
Abstract
Purpose of Review
This review paper provides step-by-step instructions on the fundamental process, from handling fastq datasets to illustrating plots and drawing trajectories.
Recent Findings
The number of studies using single-cell RNA-seq (scRNA-seq) is increasing. scRNA-seq revealed the heterogeneity or diversity of the cellular populations. scRNA-seq also provides insight into the interactions between different cell types. User-friendly scRNA-seq packages for ligand-receptor interactions and trajectory analyses are available. In skeletal biology, osteoclast differentiation, fracture healing, ectopic ossification, human bone development, and the bone marrow niche have been examined using scRNA-seq. scRNA-seq data analysis tools are still being developed, even at the fundamental step of dataset integration. However, updating the latest information is difficult for many researchers. Investigators and reviewers must share their knowledge of in silico scRNA-seq for better biological interpretation.
Summary
This review article aims to provide a useful guide for complex analytical processes in single-cell RNA-seq data analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The number of research articles using single-cell RNA-seq (scRNA-seq) is increasing. scRNA-seq has become a core technique in biology in the last 10 years [1]. scRNA-seq enabled us to determine the quantity of each type of mRNA at a single-cell resolution. There are two major reasons why the use of scRNA-seq has spread worldwide. First, scRNA-seq clarifies the heterogeneity or diversity of cell populations from the perspective of gene expression patterns. Second, scRNA-seq can predict the interactions and connectivity between cells, which cannot be easily specified in traditional ways.
scRNA-seq has been used in skeletal biology [2, 3]. For example, we can determine the cell differentiation stages of osteoclasts [4] and their interspecies differences [5•]. Ligand receptor analysis can predict drug repositioning candidates for fracture healing [6] and clarify the hidden mechanisms of ectopic ossification [7]. Gene regulatory analysis has also revealed epigenetic properties in a model of human bone development [8•] and the bone marrow niche [9]. Furthermore, a new subcellular sequencing tool to identify therapeutic targets has been proposed in the field of skeletal biology [10•].
Currently, in silico analysis is necessary not only for computational biologists but also for wet-lab biologists and well-established reviewers [11, 12]. In this review, we have summarized the in silico scRNA-seq framework. Active learners can understand the standard workflow and pitfalls of in silico analysis. In addition, this review may be useful to busy reviewers. This article provides a list of points for reviewing of scRNA-seq studies.
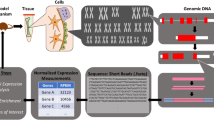
The standard workflow of in silico scRNA-seq analysis is summarized in Fig. 1. The scRNA-seq packages and tools recommended by the authors are summarized in Table 1. These tools were selected primarily because of their usability and widespread use.

Step 1. Obtain FASTQ files from wet experiments and public database
There are two ways to obtain FASTQ files. One is to utilize public resources and the other is to perform wet experiments. The combinatorial approach has become more common with the increase in public datasets.
The first method for obtaining FASTQ files is to download them from a public database. sequence read archive (SRA), European nucleotide archive (ENA), and DDBJ sequence read archive (DRA) are the three major archives of sequencing datasets. The DDBJ search website is useful for finding the SRR number of datasets related to the project. The same project in one’s own experiments sometimes spares you from conducting expensive reproductive experiments. Public datasets were also used to increase the scale of scRNA-seq experiments. Integrative analyses are recommended for the following two reasons. First, a greater variety of cells often makes cell annotation easier. Second, integrative analysis with datasets from other research groups reduces biases related to the procedures of the group and increases the external validity of the experiments.
It takes considerable time and computer resources to download heavy FASTQ files of tens of gigabytes. fasterq-dump in the SRA toolkit [16], which is a fast version of fastq-dump, is commonly used to speed up downloading. parallel-fastq-dump [17] splits fastq files and downloads them by palletization of the process.
The second method is to perform a scRNA-seq experiment on one’s own. Although wet experiments are beyond the scope of this article, attention should be paid to the process of preparing cell suspensions. This is because the selection bias in wet experiments affects the results of in silico scRNA-seq. Less bias between samples makes the integration of scRNA-seq datasets easier.
Cell suspension preparation is the first step of droplet-based scRNA-seq. To smoothen the preparation step, we should repeatedly practice the entire preparation process and stabilize protocols.
When dissociating cells from solid tissue, a variety of healthy cells should be maintained and as many dead cells should be excluded as possible. Cell-sorting techniques using flow cytometry and magnetic devices are useful. After making a cell suspension, cell aggregation often occurs, which may cause problems in fluid-based sorting and should be loosened by pipetting.
After making cell suspensions, we performed highly elaborate library construction according to the manufacturer’s protocol like chromium (10 × genomics, Pleasanton, CA, U.S.). The number of living cells is important for the first chromium step. Instead of droplet-based sequencing, traditional plate-based sequencing after cell sorting, for example, SMART-seq® Single Cell Kit (Takara Bio, San Jose, CA, U.S.) [18], and RT-RamDA® cDNA Synthesis Kit (TOYOBO, Osaka, Japan) [19], are powerful tools for full-length total sequencing. After sequencing the constructed library, the FASTQ files are obtained.
Step 2. Quality check and mapping to the reference genome
Cell Ranger is a useful pipeline to align outputted fastq files by chromium, on the prebuilt reference genome and make the folder of ready-to-use matrix files for the downstream analysis [20]. The Cell Ranger version and the reference genome used should be included in the manuscript to help reproduce the analysis. Cell Ranger consumes substantial computer memory; therefore, enough memory and storage more than the required level should be prepared. When using a public computer, the impact of load on the common space should be considered.
The authors preferred STARsolo [21], which is a single-cell version of the common aligner STAR. SMART-seq or Drop-seq datasets, other than chromium, can be processed using the same protocol. In addition, the same reference genome as that used in bulk RNA-seq can be used with simple arguments by STARsolo. Unlike Cell Ranger, the library construction protocol, including the chromium chemistry version, should be specified as a variable.
STARSolo offers three advantages. First, STARsolo can be adapted for other scRNA-seq datasets such as SMART-seq. Second, the mapping time is shorter than that of CellRanger. Third, this is the most important reason, the same reference genome as the usual bulk RNA-seq can be used.
When performing integrative analysis with other experiments of yours or public datasets by other groups, the same mapping protocol should be performed to avoid a mismatch of the reference genome. Repeat mapping on the reference genome is frequently performed. This is partially because Cell Ranger is frequently updated.
Step 3. Preparation environment for the in silico analysis
Some vendors have provided browser-based analytical tools. These readily available tools are useful for checking quickly whether wet experiments are successful. However, it is too difficult to perform advanced analysis, including cell interaction, and to produce images with publication quality using only these tools. This is why researchers and reviewers should be familiar with in silico analysis.
R language [22] is commonly used in statistical science and bioinformatics analyses. Tidyverse project [23] provides several powerful toolkits for handling datasets with simple syntax. In particular, ggplot2 [24] increases the visibility of graphs and ensures reproducibility, which is important in science.
Updating R and the packages sometimes yields different UMAP or clustering results. Major R updates require package reinstallation. Although we do not want to update these versions, version conflicts between packages do not allow us to change only problematic packages, but enforce updating all packages. The results without big picture changes with different versions, in which minor detailed changes are allowed, should be stated in the manuscript.
Memory usage should be cared for when using R. Regardless of the PC setup, the memory consumption of R can cause sudden crashes. This is a good practice for saving files and images. It is also important to delete the unused variables and intermediate files.
Taking fashionable machine learning methods into the study, converting platform R to Python3 is considered because of the large memory requirement [25]. The main feature of Python is its numerous modules, including deep learning. Another feature is the creation of a virtual environment with modules required for each project to avoid version conflicts between the packages. Matplotlib [26] and Seaborn [27] support data visualization. Based on the author’s experience, switching from R to Python requires practice.
Step 4. Preprocess of datasets
Seurat [28] is a core package for processing and normalizing scRNA-seq datasets. Seurat has been developed to integrate multiple datasets. In the latest version 5 [29••], we can choose an integration method including sctransform [30]. In Python, Scanpy [31] in scverse project [32] is the core package for processing the datasets. It is not difficult to handle fundamental packages in both R and Python because tutorials are available online and many virtual workshops are available. Core preprocessing: Filtering and normalization are almost the same regardless of the package.
The total number of detected genes, percentage of counts on mitochondrial genes, and sometimes ribosomal genes are common indicators for cutting off dead cells or poorly sequenced cells. These cutoff values should be listed in the manuscript. Different technologies for library construction typically result in different levels of these indicators. Different levels are often observed, even with the same technology. The same cutoff value is recommended for making posterior calculation easy; however, different cutoff values may be accepted for each dataset.
The selected cells are then normalized to compare RNA expression between cells. Additional normalization of the total number of reads should be performed because of the low sensitivity of single-cell sequencing from a low amount of RNA. Percentages of mitochondrial gene count and cell cycle scores are usually regressed out for normalization. Although cell cycle scoring and assignment of the cell cycle state for each cell are performed routinely by Seurat, cell cycles are predicted by comparing the mRNA expression of cell cycle genes. When analyzing datasets in which the cell cycle is highly activated, cell cycle regression may be unnecessary.
Step 5. Dataset integration
It is common to handle multiple datasets; however, it is difficult to integrate datasets with different expression levels. Various integration methods have been devised and are currently under development. The latest method should be considered at the time of submission because the choice of method leads to different results. Benchmark studies on integration are useful for selecting packages [33•]. The latest version of Seurat allows the selection of various integration methods [29••]. Scverse projects provide scvi-tools to perform probabilistic analysis, particularly when integrating datasets [34•]. Whether the results of the integration are correct should be examined from the perspective of wet scientists.
Step 6. Unbiased clustering
To understand the heterogeneous gene expression patterns at a glance, dimension reduction with tSNE [35] and UMAP [36] is performed. The distribution of datasets, mitochondrial percentage, and cell cycle are indicators of the successful integration of the datasets. Unbiased clustering after dimension reduction makes it possible to depict the cell populations. The resolution must be tuned by the authors to adapt the assumed cell types, although some tools for the automatic determination of cluster numbers have been proposed [37].
Maps derived from scRNA-seq datasets are built based on RNA expression patterns and do not always fit the standard biological view. Not only local structures but also the whole picture often change according to the method. For example, small islands and their connectivity to a main island can be easily transformed. The robustness of in silico results should be examined, especially when discussing minor cell populations. Insufficient batch-effect elimination often yields distinct clusters. In some cases, more cells are required to reach a conclusion. Public datasets are used as external references to reduce researcher bias.
Step 7. Regular visualization and functional annotation
In scRNA-seq, gene expression and cellular functions are explained at two levels: cell and cluster or group. Feature plots explain gene expression cell by cell on the same map. Continuous changes in the levels are easily depicted. Dot plots and violin plots are used to summarize the expression levels group by group. Dot plots are recommended for showing sets of genes within a limited space.
Cell types and their functions are determined by considering sets of gene expression, sometimes called gene set activities. AUCell is useful for calculating the gene set activity score [38], and with the same algorithm, we can predict upstream transcription factor activity using SCENIC or pySCENIC [39, 40]. decoupleR [41•] enables the use of multiple ensemble annotative methods including AUCell. Automatic cell annotation using the deep learning method is implemented using CellAssign [42].
Intercellular interactions can be predicted using paired gene expression in different groups of cells, including ligand receptor (LR) interactions. Several types of tools have been proposed [43]. Nitchenet [44] considers downstream pathways including receptors to transcriptional factors. The LR pair database directly affects the results of the LR analysis. Omnipath project provides large well-organized references [45]. With abundant computer memory, tensor-based cell communication analysis between more than two cell types provides further LR relationships [46].
Step 8. Trajectory analysis
RNA velocity is a well-known concept for predicting local changes in the cellular state from the spliced/unspliced ratio of sequenced reads and is implemented as Velocyto [47]. Streamline visualization can be illustrated by a generalized version of Velocyto called scVelo [48•]. Plots with arrows are attractive; however, RNA velocity tools are still under development [49]. Trajectory analysis provides just supportive evidence for cellular pathways.
Monocle2 or monocle3 in R [50], and PAGA in Python [51] are commonly used to draw global trajectory lines based on gene expression. To choose the appropriate tool for each analysis, rough topological characteristics, such as cycle, linear, branch, tree, and disconnection, should be presumed before trajectory analysis. Dynguidelines project provides a clue for selecting appropriate trajectory tools for each analysis [52•].
Conclusion
scRNA-seq provides insight into the diversity of cell populations. However, the preprocessing and integrative steps for multiple datasets remain controversial. The honest manifestation of the fundamental steps makes the study reproducible.
Reviewers of scRNA-seq research should first check these basic points (summarized in Table 2) and then discuss whether the biological interpretation is reasonable. There have been scRNA-seq studies with insufficient replicates. However, in the era when there are plenty of public scRNA-seq fastq files, investigators’ procedures should be examined for propriety and external validity.
The use of scRNA-seq analysis has continued to evolve rapidly. Discussion between investigators and reviewers should be performed within the scope of the methods at the time of submission. Better use of this innovative technique will enhance our biological knowledge.
Change history
19 February 2024
A Correction to this paper has been published: https://doi.org/10.1007/s11914-024-00861-7
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Method of the year 2013. Nat Methods. 2014;11(1):1. https://doi.org/10.1038/nmeth.2801.
Greenblatt MB, Ono N, Ayturk UM, Debnath S, Lalani S. The unmixing problem: a guide to applying single-cell RNA sequencing to bone. J Bone Miner Res. 2019;34(7):1207–19. https://doi.org/10.1002/jbmr.3802.
Ono N, Taipaleenmaki H, Veis DJ. Single-cell RNA-sequencing leading to breakthroughs in musculoskeletal research. JBMR Plus. 2022;6(7):e10652. https://doi.org/10.1002/jbm4.10652.
Tsukasaki M, Huynh NC, Okamoto K, Muro R, Terashima A, Kurikawa Y, Komatsu N, Pluemsakunthai W, Nitta T, Abe T, Kiyonari H, Okamura T, Sakai M, Matsukawa T, Matsumoto M, Kobayashi Y, Penninger JM, Takayanagi H. Stepwise cell fate decision pathways during osteoclastogenesis at single-cell resolution. Nat Metab. 2020;2(12):1382–90. https://doi.org/10.1038/s42255-020-00318-y.
Omata Y, Okada H, Uebe S, Izawa N, Ekici AB, Sarter K, Saito T, Schett G, Tanaka S, Zaiss MM. Interspecies single-Cell RNA-Seq analysis reveals the novel trajectory of osteoclast differentiation and therapeutic targets. JBMR Plus. 2022;6(7):e10631. https://doi.org/10.1002/jbm4.10631. Interspecies difference in osteoclast differentiation path was depicted using single cell RNA-seq.
Nakayama M, Okada H, Seki M, Suzuki Y, Chung UI, Ohba S, Hojo H. Single-cell RNA sequencing unravels heterogeneity of skeletal progenitors and cell-cell interactions underlying the bone repair process. Regen Ther. 2022;21:9–18. https://doi.org/10.1016/j.reth.2022.05.001.
Tachibana N, Chijimatsu R, Okada H, Oichi T, Taniguchi Y, Maenohara Y, Miyahara J, Ishikura H, Iwanaga Y, Arino Y, Nagata K, Nakamoto H, Kato S, Doi T, Matsubayashi Y, Oshima Y, Terashima A, Omata Y, Yano F, Maeda S, Ikegawa S, Seki M, Suzuki Y, Tanaka S, Saito T. RSPO2 defines a distinct undifferentiated progenitor in the tendon/ligament and suppresses ectopic ossification. Sci Adv. 2022;8(33):eabn2138. https://doi.org/10.1126/sciadv.abn2138.
Tani S, Okada H, Onodera S, Chijimatsu R, Seki M, Suzuki Y, Xin X, Rowe DW, Saito T, Tanaka S, Chung UI, Ohba S, Hojo H. Stem cell-based modeling and single-cell multiomics reveal gene-regulatory mechanisms underlying human skeletal development. Cell Rep. 2023;42(4):112276. https://doi.org/10.1016/j.celrep.2023.112276. Transcriptomic and epigenetic human bone development was illustrated by a novel multi-omics approach.
Kanazawa S, Okada H, Hojo H, Ohba S, Iwata J, Komura M, Hikita A, Hoshi K. Mesenchymal stromal cells in the bone marrow niche consist of multi-populations with distinct transcriptional and epigenetic properties. Sci Rep. 2021;11(1):15811. https://doi.org/10.1038/s41598-021-94186-5.
Okada H, Terui Y, Omata Y, Terashima A, Seki M, Tani S, Kanazawa S, Hosonuma M, Miyahara J, Makabe K, Onodera S, Yano F, Kajiya H, Gori F, Saito T, Suzuki Y, Okabe K, Baron R, Chung UI, Tanaka S, Hojo H. Inclusive living subcellular sequencing rendering physical physiological and human pathological features in osteoimmune diversity. bioRxiv. 2022.09.05.506360. https://doi.org/10.1101/2022.09.05.506360. The technology for live subcellular sequencing was achieved.
Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol. 2019;15(6):e8746. https://doi.org/10.15252/msb.20188746.
Andrews TS, Kiselev VY, McCarthy D, Hemberg M. Tutorial: guidelines for the computational analysis of single-cell RNA sequencing data. Nat Protoc. 2021;16(1):1–9. https://doi.org/10.1038/s41596-020-00409-w.
Baccin C, Al-Sabah J, Velten L, Helbling PM, Grunschlager F, Hernandez-Malmierca P, Nombela-Arrieta C, Steinmetz LM, Trumpp A, Haas S. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat Cell Biol. 2020;22(1):38–48. https://doi.org/10.1038/s41556-019-0439-6.
Wang JS, Kamath T, Mazur CM, Mirzamohammadi F, Rotter D, Hojo H, Castro CD, Tokavanich N, Patel R, Govea N, Enishi T, Wu Y, da Silva Martins J, Bruce M, Brooks DJ, Bouxsein ML, Tokarz D, Lin CP, Abdul A, Macosko EZ, Fiscaletti M, Munns CF, Ryder P, Kost-Alimova M, Byrne P, Cimini B, Fujiwara M, Kronenberg HM, Wein MN. Control of osteocyte dendrite formation by Sp7 and its target gene osteocrin. Nat Commun. 2021;12(1):6271. https://doi.org/10.1038/s41467-021-26571-7.
Zhong L, Yao L, Tower RJ, Wei Y, Miao Z, Park J, Shrestha R, Wang L, Yu W, Holdreith N, Huang X, Zhang Y, Tong W, Gong Y, Ahn J, Susztak K, Dyment N, Li M, Long F, Chen C, Seale P, Qin L. Single cell transcriptomics identifies a unique adipose lineage cell population that regulates bone marrow environment. Elife. 2020;9:e54695. https://doi.org/10.7554/eLife.54695.
The SRA Toolkit Development Team: SRA toolkit. edn 3.0.3. Edited by; 2023.
Valieris R, Fukushima K, Homer N. parallel-fastq-dump. edn 0.6.7. Edited by; 2021.
Ramskold D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, Daniels GA, Khrebtukova I, Loring JF, Laurent LC, Schroth GP, Sandberg R. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30(8):777–82. https://doi.org/10.1038/nbt.2282.
Hayashi T, Ozaki H, Sasagawa Y, Umeda M, Danno H, Nikaido I. Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs. Nat Commun. 2018;9(1):619. https://doi.org/10.1038/s41467-018-02866-0.
Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, Bielas JH. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. https://doi.org/10.1038/ncomms14049.
Kaminow B, Yunusov D, Dobin A. STARsolo: accurate, fast and versatile mapping/quantification of single-cell and single-nucleus RNA-seq data. bioRxiv. 2021.05.05.442755. https://doi.org/10.1101/2021.05.05.442755.
R Core Team. _R: a language and environment for statistical computing_. R Foundation for Statistical Computing, Vienna, Austria; 2023.
Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, Grolemund G, Hayes A, Henry L, Hester J, et al. Welcome to the Tidyverse. J Open Source Softw. 2019;4(43):1686 10.21105/joss.01686.
Wickham H. ggplot2: elegant graphics for data analysis: Springer-Verlag New York; 2009.
Van Rossum G, Drake FL. Python 3 reference manual: CreateSpace; 2009.
Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng. 2007;9:90–5. https://doi.org/10.1109/MCSE.2007.55.
Waskom M. seaborn: statistical data visualization. J Open Source Softw. 2021;6:3021. https://doi.org/10.21105/joss.03021.
Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R. Integrated analysis of multimodal single-cell data. Cell. 2021;184(13):3573–87.e29. https://doi.org/10.1016/j.cell.2021.04.048.
Hao Y, Stuart T, Kowalski MH, Choudhary S, Hoffman P, Hartman A, Srivastava A, Molla G, Madad S, Fernandez-Granda C, Satija R. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat Biotechnol. 2023. https://doi.org/10.1038/s41587-023-01767-y. The fundamental package Seurat in scRNA-seq analysis was updated to version 5.
Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20(1):296. https://doi.org/10.1186/s13059-019-1874-1.
Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 2018;19(1):15. https://doi.org/10.1186/s13059-017-1382-0.
Virshup I, Bredikhin D, Heumos L, Palla G, Sturm G, Gayoso A, Kats I, Koutrouli M; Scverse Community; Berger B, Pe'er D, Regev A, Teichmann SA, Finotello F, Wolf FA, Yosef N, Stegle O, Theis FJ. The scverse project provides a computational ecosystem for single-cell omics data analysis. Nat Biotechnol. 2023;41(5):604–6. https://doi.org/10.1038/s41587-023-01733-8.
Luecken MD, Buttner M, Chaichoompu K, Danese A, Interlandi M, Mueller MF, Strobl DC, Zappia L, Dugas M, Colome-Tatche M, Theis FJ. Benchmarking atlas-level data integration in single-cell genomics. Nat Methods. 2022;19(1):41–50. https://doi.org/10.1038/s41592-021-01336-8. This benckmark study provided a clue to choose the appropriate integration method of scRNA-seq.
Gayoso A, Lopez R, Xing G, Boyeau P, Valiollah Pour Amiri V, Hong J, Wu K, Jayasuriya M, Mehlman E, Langevin M, Liu Y, Samaran J, Misrachi G, Nazaret A, Clivio O, Xu C, Ashuach T, Gabitto M, Lotfollahi M, Svensson V, da Veiga Beltrame E, Kleshchevnikov V, Talavera-López C, Pachter L, Theis FJ, Streets A, Jordan MI, Regier J, Yosef N. A Python library for probabilistic analysis of single-cell omics data. Nat Biotechnol. 2022;40(2):163–6. https://doi.org/10.1038/s41587-021-01206-w. Quality of automatic cell annotation was improved with machine learning technique.
van der Maaten L, Hinton G. Visualizing data using t-SNE. J Mach Learn Res. 2008:2579–2605.
Leland McInnes JH, Melville J. UMAP: uniform manifold approximation and projection for dimension reduction. arXiv. 2020. https://doi.org/10.48550/arXiv.1802.03426.
Gorzalczany MB, Rudzinski F. Generalized self-organizing maps for automatic determination of the number of clusters and their multiprototypes in cluster analysis. IEEE Trans Neural Netw Learn Syst. 2018;29(7):2833–45. https://doi.org/10.1109/TNNLS.2017.2704779.
Aibar S, Aerts S. AUCell. 2016.
Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, van den Oord J, Atak ZK, Wouters J, Aerts S. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083–6. https://doi.org/10.1038/nmeth.4463.
Van de Sande B, Flerin C, Davie K, De Waegeneer M, Hulselmans G, Aibar S, Seurinck R, Saelens W, Cannoodt R, Rouchon Q, Verbeiren T, De Maeyer D, Reumers J, Saeys Y, Aerts S. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat Protoc. 2020;15(7):2247–76. https://doi.org/10.1038/s41596-020-0336-2.
Badia IMP, Velez Santiago J, Braunger J, Geiss C, Dimitrov D, Muller-Dott S, Taus P, Dugourd A, Holland CH, Ramirez Flores RO, et al. decoupleR: ensemble of computational methods to infer biological activities from omics data. Bioinform Adv. 2022;2:vbac016. decoupleR proveids ensemble methods of various annotation tools.
Zhang AW, O’Flanagan C, Chavez EA, Lim JLP, Ceglia N, McPherson A, Wiens M, Walters P, Chan T, Hewitson B, Lai D, Mottok A, Sarkozy C, Chong L, Aoki T, Wang X, Weng AP, McAlpine JN, Aparicio S, Steidl C, Campbell KR, Shah SP. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat Methods. 2019;16(10):1007–15. https://doi.org/10.1038/s41592-019-0529-1.
Armingol E, Officer A, Harismendy O, Lewis NE. Deciphering cell-cell interactions and communication from gene expression. Nat Rev Genet. 2021;22(2):71–88. https://doi.org/10.1038/s41576-020-00292-x.
Browaeys R, Saelens W, Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods. 2020;17(2):159–62. https://doi.org/10.1038/s41592-019-0667-5.
Turei D, Valdeolivas A, Gul L, Palacio-Escat N, Klein M, Ivanova O, Olbei M, Gabor A, Theis F, Modos D, Korcsmáros T, Saez-Rodriguez J. Integrated intra- and intercellular signaling knowledge for multicellular omics analysis. Mol Syst Biol. 2021;17(3):e9923. https://doi.org/10.15252/msb.20209923.
Tsuyuzaki K, Ishii M, Nikaido I. Uncovering hypergraphs of cell-cell interaction from single cell RNA-sequencing data. bioRxiv. 566182. https://doi.org/10.1101/566182.
La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, Fan J, Borm LE, Liu Z, van Bruggen D, Guo J, He X, Barker R, Sundström E, Castelo-Branco G, Cramer P, Adameyko I, Linnarsson S, Kharchenko PV. RNA velocity of single cells. Nature. 2018;560(7719):494–8. https://doi.org/10.1038/s41586-018-0414-6.
Bergen V, Lange M, Peidli S, Wolf FA, Theis FJ. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol. 2020;38(12):1408–14. https://doi.org/10.1038/s41587-020-0591-3. scVelo provides characteristic streamline trajectories on scRNA-seq maps.
Qiu X, Zhang Y, Martin-Rufino JD, Weng C, Hosseinzadeh S, Yang D, Pogson AN, Hein MY, Hoi Joseph Min K, Wang L, Grody EI, Shurtleff MJ, Yuan R, Xu S, Ma Y, Replogle JM, Lander ES, Darmanis S, Bahar I, Sankaran VG, Xing J, Weissman JS. Mapping transcriptomic vector fields of single cells. Cell. 2022;185(4):690–711.e45. https://doi.org/10.1016/j.cell.2021.12.045.
Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32(4):381–6. https://doi.org/10.1038/nbt.2859.
Wolf FA, Hamey FK, Plass M, Solana J, Dahlin JS, Göttgens B, Rajewsky N, Simon L, Theis FJ. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019;20(1):59. https://doi.org/10.1186/s13059-019-1663-x.
Saelens W, Cannoodt R, Todorov H, Saeys Y. A comparison of single-cell trajectory inference methods. Nat Biotechnol. 2019;37(5):547–54. https://doi.org/10.1038/s41587-019-0071-9. dynguidelines provide a clue to choose the appropriate method of trajectory tools.
Funding
JSPS KAKENHI grant numbers JP 22KK0272, JP 23K15736 (HO) and JP 22H04925 (HO, PAGS ver.2)
Takeda Science Foundation 2022 (HO)
Mochida Memorial Foundation for Medical and Pharmaceutical Research 2022 (HO)
Author information
Authors and Affiliations
Contributions
Conceptualization: HO.
Visualization: HO
Funding acquisition: HO, UC, and HH
Project administration: HO and HH
Supervision: UC and HH
Writing—original draft: HO
Writing—review and editing: HO, UC, and HH
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: In Table 1 of this article, each ref. number is off by one less than the correct number.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Okada, H., Chung, Ui. & Hojo, H. Practical Compass of Single-Cell RNA-Seq Analysis. Curr Osteoporos Rep (2023). https://doi.org/10.1007/s11914-023-00840-4
Accepted:
Published:
DOI: https://doi.org/10.1007/s11914-023-00840-4