and observation. The results showed that tocilizumab produced favorable mRS scores 2 months from treatment and at follow-up, and the majority of patients maintained a long-term favorable response. In this study, 28.6% of patients had antibodies to NMDA receptors, 3.3% to LGI-1, and 2.2% to amphiphysin [39]. Jaafar et al. published a case of an 8-year-old with refractory anti-GAD65 AE who responded well to tocilizumab [41]. The CIELO trial (clinicaltrials.gov, NCT05503264) is an ongoing Phase 3 study on the efficacy of satralizumab, a monoclonal antibody directed at soluble and membrane bound IL-6 receptors, for adults with NMDAR or LGI1 AE. Pending the results of these adult clinical trials, anti-IL6 therapy may be recommended earlier in the AE treatment algorithm.
Bortezomib is a proteosome inhibitor targeting plasma cells. Its use in pediatric patients is mostly published in refractory anti-NMDARE cases [7, 42,43,44,45,46,47,48]. In a study of both pediatric and adult patients (n = 5) treated with bortezomib for refractory NMDARE, all patients had symptom improvement and at 1-month follow-up improved modified Rankin Scale (mRS) scores. One patient reported mild diarrhea [45]. Cordani et al. published a case of an 8-year-old female with relapsing NMDARE and relapsing status epilepticus who received bortezomib and had symptom resolution 18 months after the initial attack [42]. Other disease-modifying therapies that have been used for refractory AE with very limited pediatric data include daratumumab, tofacitinib, inebilizumab, intrathecal methotrexate, natalizumab, and rapamycin [31, 39, 49]. Schiebe et al. described a 16-year-old patient with refractory anti-NMDARE who was treated with daratumumab with improvement in mRS score although with side effects of fever and tracheobronchitis [50].
Chronic immunotherapy and treating relapsing disease
The decision to start maintenance immunotherapy is still debated among experts [51]. Patients who recover well after their initial attack may be monitored off maintenance immunotherapy. However, for patients who were refractory or had a severe initial course (e.g., ICU admission), maintenance immunotherapy may be considered immediately following the initial attack to reduce the risk for relapse regardless of tumor status [7]. While there are case reports of relapses occurring up to 13 years after the initial episode [17], most occur within 2–3 years and less commonly earlier [6••, 17, 18]. Thus, chronic immunotherapy may not be necessary beyond the first 2–3 years. However, more research is needed to identify patients at risk for relapsing disease.
It should be noted that data for treating relapsing disease in children is limited. Relapse treatment follows a similar algorithm as acute treatment and refractory disease (Fig. 1). Current international consensus recommendations for treatment of NMDARE based on expert opinion recommend a similar treatment approach in both relapsing and refractory disease [7]. If a patient relapsed on a particular maintenance therapy, an alternative immunosuppression agent with a different mechanism should be used. Third-line agents can again be considered if a patient fails to improve within 1–3 months. Patients with relapses are likely to require a longer course of chronic immunotherapy, generally at least two years [7].
Several studies suggest that a lower relapse rate is observed in patients who receive earlier second-line treatment [17, 19, 30]. A more recent study using a survival model demonstrated that rituximab treatment during the initial attack reduces the risk of relapse by 51%, combining pediatric and adult cases [6••]. In the pediatric population, rituximab was associated with a reduced hazard ratio of 0.30 (95% CI 0.05–1.69) for time to first relapse after adjusting for age of onset, sex and presence of tumor. Rituximab was associated with a lower HR of 0.37 (95% CI 0.09–1.50) for recurring relapses after adjusting for IV steroid use, time to treatment and presence of tumor. The large confidence intervals for the pediatric population is likely due to lower sample size (n = 30) and the fact that 76% of the pediatric antibody-positive cases were anti-NMDARE [6••]. A meta-analysis of NMDARE (adult and pediatric combined) demonstrated a lower odds of relapse in patients who were treated with rituximab (OR 0.17, 95% CI 0.05–0.42) and IVIG for > 6 months (OR 0.16, 95% CI 0.07–0.33) [27].
Treatment outcomes
Treatment outcomes in pediatric cohorts are largely published in the context of anti-NMDARE. Studies with both pediatric and adult patients with NMDARE demonstrate improved outcomes with early immunotherapy [17, 19]. In a study evaluating prognosis in patients with anti-NMDARE within the first 24 months of diagnosis, 78.6% of patients had an mRS of 0–2, though 5.9% died from the disease [19]. Patients continued to improve for 18 months. Predictors of good outcome in patients with NMDARE included no intensive care unit admission and early treatment in both pediatric and adult patients [19]. A recent multi-center study on MRI features and correlation to clinical outcomes in pediatric NMDARE demonstrated that patients with T2-hyperintense lesions on initial MRI particularly in the frontal and occipital lobes had poorer outcomes (mRS greater than or equal to 3) at 1-year follow-up [52]. The same study showed that poorer outcomes at 1 year were associated with prolonged hospital stay, ICU admission, intubation, gastrostomy placement, treatment with PLEX, and/or with second-line therapies (rituximab, cyclophosphamide) and no improvement < 4 weeks from symptom onset.
Tumor risk varies depending on the type of autoantibody and overall is lower in children than adults. For example, anti-Ma2 AE caused by antibodies directed at an intracellular antigen is commonly a paraneoplastic syndrome in adults [53]. However, pediatric cases do not have a strong paraneoplastic association and may have partial response to immunotherapy [54]. Nevertheless, if a pediatric patient has a clinical relapse, even if no primary malignancy or growth was found during the initial attack, repeat tumor surveillance should be done 1–2 years after initial episode [5]. If found, treatment for any oncologic process that could be contributing should be pursued [32]. In refractory or severe cases, positron emission tomography (PET) scans can be considered in collaboration with radiology and oncology [5].
Conclusion
Pediatric AE is an increasingly recognized as a treatable cause of encephalitis with anti-NMDARE being the most common subtype. While there is strong data to support early immunotherapy to improve clinical outcomes, there is less data on long-term management and treatment strategies to reduce the risk for relapsing disease. Maintenance immunotherapy after the initial episode depends on severity of the clinical course, but there is evidence suggesting that early treatment with second-line B cell depleting agents may reduce relapse risk. Additional studies with larger cohorts are needed to further evaluate epidemiological and social risk factors for relapses in children as well as the impact of immunotherapy in children for both anti-NMDARE and non-NMDARE.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Cellucci T, Van MH, Graus F, Muscal E, Gallentine W, Klein-Gitelman MS, et al. Clinical approach to the diagnosis of autoimmune encephalitis in the pediatric patient. Neurol Neuroimmunol Neuroinflamm. 2020;7(2):e663.
Barbagallo M, Vitaliti G, Pavone P, Romano C, Lubrano R, Falsaperla R. Pediatric Autoimmune Encephalitis. J Pediatr Neurosci. 2017;12(2):130–4.
Dubey D, Pittock S, Kelly C, McKeon A, Lopez-Chiriboga A, Lennon V, et al. Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann Neurol. 2018;83(1):166–77.
de Bruijn M, Bruijstens A, Bastiaansen A, van Sonderen A, Schreurs M, Smitt PS, et al. Pediatric autoimmune encephalitis: recognition and diagnosis. Neurol Neuroimmunol Neuroinflamm. 2020;7(3):e682.
Yang J, Graves J. Anti-NMDA receptor encephalitis. In: Handbook of pediatric epilepsy case studies. 2nd ed. CRC Press; 2023.
•• Yang JH, Liu EN, Nguyen L, Dunn-Pirio A, Graves J. Survival analysis of immunotherapy effects on relapse rate in pediatric and adult autoimmune encephalitis. Neurology. 2023. https://doi.org/10.1212/WNL.0000000000207746. A single center cohort study demonstrating an overall lower relapse risk with rituximab use at the time of initial presentation of autoimmune encephalitis. However, the data for relapse reduction in children and in anti-NMDARE specifically are unclear.
Nosadini M, Thomas T, Eyre M, Anlar B, Armangue T, Benseler SM, et al. International consensus recommendations for the treatment of pediatric NMDAR antibody encephalitis. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1052.
Dalmau J, Armangué T, Planagumà J, Radosevic M, Mannara F, Leypoldt F, et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: mechanisms and models. Lancet Neurol. 2019;18(11):1045–57.
Boesen M, Born A, Lydolph M, Blaabjerg M, Børresen M. Pediatric autoimmune encephalitis in Denmark during 2011–17: a nationwide multicenter population-based cohort study. Eur J Paediatr Neurol. 2019;23(4):639–52.
Cucuzza M, Pavone P, D’Ambra A, Finocchiaro M, Greco F, Smilari P, et al. Autoimmune encephalitis and CSF anti-AMPA GluR3 antibodies in childhood: a case report and literature review. Neurol Sci. 2022;43(9):5237–41.
Dalmau J, Rosenfeld M. Autoimmune encephalitis update. Neuro Oncol. 2014;16(6):771–8.
Theroux LM, Goodkin HP, Heinan KC, Quigg M, Brenton JN. Extreme delta brush and distinctive imaging in a pediatric patient with autoimmune GFAP astrocytopathy. Mult Scler Relat Disord. 2018;26:121–3.
Gravier-Dumonceau A, Ameli R, Rogemond V, Ruiz A, Joubert B, Muñiz-Castrillo S, et al. Glial fibrillary acidic protein autoimmunity: a French cohort study. Neurology. 2022;98(6):e653–68.
Flanagan EP, Hinson SR, Lennon VA, Fang B, Aksamit AJ, Morris PP, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: Analysis of 102 patients. Ann Neurol. 2017;81(2):298–309.
Dubey D, Hinson SR, Jolliffe EA, Zekeridou A, Flanagan EP, Pittock SJ, et al. Autoimmune GFAP astrocytopathy: prospective evaluation of 90 patients in 1 year. J Neuroimmunol. 2018;15(321):157–63.
Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. Lancet Neurol. 2023;22(3):268–82.
Gabilondo I, Saiz A, Galán L, González V, Jadraque R, Sabater L, et al. Analysis of relapses in anti-NMDAR encephalitis. Neurology. 2011;77(10):996–9.
Florance N, Davis R, Lam C, Szperka C, Zhou L, Ahmad S, et al. Anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis in children and adolescents. Ann Neurol. 2009;66(1):11–8.
Titulaer M, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-N-methyl-D-Aspartate (NMDA) receptor encephalitis: a cohort study. Lancet Neurol. 2013;12(2):157–65.
Carvajal-González A, Leite MI, Waters P, Woodhall M, Coutinho E, Balint B, Lang B, Pettingill P, Carr A, Sheerin UM, Press R, Press R, Lunn MP, Lim M, Maddison P, Meinck HM, Vandenberghe WVA. Glycine receptor antibodies in PERM and related syndromes: characteristics, clinical features and outcomes. Brain. 2014;137(Pt 8):2178–92.
Armangue T, Olivé-Cirera G, Martínez-Hernandez E, Sepulveda M, Ruiz-Garcia R, Muñoz-Batista M, et al. Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: a multicentre observational study. Lancet Neurol. 2020;19(3):234–46.
Hardy D. Autoimmune Encephalitis in Children. Pediatr Neurol. 2022;132:56–66.
Fang B, McKeon A, Hinson SR, Kryzer TJ, Pittock SJ, Aksamit AJ, et al. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol. 2016;73:1297–307.
•• Nosadini M, Granata T, Matricardi S, Freri E, Ragona F, Papetti L, et al. Relapse risk factors in anti-N-methyl-D-aspartate receptor encephalitis. Dev Med Child Neurol. 2019;61:1101–7. This is a meta-analysis of anti-NMDAR encephalitis demonstrating that disease onset in adolescence was associated with an increased odds of relapse whereas treatment with rituximab and IVIG for 6 months or longer were associated with a non-relapsing course. However, no specific pediatric sub-analyses were reported.
Gastaldi M, Mariotto S, Giannoccaro MP, Iorio R, Zoccarato M, Nosadini M, et al. Subgroup comparison according to clinical phenotype and serostatus in autoimmune encephalitis: a multicenter retrospective study. Eur J Neurol. 2020;27(4):633–43.
Lee S, Kim HD, Lee JS, Kang H. Clinical features and treatment outcomes of seronegative pediatric autoimmune encephalitis. J Clin Neurol. 2021;17(2):300–6.
Nosadini M, Eyre M, Molteni E, Thomas T, Irani SR, Dalmau J, et al. Use and safety of immunotherapeutic management of N-methyl-d-aspartate receptor antibody encephalitis: a meta-analysis. JAMA Neurol. 2021;78(11):1333–44.
Nosadini M, Mohammad S, Ramanathan S, Brilot F, Dale R. Immune therapy in autoimmune encephalitis: a systemic review. Expert Rev Neurother. 2015;15(12):1391–419.
van Sonderen A, Thijs RD, Coenders EC, Jiskoot LC, Sanchez E, de Bruijn MA, et al. Anti-LGI1 encephalitis: clinical syndrome and long-term follow-up. Neurology. 2016;87(14):1449–56.
Lee WJ, Lee ST, Byun JI, Sunwoo JS, Kim TJ, Lim JA, et al. Rituximab treatment for autoimmune limbic encephalitis in an institutional cohort. Neurology. 2016;86(18):1683–91.
Yang J, Liu X. Immunotherapy for refractory autoimmune encephalitis. Front Immunol. 2021;12:790962.
Goenka A, Chikkannaiah M, Kumar G. Pediatric auto-immune encephalitis. Curr Probl Pediatr Adolesc Health Care. 2021;51:101031.
Co DO, Kwon JM. Autoimmune encephalitis: distinguishing features and specific therapies. Crit Care Clin. 2022;38:393–412.
Stingl C, Cardinale K, Van Mater H. An update on the treatment of pediatric autoimmune encephalitis. Curr Treatm Opt Rheumatol. 2018;4:14–28.
Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–88.
McAtee CL, Lubega J, Underbrink K, Curry K, Msaouel P, Barrow M, et al. Association of rituximab use with adverse events in children, adolescents, and young adults. JAMA Netw Open. 2021;4(3):e2036321.
Oh M, Lee H. A study of hepatitis B virus reactivation associated with rituximab therapy in real-world clinical practice: a single-center experience. Clin Mol Hepatol. 2013;19(1):51–9.
Dinoto A, Ferrari SMS. Treatment options in refractory autoimmune encephalitis. CNS Drugs. 2022;36(9):919–31.
Lee WJ, Lee ST, Moon J, Sunwoo JS, Byun JI, Lim JA, et al. Tocilizumab in Autoimmune encephalitis refractory to rituximab: an institutional cohort study. Neurotherapeutics. 2016;13(4):824–32.
Lee WJ, Lee ST, Shin YW, Lee HS, Shin HR, Kim DY, et al. teratoma removal, steroid, IVIG, rituximab and tocilizumab (T-SIRT) in anti-NMDAR encephalitis. Neurotherapeutics. 2021;18(1):474–87.
Jaafar F, Haddad L, Koleilat N, Sharara-Chami R, Shbarou R. Super refractory status epilepticus secondary to anti-GAD antibody encephalitis successfully treated with aggressive immunotherapy. Epilepsy Behav Rep. 2020;14:100396.
Cordani R, Micalizzi C, Giacomini T, Gastaldi M, Franciotta D, Fioredda F, et al. Bortezomib-responsive refractory anti-N-methyl-D-asparte receptor encephalitis. Pediatr Neurol. 2020;103:61–4.
Schroeder C, Back C, Koc U, Strassburger-krogias K, Reinacher-schick A, Gold R, et al. Breakthrough treatment with bortezomib for a patient with anti-NMDAR encephalitis. Clin Neurol Neurosurg. 2018;172:24–6.
Lazzarin SM, Vabanesi M, Cecchetti G, Fazio R, Fanelli GF, Volonté MA, et al. Refractory anti-NMDAR encephalitis successfully treated with bortezomib and associated movements disorders controlled with tramadol: a case report with literature review. J Neurol. 2021;268(2):741–2.
Wang T, Wang B, Zeng Z, Li H, Zhang F, Ruan X, et al. Efficacy and safety of bortezomib in rituximab-resistant anti-N-methyl-d-aspartate receptor (anti-NMDAR) encephalitis as well as the clinical characteristics: an observational study. J Neuroimmunol. 2021;354:577527.
Keddie S, Crisp SJ, Blackaby J, Cox A, Coles A, Hart M, Church AJ, Vincent A, Zandi MLM. Plasma cell depletion with bortezomib in the treatment of refractory N-methyl-d-aspartate (NMDA) receptor antibody encephalitis. Eur J Neurol. 2018;25(11):1384–8.
Scheibe F, Prüss H, Mengel AM, Kohler S, Nümann A, Köhnlein M, et al. Bortezomib for treatment of therapy-refractory anti-NMDA receptor encephalitis. Neurology. 2017;88(4):366–70.
Behrendt V, Krogias C, Reinacher-Schick A, Gold RKI. Bortezomib treatment for patients with anti-N-methyl-d-aspartate receptor encephalitis. JAMA Neurol. 2016;73(10):1251–3.
Wong K-H, Day G, Titulaer M, Torner J, Cudkowicz M, Coffey C, et al. The ExTINGUISH trial: a Phase-2B randomized placebo-controlled trial of inebilizumab in anti-NMDA receptor encephalitis. Neurology. 2022;99:S39–40.
Scheibe F, Ostendorf L, Prüss H, Radbruch H, Aschman T, Hoffmann S, et al. Daratumumab for treatment-refractory antibody-mediated diseases in neurology. Eur J Neurol. 2022;29(6):1847–54.
Abboud H, Probasco JC, Irani S, Ances B, Benavides DR, Bradshaw M, et al. Autoimmune encephalitis: proposed best practice recommendations for diagnosis and acute management. J Neurol Neurosurg Psychiatry. 2021;92(7):757–68.
Gombolay G, Brenton JN, Yang JH, Stredny CM, Kammeyer R, Otten CE, et al. MRI features and their association with outcomes in children with anti-NMDA receptor encephalitis. Neurol Neuroimmunol Neuroinflamm. 2023;10:e200130.
Dalmau J, Graus F, Villarejo A, Posner JB, Blumenthal D, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain. 2004;127(Pt 8):1831–44.
Kim H, Yum MS, Kim MJ, Ko TS. A Rare Case of Anti-Ma2 antibody-mediated autoimmune encephalomyelitis in childhood. Ann Child Neurol. 2022;30(3):148–51.
Author information
Authors and Affiliations
Contributions
M.Y. and J.Y. wrote the main manuscript text. J.Y. prepared Fig. 1. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Conflict of Interest
Maayan J. Yakir declares that he has no conflict of interest. Jennifer H. Yang declares that she has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yakir, M.J., Yang, J.H. Treatment Approaches in Pediatric Relapsing Autoimmune Encephalitis. Curr Treat Options Neurol 26, 139–149 (2024). https://doi.org/10.1007/s11940-024-00786-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11940-024-00786-7