
Abstract
Purpose of Review
Psoriatic arthritis and ankylosing spondylitis belong to a family of rheumatological diseases that lead to painful joint inflammation that impacts on patient function and quality of life. Recent studies have shown that the pro-inflammatory cytokine IL-17 is involved in the inflammatory joint changes in spondyloarthritides. We will review the pathophysiology of IL-17 and review the biological therapies targeting IL-17.
Recent Findings
IL-17 is produced and released from T cells and is dependent on multiple upstream cytokines, which include IL-23. There are six members of the IL-17 family that are secreted from multiple populations of T cells. The initial biologic medications have been developed against IL-17A, which is the best-studied member of this family. These medications appear to be effective in controlling joint inflammation, improving patient quality of life, and are generally well tolerated. More recently, medications have been developed that target both IL-17A and IL-17F. In addition, brodalumab, an antibody targeting the IL-17 receptor, has had a resurgence after initial concerns for an increased risk of suicide.
Summary
IL-17 is an inflammatory cytokine that is critical in the pathobiology of axial spondyloarthritides. Recent biological therapies targeting IL-17A are effective and well tolerated in patients with axial spondyloarthritis. Specific targeting of the Il-17A/F heterodimer is also effective and provides another viable option in the clinician’s armamentarium.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction to IL-17
Interleukin (IL)-17 is a family of pro-inflammatory cytokines, of which there are six known members (IL-17 A–F). IL-17A is the founding, and best-studied, member of the family, and was initially detected in murine lymphoid cells [1]. The functions of the IL-17 cytokines are varied and complex. While many IL-17 family members are pro-inflammatory and act in pathogen defence and immune-mediated disease, they also have roles in mucosal integrity, allergen response, anti-inflammation, and regulation of lymphocyte function. They can act in an autocrine manner and can influence the function of other cytokines, including other Il-17 family members. These IL-17 cytokine functions are briefly summarized in Table 1 and are fully reviewed by Zhang and colleagues [2].
The functions of IL-17A and IL-17F are the best understood, and as such are the main therapeutic targets. IL-17A and IL-17F can form homo- and heterodimers that bind to the IL-17 receptor to activate downstream signalling pathways leading to the release of pro-inflammatory cytokines [2–9]. The IL-17 receptor is formed by homo- or heterodimers of IL-17 receptor A and IL-17 receptor C (IL-17RA and IL-17RC, respectively). IL-17 receptor activation leads to canonical signalling and gene expression via the NF-κB, MAP kinase, and CCAAT/enhancer binding protein-B signalling pathways [10, 11••]. Activated IL-17 receptors can also interact with other cell surface receptors (EGF receptor, FGF receptor, NOTCH1, and C-type lectin receptor) to promote cell proliferation and tissue repair [12]. Functionally, IL-17 is involved in the body’s response to bacterial and fungal infection. IL-17 has been shown to be involved in fracture repair and osteogenesis [13, 14]. Indeed, the anatomical sites affected in axial spondylarthritis (axSpA) are sites of repeated micro-trauma [15]. However, dysregulation of IL-17 can lead to autoimmune disease [16, 17].
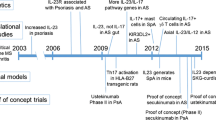
IL-17 is released from several cells, including Th17 helper cells, γ/δ T cells, interstitial lymphoid cells, neutrophils, and mast cells [18•, 19, 20]. The major source of IL-17 appears to be Th17 cells [21, 22]. The production and secretion of IL-17 cells appears to be dependent on the IL-23 cytokine, a cytokine released from antigen-presenting cells [23–27]. The manufacture and secretion of IL-17 from Th17 cells appears to be dependent upon differentiation of naïve T cells into Th17 cells and the stimulation of these cells to secrete IL-17. Th17 cell differentiation depends upon the local milieu of cytokines. Indeed, several studies have implicated a cytokine concoction, consisting of at least IL-23, TGFβ, IL-6, IL-1β, and IL-21, that can induce differentiation of naïve T cells into Th17 cells [23–27]. Conversely, the presence of IFNγ, IL-12, and IL-4 inhibits Th17 differentiation and commits naïve T cells to either Th1 or Th2 cell lineages. While IL-23 appears to have a role in Th17 differentiation, IL-23 is thought to stimulate activated Th17 cells to release IL-17 [23, 25–28]. Proof of concept comes from a mouse model of spondylarthritis, where mice treated with anti-IL-23 antibody before the onset of disease, but not after, can be protected from developing spondylarthritis [29]. Despite this evidence, IL-23 inhibitors have largely proven ineffective in treating axSpA in human trials, suggesting other cytokines like IL36, IL-1β, IL-6, and IL-7 may have important roles in IL-17 production [30].
While the release of IL-17 from Th17 cells appears to be at least in part dependent on IL-23, IL-17 production in γ/δ T cells may also be IL-23 independent. γ/δ T cells are resident cells in the ciliary body, aortic root, colon, and enthesis [31–33]. Interestingly, these anatomical locations are commonly affected in axial spondyloarthritis and psoriatic arthritis. A portion of these cells are dependent on IL-23 [33]. However, other γ/δ T cells function independently of IL-23 and have been demonstrated to participate in physiological functions such as angiogenesis and gastric epithelial integrity [31]. The IL-23-independent and IL-23-dependent pathways may both be involved in the aetiology of SpA, which includes axial spondyloarthritis and psoriatic arthritis.
Due to the IL-23/IL-17 axis’s pro-inflammatory role, there is interest in its role in pathogenesis and as a potential treatment target. The IL-23/IL-17 axis has been implicated in several rodent models of disease (rheumatoid arthritis, inflammatory bowel disease, spondylarthritis, experimental autoimmune encephalitis, asthma) [24, 27, 34–37]. These cytokines are also elevated in serum and synovial fluid from patients with lupus, rheumatoid arthritis, and ankylosing spondylitis, [38–40]. The serum of patients with psoriasis and axSpA also shows increased levels of differentiated Th17 cells [41]. This axis has also been targeted clinically with varying success in spondylarthritis (reviewed by Siebert and colleagues [42••]). The most recent agent in this class of medications is bimekizumab, which targets IL-17A and IL-17F. Below, we will review the role of the IL-23/IL-17 axis in the pathology of AxSpA including ankylosing spondylitis and psoriatic arthritis with a particular focus on IL-17A and IL-17F.
IL-17 in the Pathobiology of Ankylosing Spondylitis and Psoriatic Arthritis
Axial spondylarthritis, both radiographic and non-radiographic, is characterized by pain, stiffness, and limited range of motion of the axial skeleton and is associated with inflammation at the sacroiliac joint [43, 44]. Similarly, psoriatic arthritis is a related immune-mediated disease that affects the peripheral and axial skeleton, as well as the skin and nails. The IL-23/IL-17 axis appears to be involved in the pathogenesis of axial spondylarthritis and psoriatic arthritis. The levels of IL-17 producing cells are raised in the blood, enthesis, and synovial fluid in patients with spondylarthritis [39, 41, 45–47]. Single-nucleotide polymorphisms in the IL-23/IL-17 signalling axis are linked to ankylosing spondylitis and psoriatic arthritis [45, 48, 49]. Furthermore, targeting the IL-23/IL-17 signalling pathway with medication has shown improvement in patients with axSpA [42••, 45].
While genomewide associations have uncovered multiple proteins (including ERAP and IL-23 receptor) associated with axSpA, the strongest genetic association with ankylosing spondylitis and psoriatic arthritis is HLA-B27 [50, 51]. Interestingly, HLA-B27 may cause the presentation of arthritogenic peptides on the cell surface [52]. In addition, the HLA-B27 mutation delays the folding of the HLA protein, leading to an unfolded protein response [53]. The unfolded protein response promotes IL-23 secretion [54–56]. Therefore, patients with the HLA-B27 mutation may have increased inflammation at baseline, predisposing these patients to arthritis and other manifestations of autoimmune diseases.
Another theory (the joint-gut axis theory) posits that the increased inflammation in axSpA may begin in the intestinal tract [57]. Ankylosing spondylitis and psoriatic arthritis are associated with inflammatory bowel disease. It has been shown that axSpA patients without overt inflammatory bowel disease are found to have subclinical inflammatory changes in the bowel [58, 59]. For example, the intestinal lamina propria of ankylosing spondylitis has increased levels of mononuclear cells [60]. IL-23, but not IL-17A, levels are increased in intestinal biopsies of patients with ankylosing spondylitis [61]. This inflammation can lead to translocation of bacteria and bacterial products across the intestinal wall into the blood, which can be reversed with antibiotics [62]. The IL-23 released from cells activated in the intestines is thought to circulate and lead to the release of IL-17 [11••, 63, 64]. Indeed, in the HLA-B27 transgenic mouse model for ankylosing spondylitis, animals do not develop ankylosing spondylitis when raised in a germ-free environment [65]. Innate natural killer T cells and mucosal activated invariant T cells activated by mucosal inflammation can produce IL-17, and these cells can be found in the peripheral blood and synovial fluid [66, 67]. Therefore, IL-23 and IL-17 derived from intestinal inflammation may lead to the joint inflammation in axSpA.
As discussed above, IL-17 may be released in an IL-23-dependent or IL-23-independent manner. Clinical trials in blocking IL-23 signalling in patients with ankylosing spondylitis have proven to be ineffective [30]. One possibility is that cells responsible for releasing IL-17 have been differentiated and are in situ by the time therapy has begun. van Tok and colleagues demonstrated this in a mouse model of ankylosing spondylitis. Mice over-expressing human HLA-B27 developed arthritis if IL-23 antibodies were given after arthritis developed, but not if these antibodies were injected prophylactically [29]. Indeed, resident entheseal T cells were found to release IL-17 in an IL-23-dependent manner [33]. Interestingly, Cuthbert and colleagues have also demonstrated a population of T cells that release IL-17 independently of IL-23 [32].
Taken together, these lines of evidence suggest there is an environmental trigger that leads to an imbalance in the cytokine milieu in the joint leading to inflammation and potentially overzealous repair.
Targeting IL-17 in Ankylosing Spondylitis and Psoriatic Arthritis
As IL-17 is central to the pathogenesis of axSpA, several biological agents have been developed against IL-17 or its target receptors [68, 69]. The biologics and their targets are summarized in Table 2.
In randomized control trials, secukinumab was shown to be effective in radiographic axSpA (ankylosing spondylitis) on multiple measures of disease activity (ASAS20, ASAS40, BASDAI, ASDAS) and these improvements were durable for 5 years on extension of these trials [70–75]. Encouragingly, improvements in disease control were also noted in patients who failed previous TNF-α inhibitor treatment [72].
Without treatment, the erosion and inflammation of joints in ankylosing spondylitis can lead to pain and decreased mobility. Several clinical trials have demonstrated that secukinumab prevents or reduces the progression of radiographic change over 2 years [75–77]. Secukinumab also can decrease inflammation that can only be detected on MRI [70].
Ankylosing spondylitis can negatively impact the patient’s quality of life [78, 79]. In addition to objective measures of disease control, there are various patient-reported outcomes (PROs) that measure subjective improvements in the disease process. In these studies, patients answer surveys that address their pain, fatigue, mood/mental health, general health, sleep, and impact on activities of daily living. Post hoc analysis of secukinumab randomized clinical trials showed that secukinumab improved mental and physical health, as well as their perceived quality of life [80•, 81–83]. In addition to their improved quality of life, these patients reported less work absenteeism and increased work productivity [81]. Patients treated with secukinumab also reported less fatigue [81, 84]. The above reported outcomes were noted after 16 weeks of treatment and persisted to the end of the follow-up period (52 weeks).
Secukinumab was well tolerated, with the most common side effects being nasopharyngitis, headache, diarrhoea, and upper respiratory tract infection (URTI). There was only one severe infection for every 100 patient years [70, 71, 73]. Braun and colleagues showed similar findings for secukinumab in a 4-year trial where the most common adverse effect was viral URTIs and there was only one case of tuberculosis [74].
Secukinumab was also effective in psoriatic arthritis. Through 16 weeks of treatment, Mease and colleagues found that patients treated with secukinumab had significant improvements across musculoskeletal domains and striking improvements in psoriasis, with PASI 100 scores of up to 40–50% [85]. Moreover, these improvements were maintained for 2 years of treatment [86, 87]. Similar results were found in trials with various doses and delivery methods [88–91]. One of the cardinal features of psoriatic arthritis is tendon inflammation which manifests as enthesitis and dactylitis. Secukinumab treatment results in resolution of enthesitis and dactylitis in 40–60% of patients who had these domains involved at baseline [87, 88, 90, 91]. Secukinumab treatment also results in minimal radiographic progression in affected joints up to 3 years [87, 91–93]. This was also reflected in MRI imaging showing decreased joint inflammation [85]. As with ankylosing spondylitis, patient-reported outcomes are also important for psoriatic arthritis. A post hoc analysis of randomized controlled trials showed that secukinumab improved patient-reported outcomes on multiple facts including quality of life, fatigue, global assessment of disease, and overall health status [94, 95]. Again, secukinumab was well tolerated with few severe side effects and was also effective in patients that had failed previous treatment with TNF-α inhibitors [96].
The EXCEED trial has compared secukinumab to adalimumab monotherapy in a head-to-head trial over 52 weeks in patients with psoriatic arthritis [97•]. The primary outcome was the American College of Rheumatology (ACR) 20 response, which showed a higher percentage of responders with secukinumab. However, this was not significantly increased as compared to adalimumab using intention to treat analysis. This did not reach significance if the stricter non-responder imputation was used. Although secukinumab was not significantly better than adalimumab based on musculoskeletal measures, secukinumab was associated with better skin responses as measured by the PASI score. There were no differences in adverse effects and tolerability between these medications. This may steer providers to treat patients with secukinumab if there is significant skin involvement.
Similarly, ixekizumab was designed to target IL-17A as well. In a randomized control trial (COAST-V) of nearly 400 biological DMARD-naïve radiographic axial spondyloarthritis patients, ixekizumab had greater proportion of patients achieving ASAS40 within the first 2 weeks of the trial; this plateaued at around 50% of patients in the trial [98]. Furthermore, there were improvements on other measures of disease activity as well as decreased biochemical and MRI signs of inflammation [98]. This trial included an active comparator arm of patients treated with adalimumab that performed consistently with previous trials with adalimumab. Although the trial was not powered to compare head to head with adalimumab with ixekizumab, ixekizumab was non-inferior to adalimumab. A similar trial (COAST-W) was carried out in patients with previous inadequate response to TNF-α inhibitors. Patients enrolled in this trial showed similar results to COAST-V after 16 weeks of treatment [99]. After 16 weeks, the participants of the COAST-V and COAST-W trials were pooled and followed for a 2-year extension trial. The results after 52 weeks showed that the initial improvements noted above were maintained. Post hoc analysis of the COAST-X, COAST-W, and COAST-V trials shows that patient-reported outcomes relating to quality of life, fatigue, function, health status, and work productivity improved with ixekizumab [100–103]. Interestingly, these improvements in patient-reported outcomes were correlated with the patients’ ASAS20/40 response [100, 102]. Further analysis of the clinical trial data showed that 75% of patient had no radiographic progression over 2 years [104]. In addition, of patients with available MRI data, 96% had minimal worsening of inflammation [104]. Furthermore, ixekizumab was well tolerated with the most common side effect being nasopharyngitis. Impressively, 88% of patients remained on the study medication at the end of 52 weeks [105]. In addition, ixekizumab was also shown to be effective and well tolerated in a trial with non-radiographic axSpA [106].
As with secukinumab, ixekizumab was also trialled in patients with psoriatic arthritis. There was improvement in joint disease, dactylitis, and skin disease [85, 96, 107, 108]. These improvements were also reflected with minimal radiographic changes even up to 3 years of treatment [109, 110]. There was a similar improvement in patient-reported outcomes [111–113]. Long-term follow-up of both medications showed that these medications had long-term efficacy in psoriatic arthritis and were well tolerated [86, 96, 110]. The side effects were consistent with those found in ankylosing spondylitis patients treated with these medications [86, 96, 114].
As with secukinumab, ixekizumab was directly compared with adalimumab in patients with psoriatic arthritis in the open-label SPIRIT-H2H trial [115, 116•]. Unlike EXCEED, SPIRIT-H2H compared patients who were on biological medications alone or concomitantly with methotrexate. The SPIRIT-H2H primary outcome was a composite of PASI 100 and ACR 50. Ixekizumab was significantly better beginning in week 8 in patients only on biological medications. This advantage was held through to the end of the study (52 weeks). While improvements in the primary outcome were driven primarily by PASI 100, a further analysis of the same subgroup of patients (biological medications only) showed that ixekizumab also improved on some musculoskeletal outcomes. Ixekizumab had a better ACR 70 response as compared to adalimumab. Furthermore, as per the Disease Activity in Psoriatic Arthritis (DAPSA) score, significantly more patients treated with ixekizumab had mild or very low disease. In addition, patients with ixekizumab performed better on the Health Assessment Questionnaire Disability Index (HAQ-DI), which measures patient performance on activities of daily living. However, the advantage described above for ixekizumab was seen when methotrexate was used in combination with the biological medications. These findings suggest that ixekizumab may outperform adalimumab if used as first-line monotherapy in psoriatic arthritis.
While secukinumab and ixekizumab block signalling through IL-17A, it does not affect the signalling through the other family members in the IL-17 cytokine family. As described previously, IL-17A and IL-17F can form a heterodimer that signals through a receptor consisting of at least IL-17RA and IL-17RC [7]. As with IL-17A, IL-17F is released from activated T cells and monocytes [9] and has been shown to be involved inflammation in in vitro and mouse models of disease [5, 117]. Furthermore, IL-17A, IL-17F, and their receptors are found in the inflamed synovium of patients with rheumatoid arthritis and psoriatic arthritis [118].
Brodalumab was the first biologic that targeted other IL-17 family members in addition to IL-17A by targeting the IL-17 receptor complex IL-17RA/IL-17C and preventing its activation [119]. This medication was first trialled in psoriasis patients; however, these trials reported an increase in psychiatric adverse events, including depression, anxiety, and suicidal ideation. In the sponsor product information, there was a clear signal for suicidal ideation and behaviour. In 6243 patients for a total of 10,438 patient years, there were 39 patients with suicidal ideation or behaviour events (0.37/100 patient years). Of these events, 18 patients had suicidal behaviour and 6 patients had completed suicide [120].
However, meta-analysis of over 4000 patients treated with brodalumab for 5 years found there was no increase in psychiatric adverse events as compared to other psoriasis trials [121]. In axSpA, brodalumab was shown to be effective in treating the musculoskeletal symptoms in as early as 2 weeks [122•, 123, 124•]. In these short trials, brodalumab was well tolerated with the most common side effects being infection. Reassuringly, there was no increase in psychiatric adverse effects [122•, 123, 124•].
With all biological medications targeting the Il-17 pathway, candidia infections and the development of inflammatory bowel disease (Crohn’s disease and ulcerative colitis) are a concern. Candida infections are a specific concern because Il-17 is involved in fungal defence. In axSpA patients treated with secukinumab, the levels of candida infections were low (0.1–3.2 events/100 patient years [72–75, 87, 89, 90, 93]. Similarly, axSpA patients treated with ixekizumab and brodalumab had low levels of candida infection [98, 105, 106, 125]. A meta-analysis of randomized control trials of IL-17 inhibitors in psoriasis and psoriatic arthritis showed that only 0–5% of patients developed candida infections [126]. Encouragingly, these candida infections were mild to moderate and resolved with topical or oral treatment and did not result in discontinuation of the study medication [126]. The findings from this meta-analysis are in keeping with the trials studied here.
As described in previous sections, IL-17 is involved in repair of mucosal surfaces. Therefore, IL-17 inhibition may worsen or lead to the development of IBD [127]. As with candida infections, IBD is a rare side effect of IL-17 blockade. A meta-analysis of randomized control trials of IL-17 blockade in psoriasis and axSpA found there was no increase in IBD incidence as compared to placebo, and this risk was not increased over 2 years of treatment [128]. A real-world observational study of the French National Health database found that there was no increased risk with IL-17 inhibitor treatment when these patients were compared to patients treated with etanercept [129•].
The newest agent targeting the IL-17 pathway is bimekizumab, which was initially tested in psoriasis, but phase II trials have found it to be effective in both radiographic and non-radiographic axSpA. Bimekizumab targets both IL-17A and IL-17F; however, unlike brodalumab, it targets the cytokines directly as opposed to the receptor. In the BE AGILE trial, more than 300 patients were randomized to placebo or different doses of bimekizumab and their clinical improvement was measured by ASAS40. By the end of the 12 months, nearly over 40% of patients treated with 64 mg or more of bimekizumab showed improvement as per ASAS40. In addition to improvements based on ASAS40, there was an improvement in sacroiliac and spinal inflammation on MRI imaging. There were also improvements in patient-reported outcomes. Furthermore, these improvements were maintained for 48 weeks after re-randomization of all patients to either 160 mg or 320 mg [130]. The medication was well tolerated, with the most common side effects being nasopharyngitis, pharyngitis, bronchitis, upper respiratory infections, and oral candidiasis. For oral candidiasis, there were 7.5 events/100 patient years. These mediations were mild to moderate and resolved with topical or oral treatment. Specifically, for IBD, the rate was 0.77 exposure-adjusted incidence/100 patient years for Crohn’s and ulcerative colitis. Indeed, two out of the four total IBD cases were exacerbation of previous IBD and two were new diagnoses. Similarly, the BE ACTIVE trial was a phase IIb trial that randomly assigned patients with psoriatic arthritis to placebo or a dose of bimekizumab. Again, the results were measured at 12 weeks and the patients were re-randomized to receive either 160 mg or 320 mg for a total of 48 weeks of the medication. As with the BE AGILE trial, the patients in BE ACTIVE showed improvements in their joint function as per the ACR 50 and their skin disease as per the PASI scores. Furthermore, a greater number of bimekizumab-treated patients had enthesitis resolution as compared to placebo-treated ones. In addition to musculoskeletal and skin improvements, patients also had measurable improvements in quality of life and reduced disability with bimekizumab treatment. Furthermore, the medication was again well tolerated with nasopharyngitis, pharyngitis, bronchitis, upper respiratory infections, and oral candidiasis being the most common side effects. There were 5% of patients that developed oral candidiasis, all of which resolved with topical or oral treatment. These infections did not result in discontinuation of bimekizumab. There were no cases of IBD with bimekizumab treatment in psoriatic arthritis patients. There were few patients who ceased the trial medication due to side effects [131]
Although further research is required, these early results are promising. Bimekizumab trials in psoriasis (BE VIVID, BE READY, BE RADIANT, BE SURE) suggest that it may be more efficacious than ustekinumab (IL-12/IL-23 inhibitor), secukinumab, or adalimumab [132–136]. Unfortunately, similar comparisons have not been made for bimekizumab in psoriatic arthritis and ankylosing spondylitis. However, as the pathology of psoriasis still depends in part on IL-17, this is a promising early sign that bimekizumab may be more effective than previous treatment. Encouragingly, a recent press prelease of BE MOBILE 1 and BE MOBILE 2 studying bimekizumab in non-radiographic and radiographic axSpA showed that patients randomized to bimekizumab had positive responses as per ASAS40 and all secondary end points (i.e. BASDAI, ASDAS). Furthermore, bimekizumab was well tolerated in these patients with the safety profile in line with previous studies [137, 138].
Conclusion
The development of biologics targeting the IL-17 pathway has been exciting as they have opened a novel therapeutic target. Furthermore, these medications are exciting as they have shown to be efficacious even in patients that have failed therapies targeting TNF-α. Bimekizumab is the newest therapy targeting this pathway. The studies suggest that this therapy may be more effective than previous therapies if the studies in psoriasis can be extrapolated to ankylosing spondylitis and psoriatic arthritis. We await the publication of phase 3 trials of bimekizumab for further data elucidating the efficacy of this therapy.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Rouvier E, Luciani MF, Mattéi MG, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J Immunol. 1993;150:5445–56.
Zhang X, Angkasekwinai P, Dong C, Tang H. Structure and function of interleukin-17 family cytokines. Protein Cell. 2011;2:26–40.
Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Medicine. 1996;183:2593–603.
Monin L, Gaffen SL. Interleukin 17 family cytokines: signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harb Perspect Biol. 2017;10: a028522.
Chang SH, Dong C. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res. 2007;17:435–40.
Wright JF, Guo Y, Quazi A, Luxenberg DP, Bennett F, Ross JF, et al. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells*. J Biol Chem. 2007;282:13447–55.
Wright JF, Bennett F, Li B, Brooks J, Luxenberg DP, Whitters MJ, et al. The human IL-17F/IL-17A heterodimeric cytokine signals through the IL-17RA/IL-17RC receptor complex. J Immunol. 2008;181:2799–805.
Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–67.
Starnes T, Robertson MJ, Sledge G, Kelich S, Nakshatri H, Broxmeyer HE, et al. Cutting edge: IL-17F, a novel cytokine selectively expressed in activated t cells and monocytes, regulates angiogenesis and endothelial cell cytokine production. J Immunol. 2001;167:4137–40.
Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–67.
•• Schinocca C, Rizzo C, Fasano S, Grasso G, Barbera LL, Ciccia F, et al. Role of the IL-23/IL-17 pathway in rheumatic diseases: an overview. Front Immunol. 2021;12:637829. This is a review of the IL-23/IL-17 axis and the role of these cytokines in multiple rheumatic disease, including rheumatoid arthritis and ankylosing spondylitis.
Li X, Bechara R, Zhao J, McGeachy MJ, Gaffen SL. IL-17 receptor–based signaling and implications for disease. Nat Immunol. 2019;20:1594–602.
Ono T, Okamoto K, Nakashima T, Nitta T, Hori S, Iwakura Y, et al. IL-17-producing γδ T cells enhance bone regeneration. Nat Commun. 2016;7:10928.
Croes M, Kruyt MC, Groen WM, van Dorenmalen KMA, Dhert WJA, Öner FC, et al. Interleukin 17 enhances bone morphogenetic protein-2-induced ectopic bone formation. Sci Rep-uk. 2018;8:7269.
McGonagle D, Stockwin L, Isaacs J, Emery P. An enthesitis based model for the pathogenesis of spondyloarthropathy. Additive effects of microbial adjuvant and biomechanical factors at disease sites. J Rheumatology. 2001;28:2155–9.
McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 family of cytokines in health and disease. Immunity. 2019;50:892–906.
Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–19.
• Rosine N, Miceli-Richard C. Innate cells: the alternative source of IL-17 in axial and peripheral spondyloarthritis? Front Immunol. 2021;11:553742. This article reviews the multiple sources of IL-17 and discusses how these cell types may be involved in spondyloarthritis.
Sherlock JP, Joyce-Shaikh B, Turner SP, Chao C-C, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4-CD8- entheseal resident T cells. Nat Med. 2012;18:1069–76.
Dyring-Andersen B, Geisler C, Agerbeck C, Lauritsen JPH, Gúdjonsdottir SD, Skov L, et al. Increased number and frequency of group 3 innate lymphoid cells in nonlesional psoriatic skin. Brit J Dermatol. 2014;170:609–16.
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y-H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41.
Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17–producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32.
Aggarwal S, Ghilardi N, Xie M-H, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17*. J Biol Chem. 2003;278:1910–4.
Becker C, Dornhoff H, Neufert C, Fantini MC, Wirtz S, Huebner S, et al. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J Immunol. 2006;177:2760–4.
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8.
Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–4.
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Medicine. 2005;201:233–40.
Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rβ1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168:5699–708.
van Tok MN, Na S, Lao CR, Alvi M, Pots D, van de Sande MGH, et al. The initiation, but not the persistence, of experimental spondyloarthritis is dependent on interleukin-23 Signaling. Front Immunol. 2018;9:1550.
Siebert S, Millar NL, McInnes IB. Why did IL-23p19 inhibition fail in AS: a tale of tissues, trials or translation? Ann Rheum Dis. 2019;78:1015.
Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Gulan F, Cayatte C, et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity. 2015;43:727–38.
Cuthbert RJ, Watad A, Fragkakis EM, Dunsmuir R, Loughenbury P, Khan A, et al. Evidence that tissue resident human enthesis γδT cells can produce IL-17A independently of IL-23R transcript expression. Ann Rheum Dis. 2019;78:1559.
Reinhardt A, Yevsa T, Worbs T, Lienenklaus S, Sandrock I, Oberdörfer L, et al. Interleukin-23-dependent γ/δ T cells produce interleukin-17 and accumulate in the enthesis, aortic valve, and ciliary body in mice. Arthritis Rheumatol. 2016;68:2476–86.
Lubberts E, van den Bersselaar L, Oppers-Walgreen B, Schwarzenberger P, Roo CJJC, Kolls JK, et al. IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-κB ligand/osteoprotegerin balance. J Immunol. 2003;170:2655–62.
Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8.
Zhang Z, Zheng M, Bindas J, Schwarzenberger P, Kolls JK. Critical role of IL-17 receptor signaling in acute TNBS-induced colitis. Inflamm Bowel Dis. 2006;12:382.
Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. 2006;116:1310–6.
Zhao X-F, Pan H-F, Yuan H, Zhang W-H, Li X-P, Wang G-H, et al. Increased serum interleukin 17 in patients with systemic lupus erythematosus. Mol Biol Rep. 2009;37:81–5.
Mei Y, Pan F, Gao J, Ge R, Duan Z, Zeng Z, et al. Increased serum IL-17 and IL-23 in the patient with ankylosing spondylitis. Clin Rheumatol. 2011;30:269–73.
Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. 1999;103:1345–52.
Jandus C, Bioley G, Rivals J, Dudler J, Speiser D, Romero P. Increased numbers of circulating polyfunctional Th17 memory cells in patients with seronegative spondylarthritides. Arthritis Rheumatism. 2008;58:2307–17.
•• Siebert S, McGucken A, McInnes IB. The IL-23/IL-17A axis in spondyloarthritis: therapeutics informing pathogenesis? Curr Opin Rheumatol. 2020;32:349–56. This article reviews the role of the IL-23/IL-17A axis in spondyloarthritis and inflammatory bowel disease. It reviews the therapies targeting this axis and discusses why IL-23-targeted therapies may have been ineffective in spondyloarthritis.
Robinson PC, van der Linden S, Khan MA, Taylor WJ. Axial spondyloarthritis: concept, construct, classification and implications for therapy. Nat Rev Rheumatol. 2021;17:109–18.
Robinson PC, Sengupta R, Siebert S. Non-radiographic axial spondyloarthritis (nr-axSpA): advances in classification, imaging and therapy. Rheumatology Ther. 2019;6:165–77.
McGonagle DG, McInnes IB, Kirkham BW, Sherlock J, Moots R. The role of IL-17A in axial spondyloarthritis and psoriatic arthritis: recent advances and controversies. Ann Rheum Dis. 2019;78:1167.
Venken K, Jacques P, Mortier C, Labadia ME, Decruy T, Coudenys J, et al. RORγt inhibition selectively targets IL-17 producing iNKT and γδ-T cells enriched in spondyloarthritis patients. Nat Commun. 2019;10:9.
Cuthbert RJ, Fragkakis EM, Dunsmuir R, Li Z, Coles M, Marzo-Ortega H, et al. Brief report: Group 3 innate lymphoid cells in human enthesis. Arthritis Rheumatol. 2017;69:1816–22.
Brown MA, Kenna T, Wordsworth BP. Genetics of ankylosing spondylitis—insights into pathogenesis. Nat Rev Rheumatol. 2016;12:81–91.
Rahman P, Inman RD, Gladman DD, Reeve JP, Peddle L, Maksymowych WP. Association of interleukin-23 receptor variants with ankylosing spondylitis. Arthritis Rheumatism. 2008;58:1020–5.
Queiro R, Morante I, Cabezas I, Acasuso B. HLA-B27 and psoriatic disease: a modern view of an old relationship. Rheumatology. 2016;55:221–9.
Robinson PC, Brown MA. The genetics of ankylosing spondylitis and axial spondyloarthritis. Rheum Dis Clin N Am. 2012;38:539–53.
Evans DM, Spencer CCA, Pointon JJ, Su Z, Harvey D, Kochan G, et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet. 2011;43:761–7.
Mear JP, Schreiber KL, Münz C, Zhu X, Stevanović S, Rammensee HG, et al. Misfolding of HLA-B27 as a result of its B pocket suggests a novel mechanism for its role in susceptibility to spondyloarthropathies. J Immunol. 1999;163:6665–70.
DeLay ML, Turner MJ, Klenk EI, Smith JA, Sowders DP, Colbert RA. HLA–B27 misfolding and the unfolded protein response augment interleukin-23 production and are associated with Th17 activation in transgenic rats. Arthritis Rheumatism. 2009;60:2633–43.
Ambarus CA, Yeremenko N, Baeten DL. Altered cytokine expression by macrophages from HLA-B27-positive spondyloarthritis patients without evidence of endoplasmic reticulum stress. Rheumatology Adv Pract. 2018;2:rky014.
Goodall JC, Wu C, Zhang Y, McNeill L, Ellis L, Saudek V, et al. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc National Acad Sci. 2010;107:17698–703.
Costello M, Ciccia F, Willner D, Warrington N, Robinson PC, Gardiner B, et al. Brief report: Intestinal dysbiosis in ankylosing spondylitis. Arthritis Rheumatol. 2015;67:686–91.
Praet LV, den Bosch FEV, Jacques P, Carron P, Jans L, Colman R, et al. Microscopic gut inflammation in axial spondyloarthritis: a multiparametric predictive model. Ann Rheum Dis. 2013;72:414.
Praet LV, Jans L, Carron P, Jacques P, Glorieus E, Colman R, et al. Degree of bone marrow oedema in sacroiliac joints of patients with axial spondyloarthritis is linked to gut inflammation and male sex: results from the GIANT cohort. Ann Rheum Dis. 2014;73:1186.
Mielants H, Veys EM, Cuvelier C, Vos M de. Ileocolonoscopic findings in seronegative spondylarthropathies. Rheumatology. 1988;XXVII:95–105.
Ciccia F, Bombardieri M, Principato A, Giardina A, Tripodo C, Porcasi R, et al. Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheumatism. 2009;60:955–65.
Ciccia F, Guggino G, Rizzo A, Alessandro R, Luchetti MM, Milling S, et al. Dysbiosis and zonulin upregulation alter gut epithelial and vascular barriers in patients with ankylosing spondylitis. Ann Rheum Dis. 2017;76:1123.
Rizzo A, Guggino G, Ferrante A, Ciccia F. Role of subclinical gut inflammation in the pathogenesis of spondyloarthritis. Frontiers Medicine. 2018;5:63.
Scher JU, Ubeda C, Artacho A, Attur M, Isaac S, Reddy SM, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol. 2015;67:128–39.
Taurog JD, Richardson JA, Croft JT, Simmons WA, Zhou M, Fernández-Sueiro JL, et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Medicine. 1994;180:2359–64.
Gracey E, Qaiyum Z, Almaghlouth I, Lawson D, Karki S, Avvaru N, et al. IL-7 primes IL-17 in mucosal-associated invariant T (MAIT) cells, which contribute to the Th17-axis in ankylosing spondylitis. Ann Rheum Dis. 2016;75:2124.
Mortier C, Govindarajan S, Venken K, Elewaut D. It takes “guts” to cause joint inflammation: role of innate-like T cells. Front Immunol. 2018;9:1489.
Wang P, Zhang S, Hu B, Liu W, Lv X, Chen S, et al. Efficacy and safety of interleukin-17A inhibitors in patients with ankylosing spondylitis: a systematic review and meta-analysis of randomized controlled trials. Clin Rheumatol. 2021;40:3053–65.
He C, Xue C, Zhu G, Kang P. Efficacy and safety of interleukin-17 inhibitors in the treatment of chronic rheumatic diseases: a combined and updated meta-analysis. J Clin Pharm Ther. 2021;46:895–906.
Baeten D, Baraliakos X, Braun J, Sieper J, Emery P, van der Heijde D, et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet. 2013;382:1705–13.
Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P, et al. Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. New Engl J Medicine. 2015;373:2534–48.
Pavelka K, Kivitz AJ, Dokoupilova E, Blanco R, Maradiaga M, Tahir H, et al. Secukinumab 150/300 mg provides sustained improvements in the signs and symptoms of active ankylosing spondylitis: 3-year results from the phase 3 MEASURE 3 study. ACR Open Rheumatology. 2020;2:119–27.
Baraliakos X, Braun J, Deodhar A, Poddubnyy D, Kivitz A, Tahir H, et al. Long-term efficacy and safety of secukinumab 150 mg in ankylosing spondylitis: 5-year results from the phase III MEASURE 1 extension study. RMD Open. 2019;5: e001005.
Braun J, Baraliakos X, Deodhar A, Poddubnyy D, Emery P, Delicha EM, et al. Secukinumab shows sustained efficacy and low structural progression in ankylosing spondylitis: 4-year results from the MEASURE 1 study. Rheumatology Oxf Engl. 2019;58:859–68.
Braun J, Baraliakos X, Deodhar A, Baeten D, Sieper J, Emery P, et al. Effect of secukinumab on clinical and radiographic outcomes in ankylosing spondylitis: 2-year results from the randomised phase III MEASURE 1 study. Ann Rheum Dis. 2016;76:1070–7.
Braun J, Haibel H, de Hooge M, Landewé R, Rudwaleit M, Fox T, et al. Spinal radiographic progression over 2 years in ankylosing spondylitis patients treated with secukinumab: a historical cohort comparison. Arthritis Res Ther. 2019;21:142.
Baraliakos X, Gensler LS, D’Angelo S, Iannone F, Favalli EG, Peyrecave N de, et al. Biologic therapy and spinal radiographic progression in patients with axial spondyloarthritis: a structured literature review. Ther Adv Musculoskelet Dis. 2020;12:1759720X20906040.
Elolemy G, Aboughanima A, Ganeb S, Elziat H. Health-related quality of life in patients with ankylosing spondylitis: relationship with disease-related variables. Curr Rheumatology Rev. 2020;16:311–8.
Aissaoui N, Rostom S, Hakkou J, Ghziouel KB, Bahiri R, Abouqal R, et al. Fatigue in patients with ankylosing spondylitis: prevalence and relationships with disease-specific variables, psychological status, and sleep disturbance. Rheumatol Int. 2012;32:2117–24.
• Ho A, Younis I, Le QA. Impact of biologics on health-related quality of life in patients with ankylosing spondylitis: a systematic review and meta-analysis of randomized controlled trials. Semin Arthritis Rheu. 2022;54:151996. This article reviews trials of biological medications and performs a meta-analysis to determine the effect of these medications on patient-reported outcomes.
Deodhar AA, Dougados M, Baeten DL, Wei JC, Geusens P, Readie A, et al. Effect of secukinumab on patient-reported outcomes in patients with active ankylosing spondylitis: a phase III randomized trial (MEASURE 1). Arthritis Rheumatology Hoboken N J. 2016;68:2901–10.
Marzo-Ortega H, Sieper J, Kivitz A, Blanco R, Cohen M, Martin R, et al. Secukinumab and sustained improvement in signs and symptoms of patients with active ankylosing spondylitis through two years: results from a phase III study. Arthrit Care Res. 2017;69:1020–9.
Tahir H, Moorthy A, Chan A. Impact of secukinumab on patient-reported outcomes in the treatment of ankylosing spondylitis: current perspectives. Open Access Rheumatology Res Rev. 2020;12:277–92.
Kvien TK, Conaghan PG, Gossec L, Strand V, Østergaard M, Poddubnyy D, et al. Secukinumab and sustained reduction in fatigue in patients with ankylosing spondylitis: long-term results of two phase III randomized controlled trials. Arthrit Care Res. 2022;
Mease PJ, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, et al. Secukinumab inhibition of interleukin-17A in patients with psoriatic arthritis. New Engl J Medicine. 2015;373:1329–39.
Kavanaugh A, Mease PJ, Reimold AM, Tahir H, Rech J, Hall S, et al. Secukinumab for long-term treatment of psoriatic arthritis: a two-year followup from a phase III, randomized, double-blind placebo-controlled study. Arthrit Care Res. 2017;69:347–55.
Mease PJ, Kavanaugh A, Reimold A, Tahir H, Rech J, Hall S, et al. Secukinumab in the treatment of psoriatic arthritis: efficacy and safety results through 3 years from the year 1 extension of the randomised phase III FUTURE 1 trial. RMD Open. 2018;4: e000723.
McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2015;386:1137–46.
Mease P, van der Heijde D, Landewé R, Mpofu S, Rahman P, Tahir H, et al. Secukinumab improves active psoriatic arthritis symptoms and inhibits radiographic progression: primary results from the randomised, double-blind, phase III FUTURE 5 study. Ann Rheum Dis. 2018;77:890.
Nash P, Mease PJ, McInnes IB, Rahman P, Ritchlin CT, Blanco R, et al. Efficacy and safety of secukinumab administration by autoinjector in patients with psoriatic arthritis: results from a randomized, placebo-controlled trial (FUTURE 3). Arthritis Res Ther. 2018;20:47.
Mease PJ, Landewé R, Rahman P, Tahir H, Singhal A, Boettcher E, et al. Secukinumab provides sustained improvement in signs and symptoms and low radiographic progression in patients with psoriatic arthritis: 2-year (end-of-study) results from the FUTURE 5 study. RMD Open. 2021;7: e001600.
van der Heijde D, Landewé RB, Mease PJ, McInnes IB, Conaghan PG, Pricop L, et al. Brief report: Secukinumab provides significant and sustained inhibition of joint structural damage in a phase III study of active psoriatic arthritis. Arthritis Rheumatol. 2016;68:1914–21.
van der Heijde D, Mease PJ, Landewé RBM, Rahman P, Tahir H, Singhal A, et al. Secukinumab provides sustained low rates of radiographic progression in psoriatic arthritis: 52-week results from a phase 3 study, FUTURE 5. Rheumatology. 2019;59:1325–34.
Strand V, Mease P, Gossec L, Elkayam O, van den Bosch F, Zuazo J, et al. Secukinumab improves patient-reported outcomes in subjects with active psoriatic arthritis: results from a randomised phase III trial (FUTURE 1). Ann Rheum Dis. 2017;76:203.
Strand V, Kaeley GS, Bergman MJ, Gladman DD, Coates LC, Sherif B, et al. The effect of secukinumab on patient-reported outcomes in patients with active psoriatic arthritis in a randomised phase 3 trial. Lancet Rheumatology. 2022;4:e208–19.
Kavanaugh A, McInnes IB, Mease PJ, Hall S, Chinoy H, Kivitz AJ, et al. Efficacy of subcutaneous secukinumab in patients with active psoriatic arthritis stratified by prior tumor necrosis factor inhibitor use: results from the randomized placebo-controlled FUTURE 2 study. J Rheumatology. 2016;43:1713–7.
• McInnes IB, Behrens F, Mease PJ, Kavanaugh A, Ritchlin C, Nash P, et al. Secukinumab versus adalimumab for treatment of active psoriatic arthritis (EXCEED): a double-blind, parallel-group, randomised, active-controlled, phase 3b trial. Lancet. 2020;395:1496–505. This article provides evidence that secukinumab is non-inferior to adalimumab. This suggests that targeting IL-17A may be as effective as targeting TNF-α inhibitors in PsA.
Heijde D van der, Wei JC-C, Dougados M, Mease P, Deodhar A, Maksymowych WP, et al. Ixekizumab, an interleukin-17A antagonist in the treatment of ankylosing spondylitis or radiographic axial spondyloarthritis in patients previously untreated with biological disease-modifying anti-rheumatic drugs (COAST-V): 16 week results of a phase 3 randomised, double-blind, active-controlled and placebo-controlled trial. Lancet. 2018;392:2441–51.
Deodhar A, Mease PJ, McInnes IB, Baraliakos X, Reich K, Blauvelt A, et al. Long-term safety of secukinumab in patients with moderate-to-severe plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis: integrated pooled clinical trial and post-marketing surveillance data. Arthritis Res Ther. 2019;21:111.
Deodhar AA, Mease PJ, Rahman P, Navarro-Compán V, Strand V, Hunter T, et al. Ixekizumab improves spinal pain, function, fatigue, stiffness, and sleep in radiographic axial spondyloarthritis: COAST-V/W 52-week results. Bmc Rheumatology. 2021;5:35.
Deodhar A, Mease P, Marzo-Ortega H, Hunter T, Sandoval D, Kronbergs A, et al. Ixekizumab improves sleep and work productivity in patients with non-radiographic axial spondyloarthritis: results from the COAST-X trial at 52 weeks. Bmc Rheumatology. 2021;5:50.
Deodhar A, Mease P, Rahman P, Navarro-Compán V, Marzo-Ortega H, Hunter T, et al. Ixekizumab improves patient-reported outcomes in non-radiographic axial spondyloarthritis: results from the Coast-X trial. Rheumatology Ther. 2021;8:135–50.
Marzo-Ortega H, Mease PJ, Rahman P, Navarro-Compán V, Strand V, Dougados M, et al. Impact of ixekizumab on work productivity in patients with ankylosing spondylitis: results from the COAST-V and COAST-W trials at 52 weeks. Rheumatology Ther. 2020;7:759–74.
van der Heijde D, Østergaard M, Reveille JD, Baraliakos X, Kronbergs A, Sandoval DM, et al. Spinal radiographic progression and predictors of progression in patients with radiographic axial spondyloarthritis receiving ixekizumab over 2 years. J Rheumatology. 2021;49:265–73.
Dougados M, Wei JC-C, Landewé R, Sieper J, Baraliakos X, Bosch FV den, et al. Efficacy and safety of ixekizumab through 52 weeks in two phase 3, randomised, controlled clinical trials in patients with active radiographic axial spondyloarthritis (COAST-V and COAST-W). Ann Rheum Dis. 2020;79:176.
Deodhar A, van der Heijde D, Gensler LS, Kim T-H, Maksymowych WP, Østergaard M, et al. Ixekizumab for patients with non-radiographic axial spondyloarthritis (COAST-X): a randomised, placebo-controlled trial. Lancet. 2020;395:53–64.
Nash P, Kirkham B, Okada M, Rahman P, Combe B, Burmester G-R, et al. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: results from the 24-week randomised, double-blind, placebo-controlled period of the SPIRIT-P2 phase 3 trial. Lancet. 2017;389:2317–27.
Coates LC, Pillai SG, Tahir H, Valter I, Chandran V, Kameda H, et al. Withdrawing ixekizumab in patients with psoriatic arthritis who achieved minimal disease activity: results from a randomized, double-blind withdrawal study. Arthritis Rheumatol. 2021;73:1663–72.
Mease PJ, van der Heijde D, Ritchlin CT, Okada M, Cuchacovich RS, Shuler CL, et al. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis. 2017;76:79–87.
Chandran V, van der Heijde D, Fleischmann RM, Lespessailles E, Helliwell PS, Kameda H, et al. Ixekizumab treatment of biologic-naïve patients with active psoriatic arthritis: 3-year results from a phase III clinical trial (SPIRIT-P1). Rheumatology Oxf Engl. 2020;59:2774–84.
Tillett W, Lin C-Y, Sprabery AT, Birt J, Kavanaugh A. Clinically meaningful improvement in work productivity loss in active psoriatic arthritis: post-hoc analysis of SPIRIT-P1 and SPIRIT-P2 trials. Clin Exp Rheumatol. 2020;
Gottlieb AB, Strand V, Kishimoto M, Mease P, Thaçi D, Birt J, et al. Ixekizumab improves patient-reported outcomes up to 52 weeks in bDMARD-naïve patients with active psoriatic arthritis (SPIRIT-P1). Rheumatology Oxf Engl. 2018;57:1777–88.
Kavanaugh A, Marzo-Ortega H, Vender R, Wei C-C, Birt J, Adams DH, et al. Ixekizumab improves patient-reported outcomes in patients with active psoriatic arthritis and inadequate response to tumour necrosis factor inhibitors: SPIRIT-P2 results to 52 weeks. Clin Exp Rheumatol. 2018;
Combe B, Rahman P, Kameda H, Cañete JD, Gallo G, Agada N, et al. Safety results of ixekizumab with 1822.2 patient-years of exposure: an integrated analysis of 3 clinical trials in adult patients with psoriatic arthritis. Arthritis Res Ther. 2020;22:14.
Smolen JS, Sebba A, Ruderman EM, Schulze-Koops H, Sapin C, Gellett AM, et al. Efficacy and safety of ixekizumab with or without methotrexate in biologic-naïve patients with psoriatic arthritis: 52-week results from SPIRIT-H2H study. Rheumatology Ther. 2020;7:1021–35.
• Mease PJ, Smolen JS, Behrens F, Nash P, Leage SL, Li L, et al. A head-to-head comparison of the efficacy and safety of ixekizumab and adalimumab in biological-naïve patients with active psoriatic arthritis: 24-week results of a randomised, open-label, blinded-assessor trial. Ann Rheum Dis. 2020;79:123. This is a study of ixekizumab with adalimumab as an active comparator. It shows that ixekizumab performed better than adalimumab in PsA as per some objective and patient-reported outcomes.
Schmidt EGW, Larsen HL, Kristensen NN, Poulsen SS, Pedersen AML, Claesson MH, et al. TH17 cell induction and effects of IL-17A and IL-17F blockade in experimental colitis. Inflamm Bowel Dis. 2013;19:1567–76.
van Baarsen LG, Lebre MC, van der Coelen D, Aarrass S, Tang MW, Ramwadhdoebe TH, et al. Heterogeneous expression pattern of interleukin 17A (IL-17A), IL-17F and their receptors in synovium of rheumatoid arthritis, psoriatic arthritis and osteoarthritis: possible explanation for nonresponse to anti-IL-17 therapy? Arthritis Res Ther. 2014;16:426.
Papp KA, Leonardi C, Menter A, Ortonne J-P, Krueger JG, Kricorian G, et al. Brodalumab, an anti–interleukin-17–receptor antibody for psoriasis. New Engl J Medicine. 2012;366:1181–9.
Pharmaceuticals V. Brodalumab Food and Drug Administration Dermatologic and Ophthalmic Drugs Advisory Committee (DODAC) Meeting Date: July 19, 2016. FDA Briefing Document: Dermatologic and Ophthalmic Drugs Advisory Committee Meeting [Internet]. 2016. Available from: https://www.fda.gov/files/advisory%20committees/published/FDA-Briefing-Information-for-the-July-19--2016-Meeting-of-the-Dermatologic-and-Ophthalmic-Drugs-Advisory-Committee.pdf
Lebwohl MG, Papp KA, Marangell LB, Koo J, Blauvelt A, Gooderham M, et al. Psychiatric adverse events during treatment with brodalumab: analysis of psoriasis clinical trials. J Am Acad Dermatol. 2018;78:81-89.e5.
• Wei JC-C, Kim T-H, Kishimoto M, Ogusu N, Jeong H, Kobayashi S. Efficacy and safety of brodalumab, an anti-IL17RA monoclonal antibody, in patients with axial spondyloarthritis: 16-week results from a randomised, placebo-controlled, phase 3 trial. Ann Rheum Dis. 2021;80:1014–21. This trial demonstrated the efficacy of brodalumab in treatment of axSpA. There were no suicides or self-injury events in the treatment arm.
Mease PJ, Genovese MC, Greenwald MW, Ritchlin CT, Beaulieu AD, Deodhar A, et al. Brodalumab, an Anti-IL17RA Monoclonal Antibody, in Psoriatic Arthritis. New Engl J Medicine. 2014;370:2295–306.
• Mease PJ, Helliwell PS, Hjuler KF, Raymond K, McInnes I. Brodalumab in psoriatic arthritis: results from the randomised phase III AMVISION-1 and AMVISION-2 trials. Ann Rheum Dis. 2021;80:185–93. This report demonstrated the efficacy of brodalumab in PsA. There was only one report of suicidal ideation, which resolved in 1 day of presentation.
Papp K, Menter A, Leonardi C, Soung J, Weiss S, Pillai R, et al. Long-term efficacy and safety of brodalumab in psoriasis through 120 weeks and after withdrawal and retreatment: subgroup analysis of a randomized phase III trial (AMAGINE-1)*. Brit J Dermatol. 2020;183:1037–48.
Saunte DM, Mrowietz U, Puig L, Zachariae C. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Brit J Dermatol. 2017;177:47–62.
Fauny M, Moulin D, D’Amico F, Netter P, Petitpain N, Arnone D, et al. Paradoxical gastrointestinal effects of interleukin-17 blockers. Ann Rheum Dis. 2020;79:1132–8.
Yamada A, Wang J, Komaki Y, Komaki F, Micic D, Sakuraba A. Systematic review with meta-analysis: risk of new onset IBD with the use of anti-interleukin-17 agents. Aliment Pharm Therap. 2019;50:373–85.
• Penso L, Bergqvist C, Meyer A, Herlemont P, Weill A, Zureik M, et al. Risk of inflammatory bowel disease in patients with psoriasis and psoriatic arthritis/ankylosing spondylitis initiating interleukin-17 inhibitors: a nationwide population-based study using the French National Health Data System. Arthritis Rheumatol. 2022;74:244–52. This is a large post-market study that provides evidence that there may not be an increased risk of IBD with IL-17 inhibition.
van der Heijde D, Gensler LS, Deodhar A, Baraliakos X, Poddubnyy D, Kivitz A, et al. Dual neutralisation of interleukin-17A and interleukin-17F with bimekizumab in patients with active ankylosing spondylitis: results from a 48-week phase IIb, randomised, double-blind, placebo-controlled, dose-ranging study. Ann Rheum Dis. 2020;79:595.
Ritchlin CT, Kavanaugh A, Merola JF, Schett G, Scher JU, Warren RB, et al. Bimekizumab in patients with active psoriatic arthritis: results from a 48-week, randomised, double-blind, placebo-controlled, dose-ranging phase 2b trial. Lancet. 2020;395:427–40.
Gordon KB, Foley P, Krueger JG, Pinter A, Reich K, Vender R, et al. Bimekizumab efficacy and safety in moderate to severe plaque psoriasis (BE READY): a multicentre, double-blind, placebo-controlled, randomised withdrawal phase 3 trial. Lancet. 2021;397:475–86.
Warren RB, Blauvelt A, Bagel J, Papp KA, Yamauchi P, Armstrong A, et al. Bimekizumab versus adalimumab in plaque psoriasis. New Engl J Med. 2021;385:130–41.
Reich K, Papp KA, Blauvelt A, Langley RG, Armstrong A, Warren RB, et al. Bimekizumab versus ustekinumab for the treatment of moderate to severe plaque psoriasis (BE VIVID): efficacy and safety from a 52-week, multicentre, double-blind, active comparator and placebo controlled phase 3 trial. Lancet. 2021;397:487–98.
Reich K, Warren RB, Lebwohl M, Gooderham M, Strober B, Langley RG, et al. Bimekizumab versus secukinumab in plaque psoriasis. New Engl J Med. 2021;385:142–52.
Blauvelt A, Papp KA, Merola JF, Gottlieb AB, Cross N, Madden C, et al. Bimekizumab for patients with moderate to severe plaque psoriasis: 60-week results from BE ABLE 2, a randomized, double-blinded, placebo-controlled, phase 2b extension study. J Am Acad Dermatol. 2020;83:1367–74.
Positive top-line results for BIMZELX®▼(bimekizumab) in phase 3 ankylosing spondylitis trial. UCB News. 2021.
Positive top-line results for BIMZELX®▼(bimekizumab) in phase 3 non-radiographic axial spondyloarthritis study. UCB News. 2022.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
HJKT has no disclosures. PR reports personal fees from AbbVie, Atom Biosciences, Eli Lilly, Gilead, GlaxoSmithKline, Janssen, Kukdong, Novartis, UCB, Roche, and Pfizer; meeting attendance support from BMS, Pfizer, and UCB; and grant funding from Janssen, Novartis, Pfizer, and UCB Pharma. PN reports grants for research and clinical trials and honoraria for advice and lectures on behalf of AbbVie, BMS, Celgene, Pfizer, Roche, Samsung, Boehringer Ingelheim, Lilly, Novartis, Janssen, UCB, Gilead/Galapagos, GSK, MSD, and Sanofi.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Spondyloarthritis
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tam, H.K.J., Robinson, P.C. & Nash, P. Inhibiting IL-17A and IL-17F in Rheumatic Disease: Therapeutics Help to Elucidate Disease Mechanisms. Curr Rheumatol Rep 24, 310–320 (2022). https://doi.org/10.1007/s11926-022-01084-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11926-022-01084-4