
Abstract
Purpose of review
The purpose of this review is to highlight the molecular and clinical characteristics of the cryopyrin-associated periodic fever syndrome (CAPS) and its management. CAPS is an autosomal dominantly inherited autoinflammatory disorder associated with mutations in the NLRP3 gene, which ultimately lead to excessive production of interleukin-1β (IL-1β) and systemic inflammation. Typical systemic features include fever, urticarial rash and arthralgia, and ultimately amyloidosis. There are also multiple neurological manifestations including, but not restricted to, headache, sensorineural hearing loss, aseptic meningitis, myalgia and optic nerve involvement.
Recent findings
Since the recognition of CAPS as a single disease entity and discovery of the underlying causative gene, there has been a major breakthrough in terms of its treatment by pharmacological IL-1β inhibition. Highly targeted therapies against IL-1 have been shown to be remarkably effective in the treatment of CAPS and make early diagnosis of this condition crucial. It is hoped that starting pharmacological intervention in a timely manner will prove neuroprotective. There are three drugs licensed for treatment of CAPS; canakinumab, anakinra and rilonacept. The former two are widely used: canakinumab is a fully humanised anti-IL-1β monoclonal antibody administered as a subcutaneous injection once every 8 weeks starting at a dose of 150 mg in patients weighing more than 40 kg. Anakinra is a recombinant form of the IL-1 receptor antagonist and the adult daily dose is 100 mg subcutaneously.
Summary
CAPS is a highly debilitating disorder characterised by unregulated IL-1β production driven by autosomal dominantly inherited mutations in the NLRP3 gene. Effective therapies targeted against IL-1 are now available and are vital to prevent long-term complications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The cryopyrin-associated periodic fever syndrome (CAPS) is a multisystem genetic autoinflammatory disorder, which has a wide range of systemic as well as neurological manifestations. The cardinal symptoms of CAPS include episodes of fever, urticarial rash, red eyes and arthralgia [1], whilst neurological manifestations include headache, sensorineural hearing loss and optic nerve involvement [2••, 3, 4]. At the turn of the twenty-first century, the discovery of a common genetic locus, the NLRP3 gene on chromosome 1q44, resulted in the recognition that a number of previously described clinical syndromes, including chronic infantile neurological, cutaneous and articular syndrome (CINCA), Muckle-Wells syndrome (MWS) and familial cold autoinflammatory syndrome (FCAS) were part of a wider clinical entity. The term CAPS began to gain recognition to describe the spectrum of autosomal dominant and apparently sporadic diseases caused by mutations in NLRP3 [5, 6].
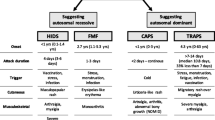
CAPS is rare and is estimated to affect one to three people per million of the population with no specific demographic predilection identified [7]. A large European registry [8••] has provided key clinical data regarding CAPS. Median age at onset was 0.8 years (interquartile range 0.1–5) with age at diagnosis of 15 years (IQR 5–36). Forty percent of patients experience a recurrent course, 40% chronic and 20% chronic with exacerbations. Most attacks resolve within 24 h (48%) but 36% can last more than 3 days. In terms of systemic features, skin involvement was the most commonly reported symptom (97%). Fever is seen in 84% and arthralgia in 86%. Patients still tend to be subdivided into FCAS, MWS and CINCA, although it is becoming increasingly clear that there is substantial overlap between these groups, and CAPS is best regarded as a continuous rather than discrete severity spectrum.
Familial cold autoinflammatory syndrome
Symptoms consistent with the FCAS phenotype most commonly develop in the first few years of life and are typified by fever, urticaria, conjunctival inflammation and arthralgia, which can occur from 30 min to 48 h following cold exposure and resolve typically after a few hours. Secondary AA amyloidosis is very rare in FCAS, but despite being the mildest CAPS phenotype, FCAS often has a severe impact on patient quality of life with symptoms precluding outdoor activities in over 95% of patients [9].
Muckle-Wells syndrome
MWS is now recognised to be an intermediate phenotype of CAPS characterised by chronic or intermittent episodes of fever, headache, urticarial like rash, red eyes, arthralgia or arthritis, often in the absence of a specific trigger [10]. Febrile attacks commence in early childhood and patients often develop progressive sensorineural hearing loss. Approximately 25% of untreated patients develop AA amyloidosis secondary to chronic inflammation [11].
Chronic infantile neurological cutaneous and articular syndrome
CINCA is viewed as the most severe CAPS phenotype [6]. In general, children present during the first days of life with a chronic urticarial rash associated with persistent low-grade fever and elevation of acute phase reactants. Patients frequently have a typical facial appearance, with frontal bossing, large cephalic perimeter and saddle-back nose [12••]. They develop permanent neurological damage with chronic aseptic meningitis, raised intracranial pressure and hydrocephalus, brain atrophy and chronic papilloedema [8••]. Chronic irritability, intellectual disability, headache, lower limb spasticity and more rarely seizures can occur [10]. There can be eye inflammation from anterior to posterior including conjunctivitis, uveitis and optic neuritis. Papilloedema due to raised intracranial pressure and optic neuritis can result in optic atrophy [13]. Cochlear inflammation leads to progressive stepwise sensorineural hearing loss in the first years of life. Approximately 60% of patients have prominent arthropathy, usually during childhood, causing deformity and degenerative arthropathy leading to contractures [12••]. Mortality is high, and death in childhood well recognised. Chronic inflammation leads to the deposition of AA amyloid and renal failure [12••].
Molecular pathogenesis of CAPS
CAPS is caused by heterozygous gain-of-function mutations within the NLRP3 gene, located on chromosome 1q44, which can be inherited in a dominant fashion with variable penetrance and expression, or arise de novo, the latter usually in CINCA. NLRP3 encodes cryopyrin, a member of the intracellular nucleotide-binding oligomerisation domain-like receptors (NLRs). NLRs are pattern recognition receptors which recognise a variety of pathogen-associated molecular pattern molecules (PAMPs) [10]. All NLRs contain a NACHT domain (common to NAIP (neuronal apoptosis inhibitory protein), CIITA (MHC class II transcription activator), HET-E (incompatibility locus protein from Podospora anserina) and TP1 (telomerase-associated protein)) that regulates autoaggregation and oligomerisation. Oligomerisation of cryopyrin results in activation of caspase-1. This catalyses the cleavage of prointerleukin-1β to interleukin-1β, and is the key step in its regulation. Interleukin-1β is a potent proinflammatory cytokine with pleiotropic effects including fever, vasodilatation and production/activation of other inflammatory cytokines [7]. Mutations in NLRP3 therefore cause overactivation of the IL-1β pathway, a cascade of complex cellular events and ultimately aberrant homeostatic tissue responses (Fig. 1) [8••, 10].

Schematic representation of the NLRP3 inflammasome. The activation of NLRP3 by a variety of pathogen-associated molecular pattern molecules interacting with the leucine-rich repeat (LRR) domain leads to aggregation and oligomerisation of a molecular complex which in turn activates caspase-1, thereby promoting the production of the proinflammatory cytokine interleukin-1β. In CAPS, gain-of-function mutations of NLRP3, often in the NACHT domain, result in overactivation of this pathway. Other domains of the assembling macromolecule labelled in the diagram are CARD (caspase activation and recruitment domain), PYD (pyrin domain) and FIIND (“function-to-find” domain).
Around 170 NLRP3 variants have been associated with CAPS. The majority of mutations identified have been observed within exon 3, which is responsible for encoding the NACHT domain, crucial for cryopyrin oligomerisation [12••]. In CINCA, mutations in other exons encoding the leucine rich repeat PAMP recognition domain are also known. Although patients with FCAS and MWS tend to show familial inheritance, CINCA usually occurs de novo. Approximately 50–65% lack mutations in NLRP3 by conventional Sanger sequencing. It is now recognised that many of these are mosaics for very damaging mutations, but other genes are also likely to be implicated in the phenotype [12••].
Neurological manifestations of CAPS
The variety of neurological manifestations reported in the literature highlights the importance of considering CAPS as a potential differential diagnosis in a wide range of neurological presentations.
A selection of articles that report the neurological manifestations of CAPS is summarised in Table 1. Key neurological features of CAPS include headache, with a notably high proportion of patients with phenotypic features consistent with migraine or chronic daily headache [2••, 3, 4]. In addition, more sinister headache phenotypes with evidence of raised intracranial pressure and aseptic meningitis have been reported [2••, 4, 8••]. Papilloedema is seen in around 30% of patients [2••, 3, 8••].
Sensorineural hearing loss occurs in approximately 40% of patients, many requiring hearing aids or cochlear implantation. As hearing loss onset is often in mid to late childhood, many of these patients acquire normal speech. Mild ocular manifestations such as conjunctivitis (66%) or uveitis (7%) are frequently reported [8••]. Severe ocular involvement, including optic atrophy, cataract and impaired vision, is less common [2••, 3, 4, 8••].
Although the numbers of patients are small, there is evidence that specific genotypes may influence the extent to which neurological symptoms manifest. Data from the cohort based in the UK suggested patients with the T348M mutation tended to have a more severe neurological phenotype with an earlier age of onset [2••]. Data from an observational study in France were consistent with the UK data and found that the R260W mutation was more likely to be associated with neurological involvement in patients compared to those with the A439V mutation [3]. The E311K mutation is reported to be associated with a higher rate of hearing loss [17].
In one interesting study, 108 patients in a neuroimmunology clinic were screened for NLRP3 mutations on the basis of systemic symptoms compatible with mild CAPS. The authors reported 17 (16%) patients had a V198M or Q703K NLRP3 variant [14]. The interpretation of these variants is contentious as they occur at a high frequency in the healthy population; V198M has an allele frequency of 0.007, and Q703K is seen in 10% of healthy controls. In vitro functional studies have suggested that V198M has an intermediate effect on inflammasome activation and IL-1β production compared to definite pathogenic mutations and wild-type controls. Many of these patients reported neurological features (e.g. headache and sensorineural hearing loss). Nine patients underwent anti-IL-1 therapy, with 5 reported to have responded to treatment [14].
A single case report recently described the diagnosis of CAPS in a patient with Tolosa-Hunt syndrome after genetic testing [16], whilst another case report highlighted chorea as a manifestation of CAPS [15]. Cognitive dysfunction may also be a feature of CAPS, with a study from a specialist centre in France reporting educational difficulties in the majority of adults (61%) and children (50%) [3].
Management of CAPS
A suggested approach to the management of CAPS is shown in Table 2. Of prime importance once a diagnosis is genetically confirmed is the institution of disease modifying therapy in the form of IL-1 inhibition. Canakinumab is a fully human anti-IL-1β monoclonal antibody administered as a subcutaneous injection once every 8 weeks starting at a dose of 150 mg. In a double-blind placebo-controlled randomised withdrawal study, there was evidence of a complete response to canakinumab in 78% of drug-naive patients [18, 19]. Monitoring of systemic disease activity in the form of serum amyloid-A protein (SAA) levels is the most sensitive marker of CAPS disease activity and predictor of the risk of systemic AA amyloidosis. Ninety percent of patients treated with canakinumab showed complete normalisation of SAA by day 8 post treatment, as well as concomitant decrease in C-reactive protein (CRP) and resolution of symptoms [18, 19].
In addition to canakinumab, anakinra, a recombinant form of the IL-1 receptor antagonist, delivered as a daily dose of 100 mg via subcutaneous injection [20], is also licensed in CAPS. Anakinra was the first commercially available anti-IL-1 agent and has been shown to be effective against IL-1-driven autoinflammatory disease [20, 21].
CAPS is a lifelong illness that affects women of child bearing age, and optimal control of symptoms during pregnancy is of utmost importance for the health of both mother and foetus. There are safety data available for anakinra in pregnancy [22]. An observational study of 15 pregnancies reported no preterm births or serious complications of pregnancy using anakinra throughout gestation, whilst providing relief of CAPS symptoms. An international collaborative study has reported that paternal exposure to IL-1 blockade appears uncomplicated in an experience of 12 cases to date. Maternal data on 23 women exposed to anakinra and 8 exposed to canakinumab are generally reassuring but the numbers remain small and there is concern about renal agenesis [23••]. Experience is that many patients are unable to discontinue treatment due to rapid recurrence of autoinflammatory disease, and individual preconception risk benefit discussion with prospective parents is strongly recommended.
As previously highlighted, headaches, and in particular migraine, are a common neurological manifestation of CAPS [2••, 3, 4]. Migraines are also common in the wider population [24] and may be unrelated to underlying CAPS pathology. It is therefore important to correlate headache and other migrainous symptoms with systemic disease activity, and in many cases migraines may resolve with disease modification [2••]. However, effective symptomatic and preventative treatments for migraine and other primary headache disorders are available and should be considered in addition to disease modification in CAPS on a patient by patient basis, using international guidelines [25].
Hearing loss is a common manifestation of CAPS. A recent study where CAPS patients underwent detailed auditory testing found that 100% of symptomatic CAPS patients had high frequency pure tone averages abnormalities, whilst 74% had an abnormal assessment focusing on standard frequencies (0.5–4 kHz) [26]. Encouragingly, treatment with IL-1 blockade resulted in improved or stable hearing in 91% of patients [26]. However, it is important to note that once sensorineural damage has occurred, hearing dysfunction is likely irreversible. This highlights the importance of regular and detailed auditory screening, as well as early institution of disease modification to prevent CAPS-related hearing loss. Beyond prevention, advances in hearing aids and cochlear implants are ongoing [27], and obtaining the relevant opinions from otolaryngology and audiology colleagues is of paramount importance in patients with established hearing loss. Similarly, input from ophthalmology colleagues is crucial as eye involvement at any level from the cornea to the optic nerve is common and can be sight threatening [2••, 4, 8••]. We would recommend annual assessment in all cases, with more frequent assessment if abnormalities are detected.
Although the majority of CAPS patients have clinically normal brain MRI [2••, 4], cranial imaging can be helpful as white matter hyperintensities are sometimes observed [2••, 3, 14, 15], and in some patients, have been reported to improve after anti-IL-1 treatment [15]. Interestingly, in the aforementioned cohort of patients from a neuroimmunology clinic, a much higher proportion of patients (88%) had MRI brain abnormalities, often in the form of white matter lesions [14].
In addition to the neurological manifestations, monitoring of systemic symptoms is important. If there is a clinical suspicion of systemic AA amyloidosis, which can be ultimately life threatening in the form of end-stage renal failure [11], a renal biopsy or serum amyloid P (SAP) nuclear medicine scan is advised [28].
Management of CAPS is therefore complex and requires significant expertise and multidisciplinary involvement. In the UK, a national service is provided by the NHS National Amyloidosis Centre based in London (http://www.ucl.ac.uk/amyloidosis/nac) and referral to such centres is key to ensuring optimal management.
Conclusions
Much is known about CAPS at a molecular and genetic level, but due to the non-specific, chronic, relapsing-remitting nature of the condition, patients’ diagnosis and treatment are often delayed. Once thought to be three separate entities, CAPS is now considered a continuous clinical spectrum of a single condition, with genotype-phenotype variations. Recurrent flares of fever, skin and joint involvement and neurological symptoms should lead a clinician to consider the diagnosis of CAPS, particularly in those with a positive family history. Neurological manifestations range from the most commonly occurring headache and sensorineural hearing impairment to severe features such as hydrocephalus and aseptic meningitis. Systemic inflammatory markers are elevated during exacerbations, as are levels of serum amyloid A protein. MR imaging may show non-specific white matter abnormalities. Diagnostic criteria with a sensitivity of 81% and specificity of 94% have recently been proposed [29]. These include raised inflammatory markers (CRP/SAA) plus at least two of six CAPS-typical symptoms (urticaria-like rash, cold-triggered episodes, sensorineural hearing loss, musculoskeletal symptoms, chronic aseptic meningitis and skeletal abnormalities) [29]. Diagnosis is confirmed by genetic testing for the NRLP3 mutation. The fully human anti-IL-1β monoclonal antibody canakinumab or the recombinant form of the IL-1 receptor antagonist anakinra are highly effective medications preventing further episodes of systemic inflammation and transforming quality of life, but will not reverse established damage such as hearing or visual impairment. Symptomatic treatment of pain, headaches and skin rash should also be provided. Identification of affected patients early in the course of the disease is crucial to prevent irreversible damage or the development of AA amyloidosis.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Touitou I, Koné-Paut I. Autoinflammatory diseases. Best Pract Res Clin Rheumatol. 2008;22(5):811–29.
•• Parker T, Keddie S, Kidd D, Lane T, Maviki M, Hawkins PN, et al. Neurology of the cryopyrin-associated periodic fever syndrome. Eur J Neurol. 2016;23(7):1145–51. A single quaternary referral centre's study on the neurological manifestations of CAPS from 38 patients, concluding that neurological symptoms are extremely common and varied in severity.
Mamoudjy N, Maurey H, Marie I, Koné-Paut I, Deiva K. Neurological outcome of patients with cryopyrin-associated periodic syndrome (CAPS). Orphanet J Rare Dis. 2017;12(1):33.
Kitley JL, Lachmann HJ, Pinto A, Ginsberg L. Neurologic manifestations of the cryopyrin-associated periodic syndrome. Neurology. 2010;74(16):1267–70.
Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29(3):301–5.
Feldmann J, Prieur A-M, Quartier P, Berquin P, Certain S, Cortis E, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71(1):198–203.
Cuisset L, Jeru I, Dumont B, Fabre A, Cochet E, Le Bozec J, et al. Mutations in the autoinflammatory cryopyrin-associated periodic syndrome gene: epidemiological study and lessons from eight years of genetic analysis in France. Ann Rheum Dis. 2011;70(3):495–9.
•• Levy R, Gérard L, Kuemmerle-Deschner J, Lachmann HJ, Koné-Paut I, Cantarini L, et al. Phenotypic and genotypic characteristics of cryopyrin-associated periodic syndrome: a series of 136 patients from the Eurofever Registry. Ann Rheum Dis. 2014;74(11):2043–9. The largest published series of CAPS patients, which used the Eurofever Registry (of autoinflammatory diseases) to identify genetic and phenotypic features of patients and identify risk factors for severe disease.
Stych B, Dobrovolny D. Familial cold auto-inflammatory syndrome (FCAS): characterization of symptomatology and impact on patients’ lives. Curr Med Res Opin. 2008;24(6):1577–82.
Posch C, Kaulfersch W, Rappersberger K. Cryopyrin-associated periodic syndrome. Pediatr Dermatol. 2012;16:1–4.
Muckle TJ. The “Muckle-Wells” syndrome. Br J Dermatol. 1979 Jan;100(1):87–92.
Finetti M, Omenetti A, Federici S, Caorsi R, Gattorno M. Chronic infantile neurological cutaneous and articular ( CINCA ) syndrome: a review. Orphanet J Rare Dis. 2016;1–11. A detailed review of CINCA syndrome, illustrated with clinical images, pathophysiological mechanisms, and radiological imaging, which summarises studies reporting clinical outcomes with anti IL-1 drugs.
Oberg TJ, Vitale AT, Hoffman RO, Bohnsack JF, Warner JE. Cryopyrin-associated periodic syndromes and the eye. Ocul Immunol Inflamm. 2013;21(4):306–9.
Schuh E, Lohse P, Ertl-Wagner B, Witt M, Krumbholz M, Frankenberger M, et al. Expanding spectrum of neurologic manifestations in patients with NLRP3 low-penetrance mutations. Neurol Neuroimmunol Neuroinflamm. 2015 Aug;2(4):e109.
Schwarzbach CJ, Schmitt WH, Szabo K, Bäzner H, Hennerici MG, Blahak C. Chorea in a patient with cryopyrin-associated periodic syndrome. Neurology. 2016;86(3):241–4.
Höhne C, Schuh E, Kümpfel T, Straube A. Cryopyrin-associated periodic fever syndrome manifesting as Tolosa-Hunt syndrome. Cephalalgia. 2016;36(14):1392–6.
Kuemmerle-Deschner JB, Lohse P, Koetter I, Dannecker GE, Reess F, Ummenhofer K, et al. NLRP3 E311K mutation in a large family with Muckle-Wells syndrome - description of a heterogeneous phenotype and response to treatment. Arthritis Res Ther. 2011;13(6):R196.
Koné-Paut I, Lachmann HJ, Kuemmerle-Deschner JB, Hachulla E, Leslie KS, Mouy R, et al. Sustained remission of symptoms and improved health-related quality of life in patients with cryopyrin-associated periodic syndrome treated with canakinumab: results of a double-blind placebo-controlled randomized withdrawal study. Arthritis Res Ther. 2011;13(6):R202.
Kuemmerle-Deschner JB, Hachulla E, Cartwright R, Hawkins PN, Tran TA, Bader-Meunier B, et al. Two-year results from an open-label, multicentre, phase III study evaluating the safety and efficacy of canakinumab in patients with cryopyrin-associated periodic syndrome across different severity phenotypes. Ann Rheum Dis. 2011;70(12):2095–102.
Koné-Paut I, Galeotti C. Anakinra for cryopyrin-associated periodic syndrome. Expert Rev Clin Immunol. 2014;10(1):7–18.
Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355(6):581–92.
Chang Z, Spong CY, Jesus AA, Davis MA, Plass N, Stone DL, et al. Brief report: anakinra use during pregnancy in patients with cryopyrin-associated periodic syndromes. Arthritis Rheumatol. 2014;66(11):3227–32.
•• Youngstein T, Hoffmann P, Gül A, Lane T, Williams R, Rowczenio DM, et al. International multi-centre study of pregnancy outcomes with interleukin-1 inhibitors. Rheumatology. 2017;149:377–84. Recently reported data from an international multi centre study covering IL-1 inhibitor use in pregnancy. Thirty-one maternal-exposed pregnancies from seven countries were studied. The data are generally reassuring, although renal agenesis was seen in a patient treated with anakinra.
Angus-Leppan H. Migraine: mimics, borderlands and chameleons. Pract Neurol. 2013;13(5):308–18.
Antonaci F, Dumitrache C, De Cillis I, Allena M. A review of current European treatment guidelines for migraine. J Headache Pain. 2010;11(1):13–9.
Kuemmerle-Deschner JB, Koitschev A, Tyrrell PN, Plontke SK, Deschner N, Hansmann S, et al. Early detection of sensorineural hearing loss in Muckle-Wells-syndrome. Pediatr Rheumatol. 2015;13(1):43.
Wilson BS. The modern cochlear implant: a triumph of biomedical engineering and the first substantial restoration of human sense using a medical intervention. IEEE Pulse. 2017;8(2):29–32.
Hawkins PN, Pepys MB. Imaging amyloidosis with radiolabelled SAP. Eur J Nucl Med. 1995;22(7):595–9.
Kuemmerle-Deschner JB, Ozen S, Tyrrell PN, Kone-Paut I, Goldbach-Mansky R, Lachmann H, et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann Rheum Dis. 2017;76(6):942–7.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
T. Parker, S. Keddie and L. Ginsberg declare that they have no conflict of interest. Dr. Helen Lachmann reports personal fees and non-financial support from SOBI, personal fees and non-financial support from Novartis.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Stephen Keddie and Thomas Parker are joint first authors.
This article is part of the Topical Collection on Neurologic Manifestations of Systemic Disease
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Keddie, S., Parker, T., Lachmann, H.J. et al. Cryopyrin-Associated Periodic Fever Syndrome and the Nervous System. Curr Treat Options Neurol 20, 43 (2018). https://doi.org/10.1007/s11940-018-0526-1
Published:
DOI: https://doi.org/10.1007/s11940-018-0526-1