
Abstract
Purpose of Review
The present review addresses most recently identified mechanisms implicated in metastasis-induced bone resorption and muscle-wasting syndrome, known as cachexia.
Recent Findings
Metastatic disease in bone and soft tissues is often associated with skeletal muscle defects. Recent studies have identified a number of secreted molecules and extracellular vesicles that contribute to cancer cell growth and metastasis leading to bone destruction and muscle atrophy. In addition, alterations in muscle microenvironment including dysfunctions in hepatic and mitochondrial metabolism have been implicated in cancer-induced regeneration defect and muscle loss. Moreover, we review novel in vitro and animal models including promising new drug candidates for bone metastases and cancer cachexia.
Summary
Preservation of bone health could be highly beneficial for maintaining muscle mass and function. Therefore, a better understanding of molecular pathways implicated in bone and muscle crosstalk in metastatic disease may provide new insights and identify new strategies to improve current anticancer therapeutics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metastasis is a multi-step process, which includes dissemination of cancer cells from the primary tumor, survival in the circulation and colonization at distant metastatic site. Bone metastases are most common in patients with prostate and breast cancer, and to a lesser extent in kidney, lung, and thyroid cancer patients [1,2]. Bone metastatic cascade is initiated by cancer cell invasion through the basement membrane and extracellular matrix and migration to the blood or lymphatic system [3]. During cancer cell invasion, important roles are attributed to matrix metalloproteinases (MMPs) due to their ability to cleave and degrade extracellular matrix (ECM) [4]. In addition, cancer cells undergo epithelial-mesenchymal transition (EMT) which enhances their invasiveness, while following homing at metastatic site they revert to epithelial phenotype by mesenchymal-epithelial transition (MET) required for metastatic outgrowth [5]. Once in the bone, metastatic cancer cells secrete cytokines including parathyroid hormone-related peptide (PTHrP), which induce bone-forming osteoblasts to produce excessive amount of receptor activator of nuclear factor kappa-B ligand (RANKL) to activate the bone-resorbing osteoclasts. Upon bone resorption, growth factors such as transforming growth factor β (TGF-ß) that further stimulate tumor growth creating a so-called “vicious cycle of bone metastasis.” In addition to osteolytic lesions, certain cancers, such as prostate cancer, result in osteoblastic metastases due to abnormal formation of woven, poor-quality bone.
Although skeletal muscle is the most abundant tissue in the vertebrate body, metastases are very rare with a prevalence range from 0.03 to 17.5% [6]. However, excessive muscle wasting, or the loss of muscle tissue, is commonly observed in metastatic cancer [7,8]. The release of soluble proteins, exosomes, and metabolites from metastatic tissues can systematically affect distant organs such as muscle and lead to muscle wasting syndrome, also known as cachexia [7,9]. Cachexia is a multi-organ wasting syndrome characterized by ongoing loss of skeletal muscle mass, decreased muscle strength, systemic inflammation, and increased basal energy expenditure that cannot be fully reversed by conventional nutritional support [10,11]. Ultimately, cachexia leads to progressive functional impairment, decreased quality of life, and increased mortality of cancer patients [12]. In general, skeletal muscle wasting has been highly associated with pancreatic, stomach, colorectum, lung, head-neck, and breast cancer patients [13]. Currently, there is no standard treatment for cancer cachexia. Thus, there is an urgent need for further investigation and better understanding of underlying mechanisms which could lead to potentially new therapeutic targets.
Muscle Defects in Metastatic Disease
Molecular Mechanisms Underlying Cancer Cachexia
The mechanisms that drive metastasis-induced cachexia are not fully understood. While muscle protein breakdown in cancer is clearly induced, changes in muscle protein synthesis are not consistent [14,15]. The main protein degradation pathway is the ubiquitin-proteasome system, which involves muscle specific E3 ligases atrogin-1/MAFbx (muscle atrophy F-box protein) and MuRF-1 (muscle-specific RING-Finger-1) [14]. In advanced cancer, transcriptionally activated E3 ligases mediate ubiquitination of muscle structural and contractile proteins, thus contributing to muscle atrophy and decreased muscle function [14]. Muscle degradation in cancer is also mediated through the autophagic-lysosomal system, which induces lysosomal-dependent degradation of cytoplasmic proteins and organelles, and calcium-dependent proteolysis composed of cysteine proteases, also known as calpains [14,15,16]. In metastatic cancer, these pathways are activated by pro-inflammatory cytokines such as interleukin 6 (IL-6), interleukin 1 (IL-1), tumor necrosis factor alpha (TNF-α), and interferon gamma (IFN-γ) [17,18,19,20]. These cytokines, secreted by cancer cells, immune cells, and other non-cancer cells within the tumor microenvironment, contribute to systemic inflammation and activate catabolic processes in muscle through transcriptional regulators such as p38 MAPK, nuclear factor kappa B (NF-κB), and STAT3 [11,17]. In addition to cytokines, other tumor-derived factors are implicated in cancer cachexia such as hormones, metal ions, microRNAs, and members of the TGF-β superfamily including TGF-ß, activin A, growth differentiation factor-11 (GDF-11), and myostatin [17,21,22]. Also, recent studies suggest that alterations in the muscle microenvironment in cancer cachexia can affect its regenerative ability [23,24]. Furthermore, mitochondrial dysfunction in skeletal muscle has been implicated in muscle catabolism and cancer-induced muscle wasting [25].
Mechanisms Underlying Muscle Defects in Bone Metastases
Metastatic bone disease is often associated with skeletal muscle weakness [22,26,27,28]. Osteolytic bone metastases stimulate bone resorption, which leads to the release of bone-derived factors, particularly TGF-β, from the bone matrix [22,29]. The mechanism by which TGF-β contributes to skeletal muscle weakness was shown to be through NADPH oxidase 4 (NOX4)-mediated oxidation of skeletal muscle proteins such as ryanodine receptor/calcium (Ca2+) release channel (RyR1) [22]. As RyR1 channels are required for muscle contraction by releasing calcium from sarcoplasmic reticulum stores into cytoplasm, their oxidation results in intracellular calcium leakage, which leads to muscle weakness. A similar mechanism was observed in mouse models of osteolytic bone metastases from breast, prostate, and lung cancers; multiple myeloma; and a syngeneic mouse model of osteolytic cancer in the bone [22,28]. Consistently, we have demonstrated the effect of osteolytic breast cancer bone metastases on muscle weakness and muscle fiber atrophy via bone-matrix–derived TGF-ß and activated p38/NF-κB signaling cascade [27••]. Pharmacological inhibition of sclerostin an inhibitor of the WNT signaling and bone formation reduced cancer progression and bone destruction and importantly improved muscle microarchitecture and function. In addition, observed expansion of Pax7-positive satellite cells in the muscle most likely contributes to declining muscle strength, as NF-κB activation usually leads to quiescent state of satellite cells and dysregulation of the myogenic program [30]. The regenerative capacity of muscle was partially restored by anti-sclerostin antibody treatment, which resulted in suppressed p38/NF-κB signaling and restored number of Pax7-positive satellite cells [27••].
In accordance with our findings, a severe muscle regeneration defect was associated with an elevated number of Pax7-positive satellite cells in a cancer cachexia model using colon-26 (C26) cancer cells [23,24]. Mechanistically, the impaired myogenesis resulted from a suppressed differentiation potential of satellite cells due to reduced neutrophil infiltration and macrophage recruitment in cachectic muscle or dysregulated IL-4-dependent signaling. Regarding the latter, IL-4 treatment counteracted cachexia and restored muscle mass by increasing muscle protein synthesis [24]. Furthermore, muscle regeneration was partially restored by IL-4 as shown by a reduced accumulation of satellite cells and fibro-adipogenic progenitors, which are non-myogenic muscle stem cells that regulate muscle homeostasis and can differentiate to adipocytes or fibroblasts in pathological and chronic conditions [31]. Finally, IL-4 administration potentially increased the immune response against the tumor as large areas of necrosis in tumors were accompanied by an increased number of cytotoxic lymphocytes and type II macrophages.
Mechanism of Muscle Defects in Soft Tissue Metastases
Recent studies have identified the role of the metal-ion transporter ZIP14 in both advanced cancer patients and metastatic breast cancer and colon cancer mouse models [7]. ZIP14 was upregulated in cachectic muscles by TGF-ß and TNF-α, leading to enhanced ZIP14-mediated zinc uptake by muscle, reduced expression of myogenic regulatory factors, and loss of myosin heavy chain. Similar mechanism of ZIP14 upregulation and increased zinc uptake in the muscle was associated with the activation of TGF-ß/SMAD signaling and progression of cachexia in metastatic models of breast cancer and pancreatic ductal adenocarcinoma [32,33]. Therefore, ZIP14 provides a potential link between zinc accumulation and metastasis-induced cachexia and serves as a potential therapeutic target.
Cachectic phenotype was also observed in advanced colorectal cancer models, including C26, MC38, and HCT116, accompanied by development of liver metastases [34••, 35, 36]. Muscle atrophy was linked with aberrant activation of STAT3, which contributed to proteolysis pathways through activation of Atrogin-1 and MuRF1 [34••,35]. STAT3-mediated muscle wasting was triggered by circulating IL-6, whereas inhibition of IL-6/STAT3 signaling rescued muscle atrophy [35]. Indeed, increased levels in total STAT3 have been shown to correlate with poor prognosis of advanced cancer patients [37]. Recently, HSP90-mediated activation of STAT3 was shown to activate ubiquitin-proteasome pathway in a FOXO1-dependent manner in cancer cachexia [38]. Furthermore, the release of HSP90 by cancer cells was responsible for Toll-like receptor 4 (TLR4) activation and promotion of muscle catabolism by activating p38/MAPK signaling cascade [39], consistent with previous data in pancreatic cancer, where ZIP4-mediated release of HSP70 and HSP90 promoted muscle atrophy [40]. Colorectal cancer progression was accompanied with bone resorption and bone loss; however, the presence of liver metastases exacerbated cachectic phenotype [34••, 35, 36]. Hepatic alterations have been associated with cancer-induced muscle wasting [41,42] and furthermore liver fibrosis-induced muscle atrophy was promoted by elevated levels of circulating IL-6, TNF-α and myostatin during progression of liver disease [43,44].
Recently, altered mitochondrial homeostasis has been implicated in cancer-induced muscle wasting [25,34••,35,45], contributing to a shift from oxidative to glycolytic metabolism, which results in intramuscular lipid accumulation and decreased muscle strength and function [34••]. Indeed, patients with metastatic melanoma demonstrated fatty infiltration in the muscle, leading to reduced skeletal muscle density and poor survival [46]. Moreover, suppression of PGC1α, the major regulator of mitochondrial biogenesis, has been linked with high circulating IL-6 levels [47] and muscle atrophy [34••,35,45]. In addition, mitochondrial dysfunction results in release of myokine FGF21 [48], which contributes to muscle loss [49]. Most recently, cachectic muscles have been characterized by reduced mitochondrial iron content accompanied with increased catabolism, while iron supplementation restored mitochondrial function, which resulted in improved muscle mass, function and strength [50]. These results establish a critical role of iron in maintaining skeletal muscle homeostasis, revealing iron metabolism as a potential therapeutic target in cancer-induced muscle atrophy.
Regarding other cytokines, IL-8 which is released from pancreatic cancer cells at high levels correlated with muscle atrophy, acting through CXCR2 receptor and activation of ERK1/2 signaling [51]. More recently, high serum concentration of IL-35 in advanced non-small lung cancer and breast cancer patients was linked with increased skeletal muscle atrophy and activation of MuRF1 and Atrogin-1 [52]. Stress-responsive cytokine GDF15 and its receptor GDNF receptor alpha like (GFRAL) have been implicated in cancer cachexia [53]. Moreover, exosomes secreted by C26 colon cancer cells were enriched with GDF-15 and found to contribute to the development of cancer cachexia by inducing muscle atrophy via regulating Bcl-2/caspase-3 pathways [54].
New cachectic and anti-cachectic factors have been identified in recent years. For instance, PAUF, which is secreted by pancreas cancer cells, functions through Atrogin-1-dependent catabolic pathways and has been associated with poor clinical outcome in pancreatic cancer patients [55]. Furthermore, IFIT2 depletion was shown to induce oral squamous cell carcinoma metastasis and skeletal muscle atrophy through IL-6 signaling [56], whereas deletion of stress-response protein REDD1 prevented chemotherapy-induced muscle atrophy via mTORC1-dependent signaling [57]. 3-MA was recently identified as an anti-cachectic and anti-tumorigenic factor in pancreatic cancer acting through inhibition of p53 apoptosis effector related to PMP22 (PERP) and suppression of pancreatic cancer cell growth [58].
MicroRNAs in Metastasis-Induced Muscle Weakness
To date, several noncoding RNAs are known to be involved in cancer-mediated muscle wasting [59]. Recently, Xie et al. demonstrated that repression of miR-29c induced leukemia inhibitory factor (LIF) in muscles that promoted muscle wasting through the JAK/STAT and MAP kinase pathways [60]. Muscle atrophy in colorectal cancer patients with metastasis associated with increased circulating levels of miR-203 secreted by metastatic tissue [21]. Mechanistically, overexpression of miR-203 resulted in suppressed proliferation and stimulated apoptosis of skeletal muscle cells via targeting BIRC5 (survivin), a negative regulator of apoptosis. While miR-181a-3p and both miR-195a-5p and miR-125b-1-3p induced muscle atrophy by activating apoptotic signaling pathways [61,62], a decrease in miR-497-5p mediated by IL-6, counteracted muscle atrophy by stimulating expression of hypertrophy-related genes in cancer cachexia [63]. Furthermore, miR-450-5p and miR-451a were found differentially expressed in skeletal muscle of cachectic lung cancer patients; however, more research is needed to better understand their regulation in muscle atrophy [64].
Models to Investigate Cancer-Induced Muscle Defects
Mechanistic studies of metastasis-induced cachexia have been limited due to the lack of animal models that recapitulate clinical features seen in humans. Therefore, development of new mouse models is needed to improve our knowledge on the mechanisms that drive the disease and possibly provide new therapeutic targets. In line with this, multiple zebrafish models of metastatic hepatocellular carcinoma have been established in recent years [65]. These models exhibit inflammation and cancer-induced skeletal muscle wasting and could thus be useful in high-throughput in vivo screening for anti-metastatic or anti-cachectic drugs. Recently, a new mouse model with metastases to the lungs has been developed by utilizing human papilloma virus (HPV) and oropharyngeal squamous cell carcinoma cell line [66]. This model recapitulates key features of cancer cachexia, as evident by progressive loss of body mass, functional disability, systemic inflammation, and muscle wasting mediated by activation of ubiquitin proteasome and autophagy pathways. A new murine model of breast cancer with spontaneous metastases has been developed recently by orthotopic injection of Bard1-deficient breast cancer cells that spontaneously metastasize to the lung [32]. Affected mice developed cancer-associated muscle atrophy, demonstrating the suitability of this model for future translational research.
Therapeutic Targeting of Cancer-Induced Muscle Defects
A number of promising drug candidates are being assessed for cancer cachexia [67,68,69,70,71,72]. Specifically, activin type 2 receptor (ActRIIB) or its ligands such as myostatin, GDF11, and activins are attractive therapeutic targets considering their important role in the regulation of muscle growth. Many myostatin inhibitors failed in clinical trials in recent years; however, new potential candidates such as IMB0901 exhibited promising results in rescuing muscle atrophy in cancer cachexia [73]. Furthermore, inhibition of ActRIIB signaling by ActRIIB-Fc effectively preserved skeletal muscle mass and strength in mice bearing advanced colorectal cancers [74]. Moreover, dual anti-ActRIIA/IIB antibody treatment reduced serum levels of IL-6 and reversed cachexia in mice, supporting a functional link between activin A and IL-6 signaling pathways observed in ovarian cancer cells [75]. In addition, blocking type I receptors ALK4/5 of the TGF-β family preserved cancer-associated muscle loss and downregulated catabolic processes in the muscle [76]. Endogenous antagonist follistatin-like 3 (FSTL3) binds to activins, GDF8, and GDF11 [77], without affecting other ligands of the TGF-β family. Indeed, systemic administration of monovalent human FSTL3 Fc-fusion protein (mono-FSTL3-Fc) resulted in increased muscle mass in mice, representing a promising therapeutic option for muscle loss due to its more specific action and less adverse effects than ActRIIB-Fc [77]. In cancer cachexia, inhibition of Activin A preserved muscle mass and MEF2C expression, which is involved in the regulation of MYHC7 expression, one of the myosin heavy-chain (MYHC) isoforms in muscle [78]. Inhibition of GDF15–GFRAL signaling by monoclonal antibody 3P10 showed beneficial effects on lipid metabolism and reversed cancer cachexia in mice [79]. Anti-cachectic and anti-tumorigenic effects of mitochondrial assembly receptor (MasR) agonist AVE 0991 were identified [80]. In addition, administration of mitochondria-targeting antioxidant mitoquinone preserved skeletal muscle mass and strength, normalized mitochondrial homeostasis, and improved oxidative metabolism, which contributed to decreased intramuscular fat infiltration in cancer cachexia [81•]. Another mitochondria-targeted peptide SS-31 had only partial effect on preventing body wasting, but improved mitochondrial activity and mostly modulated liver metabolome by rescuing the levels of glucose and glycogen that are usually reduced in cachexia[82].
Bone Defects in Metastatic Disease
Early Stages of Bone Metastasis and the Pre-Metastatic Niche
Metastasis starts in the primary site and involves cancer cell intrinsic and extrinsic events. Besides changing their phenotype to an invasive, mesenchymal-like through epithelial-mesenchymal transition (EMT), accumulating evidence suggests that primary tumors establish a permissive pre-metastatic niche within the bone by secreting tumor-derived factors such as growth factors and extracellular vesicles (EVs), by modifying extracellular matrix and recruiting bone marrow-derived cells (BMDCs) [83]. Various cytokines such as epidermal growth factor (EGF), hepatocyte growth factor (HGF), platelet-derived growth factor (PDGF), and TGF-ß are involved in EMT induction including transcription factors Snail, Slug, Twist, and ZEB1 [84]. In addition, EMT is associated with suppressed anti-tumor immune response [85]. Recently, oncogene MCT-1 was found to promote IL-6/IL-6R/STAT3 axis that leads to increased EMT process and cancer stemness but also affects the tumor immunity as seen by increased polarization of macrophages toward the immunosuppressive M2 phenotype which drive the invasiveness of breast cancer cells [86]. Tumor-associated macrophages (TAMs), M1 and M2 macrophages, participate in the formation of the tumor microenvironment, immunosuppression, and regulation of tumor growth [87]. Moreover, M2 macrophages were found to secrete chemokine CCL5, which promotes prostate cancer cell invasion, migration, and EMT via activating β-catenin/STAT3 pathway, whereas CCL5 knockdown suppresses tumor growth and bone metastases [88]. Circulating tumor cells escape detection by the immune system and settle within the bone marrow microenvironment, where they interact with the osteoblastic niche through connexin 43 gap-junctions, thereby activating calcium signaling and cancer cell growth in bone [89]. In addition, a key role of CXCL12/CXCR4 signaling axis in mediating cancer cell homing to bone has been previously established [83].
Significant advances have been made over the recent years in understanding the importance of the pre-metastatic niche. Breast cancer cell–derived EVs are recruited by the bone microenvironment where they increase the ability of osteoblasts to secrete cytokines and EVs to induce osteoclast formation and metastasis-induced osteolysis [90]. EVs also transfer miR-21 to osteoclasts and promote osteoclast differentiation via regulating programmed cell death 4 (PDCD4) expression [91•]. Xu et al. showed that novel circRNA circIKBKB promoted breast cancer bone metastases by inducing the bone pre-metastatic niche through NF-κB signaling pathway [92]. Therefore, disrupting the communication between breast cancer cells and the bone microenvironment would present an interesting future therapeutic strategy for bone metastases. Moreover, breast cancer–derived factors support attachment and survival of disseminated tumor cells to the premetastatic niche in bone by inducing changes in bone mineral properties [93]. Similarly, R-spondin 2 (RSPO2) and RANKL, secreted from breast cancer cells, are involved in the recruitment of osteoclast progenitors and formation of osteoclastic pre-metastatic niche [94]. They bind to the LGR4 receptor and regulate the expression of Dickkopf-related protein 1 (DKK1), a soluble inhibitor of Wnt signaling. Recently, cells from the immune system such as dendritic cells were shown to differentiate to osteoclast progenitors in response to T cell–mediated release of cytokines, such as RANKL, while interleukin 23 (IL23) produced by differentiated dendritic cells further maintains T cell pro-osteoclastogenic activity in the bone marrow [95]. This keeps a positive feedback loop of bone destruction and contributes to the formation of the pre-metastatic niche within the bone microenvironment before tumor cell homing.
Disseminated cancer cells can enter an extended period of proliferative dormancy within the bone metastatic niche and become reactivated by escaping cell cycle arrest [96,97,98]. Dormant breast cancer cells compete with long-term hematopoietic stem cells for the occupancy of the endosteal niche, which is enriched in spindle-shaped N-cadherin+/CD45− osteoblasts (SNOs) and keeps tumor cells in quiescent state in a Notch2-dependent manner [99]. Inhibition of the Notch2 pathway has been suggested to reactivate and mobilize dormant breast cancer cells from the endosteal niche by releasing the interaction between dormant cancer cells and SNOs. Consequently, cancer cells can exit the bone microenvironment and colonize distant sites such as the liver. Recently, N-Cadherin was found to play an essential role in maintaining breast cancer cell dormancy in the bone, by increasing their capacity to adhere to SNOs [100]. Moreover, a group of genes, such as Cfh, Gas6, Mme, and Ogn, is highly expressed in dormant breast cancer cells in bone and correlated with recurrence-free survival in breast cancer patients [101]. Disseminated breast cancer cells residing in bone marrow perivascular niche are protected from chemotherapy by their integrin-mediated interactions with molecules including von Willebrand Factor (VWF) and vascular cell adhesion molecule-1 (VCAM-1) [102]. Disruption of these interactions with integrin inhibitors sensitized cancer cells to chemotherapy and reduced bone metastases. Accordingly, integrin β3 has been shown to promote chemoresistance in bone metastases, whereas mTORC1 inhibitor therapy enhanced the chemotherapy effect as evident by decreased bone metastases and cancer-induced bone loss [103].
Cancer-Induced Bone Destruction
Adult bone is continually remodeled by the processes of bone resorption and bone formation [104]. Following successful seeding of disseminated cancer cells in the bone, tumor cells interact with the bone microenvironment leading to tumor growth, which can elicit osteoclast-mediated bone resorption or osteoblast-mediated bone formation. Most common osteolytic metastases are caused by bone resorption, where cytokines IL-1 and IL-6 and PTHrP and RANKL play crucial role in osteoclast formation and activation [83]. Continuous release of cytokines and growth factors from the bone matrix further supports osteoclast activation and tumor growth via various signaling pathways including the RANK/RANKL/osteoprotegerin (OPG)-axis, canonical WNT, and bone morphogenetic protein (BMP)/TGF-ß signaling pathways [105]. Conversely, osteoblastic metastasis in prostate cancer results from excessive bone formation activated by many factors such as endothelin-1 (ET-1), BMPs, PDGF, and TGF-β [83].
Cancer cells preferentially colonize the trabecular region of bone enriched with osteoblasts and micro-vessels [106]. Cancer cell-osteoblast interactions within the bone microenvironment are essential for bone metastasis progression, as osteoblasts are known to protect breast cancer cells from stress-induced death by both paracrine and juxtracrine signals and therefore can limit the number of cancer-supportive niches [107]. However, Kolb et al. demonstrated that a subpopulation of osteoblasts in the bone microenvironment is involved in the suppression of breast cancer cell growth via decorin and NOV (CCN3) proteins [108]. Furthermore, these osteoblasts produce EVs enriched with miR-148a-3p, which further suppresses bone metastatic breast cancer proliferation partly through extracellular signal-regulated kinase 1/2 (ERK1/2) signaling [109]. We have previously identified an important role of TG-interacting factor-1 (Tgif1) in mediating interactions between breast cancer cells and osteoblasts in the bone marrow microenvironment [110]. Absence of Tgif1 in osteoblasts resulted in suppressed breast cancer cell migration and bone metastases, which is mediated through increased Semaphorin 3E (Sema3E) expression. In addition, acidosis may contribute to the colonization of breast cancer cells in the bone by promoting extracellular matrix (ECM) organization [111] and by interfering with bone remodeling [112]. Acidic environment recruits osteoclast precursors and stimulates osteoblasts to secrete the pro-osteoclastogenic factors RANKL and macrophage colony-stimulating factor (M-CSF) and inflammatory mediators TNF, IL-6, and IL-8, which promote osteolysis. Similarly, ERK1/2 activation in both cancer cells and osteoblasts induced inflammatory phenotypic conversion of osteoblasts leading to secretion of cytokines and growth factors, which promoted osteoclastogenesis and cancer growth. This effect was reverted following ERK1/2 inhibition by trametinib [113].
Growth Factors Mediating Bone Defects
The bone microenvironment is rich in growth factors, which stimulate tumor growth and metastasis. Recently, it has been demonstrated that tumoral TGF-β signaling has a role in promoting bone metastatic progression and osteolysis in ER+ breast cancer through stimulating the secretion of osteolytic factors such as PTHrP [114]. In addition, TGF-β-induced DACT1 suppressed WNT signaling and promoted breast and prostate cancer bone metastasis [115]. In both prostate and breast cancer, TGF-β was found to stimulate tumor microenvironment by modulating the recruitment of bone marrow-derived mesenchymal stem cells (BMSCs) into the tumor, mediated by transmembrane protein neural cadherin (N-cadherin) [116,117]. More recently, prostate cancer–derived GDF15 was found to increase the osteoclastogenic potential of osteoblasts, which secrete RANKL and CCL2 to promote bone resorption [118]. In addition, prostate cancer cells induced osteocytes to secrete GDF15 into the bone microenvironment which, in turn, stimulated early growth response 1 (EGR1) expression in prostate cancer cells and promoted tumor progression [119]. BMPs are actively involved in the tumor development and bone metastatic progression by mediating interactions between cancer cells and the bone environment [120,121]. Previously, it was reported that conditional deletion of BMPR1a in myeloid cells suppresses prostate tumor growth and changes macrophage polarization [122]. In myeloma bone disease, inhibition of BMP signaling prevented bone loss by reducing osteoclastogenesis and promoted osteoblast differentiation by reducing the concentration of sclerostin the bone marrow [123]. Pharmacologic inhibition of BMP signaling by small molecule antagonist DMH1 in prostate cancer models of bone metastasis restricted cancer cell colonization to bone in immunodeficient mice; however, in mice with intact immune system, DHM1 had no effect on tumor growth and bone health [124]. Interestingly, numerous studies identified a dual role of BMPs in cancer development with BMPs acting both as tumor promoters or suppressors [125]. Recently, inhibition of the BMP pathway by LDN-193189 was shown to enhance bone metastasis development in breast cancer [126], suggesting future studies are needed to elucidate the role of BMP signaling in cancer patient treatment.
The Role of Inflammation and Immune Suppression in Bone Metastasis
Bone metastasis formation and progression are associated with systemic inflammation and immune suppression [127,128]. Numerous studies have identified key roles of immune cells such as macrophages, neutrophils, dendritic cells, natural killer cells, and T cells in the formation of bone metastatic niche. In advanced breast cancer, monocyte-derived macrophages promote bone metastasis growth in an IL4R signaling-dependent manner [129], indicating that inhibition of macrophages and IL4R may lead to a new potential therapy targeting bone metastasis. Neutrophil infiltration in bone results in enhanced survival of the metastatic cells by weakening cytotoxic CD8+ T cells responses in advanced breast cancer [130]. Mechanistically, downregulation of catenin delta 1 (CTNND1) in metastatic bone lesions promoted tumor recruitment to bone by upregulating CXCL12/CXCR4 axis via PI3K/AKT/HIF-1α pathway, while recruitment of neutrophils in the bone was stimulated by secretion of GM-CSF and IL-8. This was not in accordance with a previous report demonstrating the anti-tumor role of neutrophils in advanced prostate cancer [131]. Costanzo-Garvey et al. showed that neutrophils induce apoptosis of disseminated cancer cells; however, during bone metastasis progression, neutrophils gradually lose the cytotoxic effect on prostate cancer cells. Recently, estrogen-related receptor alpha (ERRα) was shown to inhibit metastasis progression of breast cancer cells by activating immune response in the bone [132]. Recruitment of CD8+ T cells to bone was enhanced by the production of chemokines CCL17 and CCL20, while suppression of TGF-ß, a key repressor of T cell activity, resulted in increased antitumor cytotoxic response in the bone. In addition, eosinophils were found to induce tumor cell migration and metastasis in bone through CCL6-CCR1 signaling [133].
Secreted Molecules and Extracellular Vesicles in Bone Metastases
Cytokines and chemokines are involved in bone metastatic progression at different stages [134]. IL-11 plays an essential role in breast cancer bone metastases by inducing osteoclastogenesis via JAK1/STAT3 signaling pathway independent of RANKL [135]. Accordingly, blocking of STAT3 activation reduced osteolysis and bone metastatic progression. In addition, neutrophil-derived IL-4 plays a key role in osteolysis in colorectal cancer (CRC) with bone metastases [136]. IL4/IL4Rα signaling activated ERK pathway, which further stimulated the proliferation of osteoclast precursors in bone metastases. Treatment with Ravoxertinib, an inhibitor of the ERK pathway, prevented IL4-mediated bone resorption. Bone marrow–derived IL-1ß stimulates breast cancer metastatic colonization in the bone microenvironment by promoting WNT signaling via NF-κB and CREB [137,138]. Hence, targeting IL-1β-WNT signaling by IL-1 receptor inhibitors such as Anakinra prevented colonization of disseminated cancer cells into bone and decreased the overall metastatic burden. Consistently, formation of the bone metastatic niche was recently demonstrated to be regulated through NAT1/NF-kB/IL-1ß axis [139]. Tulotta et al. demonstrated a dual role of IL1ß, where microenvironment-derived IL-1ß promoted the progression of breast cancer metastases in bone, whereas it inhibited the growth of primary tumor by recruiting innate immune cells with possible anti-tumor roles [140]. Only combined therapy of anakinra with doxorubicin and zoledronic acid exhibited anti-inflammatory effect and markedly inhibited both primary tumor growth and metastatic recurrence in bone. MSC-secreted IL-28 stimulated apoptosis of bone metastatic prostate cancer cells through STAT1 signaling; however, following chronic exposure to IL-28, certain populations of cancer cells became resistant to apoptosis and shifted to STAT3 signaling [141]. Accordingly, STAT3 inhibition resulted in a decreased prostate cancer progression in bone and may be responsible for desensitizing prostate cancer cells to chemotherapy. By using ex vivo bone metastasis culture, chemokine CXCL5 was found to promote breast cancer colonization in bone and could contribute to the switch from a dormant state [142].
Recently, EVs have been identified as important mediators of crosstalk between cancer cells and the bone microenvironment, enabling the transfer of active molecules to distant sites [143,144]. Furthermore, EVs are involved in the formation of the pre-metastatic niche, dissemination of cancer cells to metastatic sites, and cancer cell growth and survival [145]. The role of EVs has been extensively reported in bone metastatic prostate cancer. Prostate cancer–derived EVs target bone marrow cells leading to activation of NF-κB signaling and increased osteoclast differentiation, thereby further enhancing metastatic tumor burden in a cholesterol-dependent manner [146]. Interaction between the long non-coding RNA NORAD and miR-541-3p promoted bone metastases in prostate cancer by upregulating the release of EVs enriched with pyruvate kinase M2 (PKM2) from prostate cancer cells to BMSCs [147]. Moreover, exosomal PKM2 is transferred to BMSCs where it upregulates the production of CXCL12 in a HIF-1α-dependent fashion and subsequently contributes to prostate cancer growth and progression of bone metastasis [148]. In addition, multiple myeloma-derived exosomes are reported to stimulate osteoclastogenesis, acting directly through the IRE1α/XBP1 axis or via amphiregulin (AREG)-mediated activation of EGFR pathway in osteoclast progenitors followed by the release of pro-osteoclastogenic MSC-derived IL-8 [149,150].
Among the biomolecules transported by EVs, non-coding RNAs including lncRNAs and miRNAs exhibit various roles in metastatic bone disease [151,152]. Recent studies demonstrated that several miRNAs are involved in the regulation of osteoclasts and osteoblasts during bone metastasis progression in breast, prostate and colorectal cancer [153,154,155,156,157,158,159,160,161]. Furthermore, these miRNAs show a correlation with disease progression and can be used as biomarkers for cancer progression [162,163,164,165,166]. In breast cancer, the novel lncRNA DGUOK-AS1 was identified to promote cancer progression and bone metastasis by decreasing tumor suppressor miR-204-5p and stimulating the secretion of IL-11 [167]. In bone metastasis of prostate cancer, novel tumor suppressive miRNAs, miR-582-3p, and miR-582-5p inhibit bone metastases through inactivation of NF-κB signaling [168,169], whereas miR-532-3p and miR-204-5p by suppressing TGF-ß signaling activity [170], suggesting a strong potential as therapeutic target. Moreover, Dai et al. demonstrated the important role of TGF-ß-dependent double-negative feedback loop between miR-33a-5p and ZEB1 in the promotion of prostate cancer bone metastasis [171]. Exosomal miR-378a-3p promoted prostate cancer progression and osteolysis by targeting Dyrk1a/Nfatc1 pathway in bone marrow macrophages leading to increased secretion of angiopoietin like 2 (Angptl2) into the bone microenvironment [172]. Ma et al. demonstrated that prostate cancer-derived EVs deliver miR-152-3p to osteoclasts and promote bone osteolysis by targeting osteoclastogenic regulator MAFB [173]. In hepatocellular carcinoma, lnc34a was identified to promote bone metastasis acting through suppression of miR-34a, which inhibits TGF-ß/Smad signaling and its downstream targets, connective tissue growth factor (CTGF) and IL-11 [174]. In non-small cell lung cancer, exosomal lncRNA-SOX2OT promoted bone metastases by targeting TGF-β/pTHrP/RANKL signaling pathway in osteoclasts [175].
Novel Approaches to Treat Bone Metastases
Animal models of skeletal metastasis are essential for understanding the pathogenesis of cancer bone metastases. There has been an increased interest in generating new three-dimensional (3D) in vitro models including patient-derived xenograft models, organoid, and scaffold models that can mimic native bone microenvironment [176,177,178,179,180,181,182,183,184,185]. In addition, numerous new compounds have been evaluated for their effect and therapeutic potential on metastatic bone disease [186,187,196,197,198,188,189,190,191,192,193,194,195]. Currently approved therapeutic agents for bone metastases include chemotherapy, radiotherapy, bisphosphonates, and anti-RANKL therapy [199]. Recently, a series of experiments were conducted to apply the technology of induced tumor-suppressing cells (iTSCs) to bone cells, MSCs, and cancer cells, where activation of oncogenic signaling such as WNT results in production of tumor-suppressing secretomes [200]. WNT activation by LRP5/ß-catenin overexpression in osteocytes generated tumor-suppressive secretomes, which successfully suppressed tumor growth and bone destruction by downregulating chemokines CXCL1 and CXCL5, upregulating tumor suppressors such as P53 and suppressing the expression of oncogenic genes such as MMP-9, Runx2, TGFβ, and Snail [201,202]. Similar anti-tumor capability was observed in MSCs generated by overexpressing LRP5, β-catenin, Snail, or Akt [203]. Activation of WNT signaling in osteoclasts, osteoblasts, and cancer cells resulted in their conversion to tumor-suppressive cells with secretomes enriched with Hsp90ab1, enolase 1 (Eno1), moesin (MSN), and ubiquitin C (Ubc), which acted as atypical tumor-suppressors [204,205,206]. Osteoclast secretome-derived Hsp90ab1 and Eno1 inhibited tumor progression by suppressing TGF-ß signaling and interacting with CD44, facilitating tumor cell killing by natural killer (NK) cells [204,207]. They also exhibited bone-protective roles as osteoclast secretome inhibited RANKL-stimulated osteoclast differentiation and stimulated osteoblast differentiation. Collectively, novel iTSC technology and generation of anti-tumor secretomes represent a potential therapeutic approach for bone metastases. In recent years, a nanoparticle-based drug delivery system (DDS) is often used to deliver different therapeutics to bone [208,209]. Huang et al. showed that nanoparticles loaded with cisplatin and zoledronate significantly inhibited tumor growth and bone resorption in breast cancer bone metastasis [210]. Furthermore, gold clusters suppressed breast cancer-induced osteoclastogenesis and osteolysis, demonstrating their potential for treating breast cancer bone metastases [211].
Conclusions
Here, we presented an overview of recent discoveries related to metastatic bone and muscle disease. Muscle and bone share close mechanical and biochemical relationship, giving rise to muscle-bone crosstalk with both tissues releasing either muscle-derived myokines or bone-derived osteokines that positively or negatively affect bone and muscle metabolism (Fig. 1). Hence, pharmacological approaches for bone health could be efficient in preserving muscle mass and function in cancer cachexia. Recently, it was demonstrated that tumor-derived RANKL in cancer-bearing mice is associated with increased bone turnover and skeletal muscle atrophy, while anti-RANKL or bisphosphonate treatment preserved bone and partially prevented the loss of muscle mass and strength [212••]. Moreover, the administration of bisphosphonates in mice exposed to a chemotherapeutic agent had beneficial effect on muscle mass and strength, acting through bone preservation and inhibiting the release of bone-derived factors upon bone resorption [213]. Therefore, a better understanding of molecular pathways implicated in cancer-mediated bone resorption and muscle wasting may provide new insights for discovering new antiresorptive, anti-cachectic and possibly anti-cancer therapeutics.

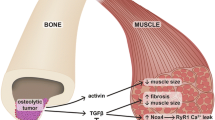
Bone-muscle crosstalk in bone metastasis. Bone-derived osteokines and muscle-derived myokines mediate the bone-muscle interactions in physiological and pathological conditions. In bone metastasis, several cytokines are released from the bone matrix (e.g., TGF-β) that impair muscle function and promote tumor growth. Furthermore, local such as RANKL promote bone destruction and reduce muscle strength. Thus, preventing pathological bone resorption could be an effective therapeutic strategy to preserve not only the bone but also muscle health in metastasis
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Huang J-F, Shen J, Li X, Rengan R, Silvestris N, Wang M, et al. Incidence of patients with bone metastases at diagnosis of solid tumors in adults: a large population-based study. Ann Transl med [internet]. AME Publishing Company. 2020;8:482 Available from: https://pubmed.ncbi.nlm.nih.gov/32395526.
Macedo F, Ladeira K, Pinho F, Saraiva N, Bonito N, Pinto L, et al. Bone metastases: an overview. In: Oncol rev [internet], vol. 11. Pavia, Italy: PAGEPress Publications; 2017. p. 321. Available from: https://pubmed.ncbi.nlm.nih.gov/28584570.
Shao H, Varamini P. Breast cancer bone metastasis: a narrative review of emerging targeted drug delivery systems. In: Cells [internet], vol. 11: MDPI; 2022. p. 388. Available from: https://pubmed.ncbi.nlm.nih.gov/35159207.
Li X, Jin L, Tan Y. Different roles of matrix metalloproteinase 2 in osteolysis of skeletal dysplasia and bone metastasis (review). Mol Med Rep [Internet]. 2020/11/25. D.A. Spandidos; 2021;23:70. Available from: https://pubmed.ncbi.nlm.nih.gov/33236155
Dongre A, Weinberg RA. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol [Internet]. 2019;20:69–84. https://doi.org/10.1038/s41580-018-0080-4.
Surov A, Hainz M, Holzhausen H-J, Arnold D, Katzer M, Schmidt J, et al. Skeletal muscle metastases: primary tumours, prevalence, and radiological features. Eur Radiol [internet]. Germany. 2010;20:649–58. https://doi.org/10.1007/s00330-009-1577-1.
Wang G, Biswas AK, Ma W, Kandpal M, Coker C, Grandgenett PM, et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat Med [Internet]. 2018;24:770–81. https://doi.org/10.1038/s41591-018-0054-2.
Biswas AK, Acharyya S. Understanding cachexia in the context of metastatic progression. Nat Rev Cancer [Internet]. 2020;20:274–84. https://doi.org/10.1038/s41568-020-0251-4.
Biswas AK, Acharyya S. Cancer-associated cachexia: a systemic consequence of cancer progression. Annu Rev Cancer Biol [Internet]. Annual Reviews; 2020;4:391–411. Available from: https://doi.org/10.1146/annurev-cancerbio-030419-033642
Raynard B, Pigneur F, Di Palma M, Deluche E, Goldwasser F. The prevalence of CT-defined low skeletal muscle mass in patients with metastatic cancer: a cross-sectional multicenter French study (the SCAN study). Support Care Cancer [Internet]. 2022;30:3119–29. https://doi.org/10.1007/s00520-021-06603-0.
da Fonseca GWP, Farkas J, Dora E, von Haehling S, Lainscak M. Cancer cachexia and related metabolic dysfunction. Int J Mol Sci [Internet]. 2020;21:2321 Available from: https://www.mdpi.com/1422-0067/21/7/2321.
Ikeda T, Ishihara H, Iizuka J, Hashimoto Y, Yoshida K, Kakuta Y, et al. Prognostic impact of sarcopenia in patients with metastatic hormone-sensitive prostate cancer. Jpn J Clin Oncol [Internet]. 2020;50:933–9. https://doi.org/10.1093/jjco/hyaa045.
Marceca GP, Londhe P, Calore F. Management of cancer cachexia: attempting to develop new pharmacological agents for new effective therapeutic options. Front Oncol [Internet]. 2020:10. https://doi.org/10.3389/fonc.2020.00298/full.
Singh A, Phogat J, Yadav A, Dabur R. The dependency of autophagy and ubiquitin proteasome system during skeletal muscle atrophy. Biophys Rev [Internet]. 2021;13:203–19. https://doi.org/10.1007/s12551-021-00789-7.
Penna F, Ballarò R, Beltrà M, De Lucia S, García Castillo L, Costelli P. The skeletal muscle as an active player against cancer cachexia. Front Physiol [Internet]. 2019:10. https://doi.org/10.3389/fphys.2019.00041/full.
Zhang Y, Wang J, Wang X, Gao T, Tian H, Zhou D, et al. The autophagic-lysosomal and ubiquitin proteasome systems are simultaneously activated in the skeletal muscle of gastric cancer patients with cachexia. Am J Clin Nutr [Internet]. 2020;111:570–9. https://doi.org/10.1093/ajcn/nqz347.
Argilés JM, López-Soriano FJ, Busquets S. Mediators of cachexia in cancer patients. Nutrition [Internet]. 2019;66:11–5 Available from: https://www.sciencedirect.com/science/article/pii/S0899900719300358.
Armstrong VS, Fitzgerald LW, Bathe OF. Cancer-associated muscle wasting—candidate mechanisms and molecular pathways. Int J Mol Sci [Internet]. 2020;21:9268 Available from: https://www.mdpi.com/1422-0067/21/23/9268.
Rupert JE, Narasimhan A, Jengelley DHA, Jiang Y, Liu J, Au E, et al. Tumor-derived IL-6 and trans-signaling among tumor, fat, and muscle mediate pancreatic cancer cachexia. J Exp Med [Internet]. 2021:218 Available from: https://rupress.org/jem/article/218/6/e20190450/211985/Tumor-derived-IL-6-and-trans-signaling-among-tumor.
Baazim H, Antonio-Herrera L, Bergthaler A. The interplay of immunology and cachexia in infection and cancer. Nat Rev Immunol [Internet]. 2021. https://doi.org/10.1038/s41577-021-00624-w.
Okugawa Y, Toiyama Y, Hur K, Yamamoto A, Yin C, Ide S, et al. Circulating miR-203 derived from metastatic tissues promotes myopenia in colorectal cancer patients. J Cachexia Sarcopenia Muscle [Internet]. 2019;10:536–48. Available from: https://onlinelibrary.wiley.com/doi/10.1002/jcsm.12403
Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S, et al. Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat Med [Internet]. 2015;21:1262–71. Available from: http://www.nature.com/articles/nm.3961
Inaba S, Hinohara A, Tachibana M, Tsujikawa K, Fukada S-I. Muscle regeneration is disrupted by cancer cachexia without loss of muscle stem cell potential. Asakura A, editor. PLoS One [Internet]. 2018;13:e0205467. Available from: https://dx.plos.org/10.1371/journal.pone.0205467
Costamagna D, Duelen R, Penna F, Neumann D, Costelli P, Sampaolesi M. Interleukin-4 administration improves muscle function, adult myogenesis, and lifespan of colon carcinoma-bearing mice. J Cachexia Sarcopenia Muscle [Internet]. 2020/02/27. John Wiley and Sons Inc.; 2020;11:783–801. Available from: https://pubmed.ncbi.nlm.nih.gov/32103619
Beltrà M, Pin F, Ballarò R, Costelli P, Penna F. Mitochondrial dysfunction in cancer cachexia: impact on muscle health and regeneration. Cells. 2021;10.
Waning DL, Guise TA. Molecular mechanisms of bone metastasis and associated muscle weakness. Clin Cancer Res [Internet]. 2014/03/27. 2014;20:3071–7. Available from: https://pubmed.ncbi.nlm.nih.gov/24677373
•• Hesse E, Schröder S, Brandt D, Pamperin J, Saito H, Taipaleenmäki H. Sclerostin inhibition alleviates breast cancer–induced bone metastases and muscle weakness. JCI Insight [Internet]. 2019;4. Available from: https://insight.jci.org/articles/view/125543. This study shows that bone matrix–derived TGF-ß contributes to reduced muscle function in breast cancer metastasis, which is abrogated with anti-sclerostin therapy. Authors suggest that anti-sclerostin therapy could be used as a promising pharmacological candidate for both muscle and bone defects in breast cancer patients by intervening with the vicious cycle of bone metastasis.
Regan JN, Mikesell C, Reiken S, Xu H, Marks AR, Mohammad KS, et al. Osteolytic breast cancer causes skeletal muscle weakness in an immunocompetent syngeneic mouse model. Front Endocrinol (Lausanne) [Internet]. 2017;8:358. Available from: http://journal.frontiersin.org/article/10.3389/fendo.2017.00358/full
Trivedi T, Guise TA. Systemic effects of abnormal bone resorption on muscle, metabolism, and cognition. Bone [Internet]. 2022;154:116245 Available from: https://www.sciencedirect.com/science/article/pii/S8756328221004117.
He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J, et al. NF-κB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest. 2013;123:4821–35.
Theret M, Rossi FM V, Contreras O. Evolving roles of muscle-resident fibro-adipogenic progenitors in health, regeneration, neuromuscular disorders, and aging [Internet]. Front. Physiol. 2021. Available from: https://www.frontiersin.org/article/10.3389/fphys.2021.673404
Shakri AR, James Zhong T, Ma W, Coker C, Hegde R, Scholze H, et al. Aberrant Zip14 expression in muscle is associated with cachexia in a Bard1 -deficient mouse model of breast cancer metastasis. Cancer Med [Internet]. 2020;9:6766–75. Available from: https://onlinelibrary.wiley.com/doi/10.1002/cam4.3242
Shakri AR, Zhong TJ, Ma W, Coker C, Kim S, Calluori S, et al. Upregulation of ZIP14 and altered zinc homeostasis in muscles in pancreatic cancer cachexia. Cancers (Basel) [Internet]. 2019;12:3. Available from: https://www.mdpi.com/2072-6694/12/1/3
•• Huot JR, Novinger LJ, Pin F, Narasimhan A, Zimmers TA, O’Connell TM, et al. Formation of colorectal liver metastases induces musculoskeletal and metabolic abnormalities consistent with exacerbated cachexia. JCI Insight [Internet]. 2020;5. Available from: https://insight.jci.org/articles/view/136687. In this study, authors report that advanced colorectal cancer mouse models with intrasplenically injected C26 tumor cells exhibit skeletal muscle wasting accompanied with bone loss, altered mitochondria homeostasis and increased myosteatosis. Moreover, liver metastases exarcerbate cachetic phenotype supporting the need to use metastatic in vivo models to faithfully mimic cancer cachexia in human patients.
Huot JR, Novinger LJ, Pin F, Bonetto A. HCT116 colorectal liver metastases exacerbate muscle wasting in a mouse model for the study of colorectal cancer cachexia. Dis Model Mech [Internet]. 2020;13. Available from: https://journals.biologists.com/dmm/article/doi/10.1242/dmm.043166/267155/HCT116-colorectal-liver-metastases-exacerbate
Huot JR, Pin F, Essex AL, Bonetto a. MC38 tumors induce musculoskeletal defects in colorectal cancer. Int J Mol Sci [Internet]. MDPI; 2021;22:1486. Available from: https://pubmed.ncbi.nlm.nih.gov/33540821
Wu P, Wu D, Zhao L, Huang L, Shen G, Huang J, et al. Prognostic role of STAT3 in solid tumors: a systematic review and meta-analysis. Oncotarget [internet]. Impact Journals LLC. 2016;7:19863–83 Available from: https://pubmed.ncbi.nlm.nih.gov/26959884.
Niu M, Song S, Su Z, Wei L, Li L, Pu W, et al. Inhibition of heat shock protein (HSP) 90 reverses signal transducer and activator of transcription (STAT) 3-mediated muscle wasting in cancer cachexia mice. Br J Pharmacol. 2021;178:4485–500.
Sin TK, Zhang G, Zhang Z, Gao S, Li M, Li Y-P. Cancer takes a toll on skeletal muscle by releasing heat shock proteins-an emerging mechanism of cancer-induced cachexia. cancers (Basel) [Internet]. MDPI. 2019;11:1272 Available from: https://pubmed.ncbi.nlm.nih.gov/31480237.
Yang J, Zhang Z, Zhang Y, Ni X, Zhang G, Cui X, et al. ZIP4 promotes muscle wasting and cachexia in mice with orthotopic pancreatic tumors by stimulating RAB27B-regulated release of extracellular vesicles from cancer cells. Gastroenterology [Internet]. 2018/10/17. 2019;156:722-734.e6. Available from: https://pubmed.ncbi.nlm.nih.gov/30342032
Rosa-Caldwell ME, Brown JL, Lee DE, Wiggs MP, Perry Jr RA, Haynie WS, et al. Hepatic alterations during the development and progression of cancer cachexia. Appl Physiol Nutr Metab [Internet]. 2019/10/16. 2020;45:500–512. Available from: https://pubmed.ncbi.nlm.nih.gov/31618604
Khamoui A V, Tokmina-Roszyk D, Rossiter HB, Fields GB, Visavadiya NP. Hepatic proteome analysis reveals altered mitochondrial metabolism and suppressed acyl-CoA synthetase-1 in colon-26 tumor-induced cachexia. Physiol Genomics [Internet]. 2020/03/09. Am Physiol Soc; 2020;52:203–216. Available from: https://pubmed.ncbi.nlm.nih.gov/32146873
Kurosawa T, Goto M, Kaji N, Aikiyo S, Mihara T, Ikemoto-Uezumi M, et al. Liver fibrosis-induced muscle atrophy is mediated by elevated levels of circulating TNFα. Cell Death Dis [Internet]. 2021;12:11. Available from. https://doi.org/10.1038/s41419-020-03353-5.
Giusto M, Barberi L, Di Sario F, Rizzuto E, Nicoletti C, Ascenzi F, et al. Skeletal muscle myopenia in mice model of bile duct ligation and carbon tetrachloride-induced liver cirrhosis. Physiol Rep [Internet]. John Wiley and Sons Inc.; 2017;5:e13153. Available from: https://pubmed.ncbi.nlm.nih.gov/28364027
Ballarò R, Beltrà M, De Lucia S, Pin F, Ranjbar K, Hulmi JJ, et al. Moderate exercise in mice improves cancer plus chemotherapy-induced muscle wasting and mitochondrial alterations. FASEB J [Internet]. John Wiley & Sons, ltd; 2019;33:5482–5494. Available from. https://doi.org/10.1096/fj.201801862R.
Chu MP, Li Y, Ghosh S, Sass S, Smylie M, Walker J, et al. Body composition is prognostic and predictive of ipilimumab activity in metastatic melanoma. J Cachexia Sarcopenia Muscle [Internet]. 2020/02/13. John Wiley and Sons Inc.; 2020;11:748–55. Available from: https://pubmed.ncbi.nlm.nih.gov/32053287
White JP, Puppa MJ, Sato S, Gao S, Price RL, Baynes JW, et al. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet Muscle [Internet]. BioMed Central; 2012;2:14. Available from: https://pubmed.ncbi.nlm.nih.gov/22769563
Favaro G, Romanello V, Varanita T, Andrea Desbats M, Morbidoni V, Tezze C, et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat Commun [Internet]. Nature Publishing Group UK. 2019;10:2576 Available from: https://pubmed.ncbi.nlm.nih.gov/31189900.
Oost LJ, Kustermann M, Armani A, Blaauw B, Romanello V. Fibroblast growth factor 21 controls mitophagy and muscle mass. J Cachexia Sarcopenia Muscle [Internet]. 2019/03/20. John Wiley and Sons Inc.; 2019;10:630–42. Available from: https://pubmed.ncbi.nlm.nih.gov/30895728
Wyart E, Hsu MY, Sartori R, Mina E, Rausch V, Pierobon ES, et al. Iron supplementation is sufficient to rescue skeletal muscle mass and function in cancer cachexia. EMBO Rep [Internet]. John Wiley & Sons, Ltd; 2022;23:e53746. Available from: https://doi.org/10.15252/embr.202153746
Callaway CS, Delitto AE, Patel R, Nosacka RL, D’Lugos AC, Delitto D, et al. IL-8 released from human pancreatic cancer and tumor-associated stromal cells signals through a CXCR2-ERK1/2 axis to induce muscle atrophy. Cancers (Basel) [Internet]. MDPI; 2019;11:1863. Available from: https://pubmed.ncbi.nlm.nih.gov/31769424
Li Z, Zhu L, Zheng H, Jiang W, Wang Y, Jiang Z, et al. Serum IL-35 levels is a new candidate biomarker of cancer-related cachexia in stage IV non-small cell lung cancer. Thorac cancer [Internet]. 2022/02/09. John Wiley & Sons Australia, Ltd; 2022;13:716–23. Available from: https://pubmed.ncbi.nlm.nih.gov/35142058
Ahmed DS, Isnard S, Lin J, Routy B, Routy J-P. GDF15/GFRAL pathway as a metabolic signature for cachexia in patients with cancer. J cancer [internet]. Ivyspring Int Publisher. 2021;12:1125–32 Available from:https://pubmed.ncbi.nlm.nih.gov/33442410.
Zhang W, Sun W, Gu X, Miao C, Feng L, Shen Q, et al. GDF-15 in tumor-derived exosomes promotes muscle atrophy via Bcl-2/caspase-3 pathway. Cell Death Discov [Internet]. 2022;8:162. Available from. https://doi.org/10.1038/s41420-022-00972-z.
Yoo W, Choi H, Son YH, Lee J, Jo S, Jung D, et al. Pancreatic cancer induces muscle wasting by promoting the release of pancreatic adenocarcinoma upregulated factor. Exp Mol Med [Internet]. 2021/03/17. Nature Publishing Group UK; 2021;53:432–445. Available from: https://pubmed.ncbi.nlm.nih.gov/33731895
Lai K-C, Hong Z-X, Hsieh J-G, Lee H-J, Yang M-H, Hsieh C-H, et al. IFIT2-depleted metastatic oral squamous cell carcinoma cells induce muscle atrophy and cancer cachexia in mice. J Cachexia Sarcopenia Muscle [Internet]. 2022/02/15. John Wiley and Sons Inc.; 2022;13:1314–28. Available from: https://pubmed.ncbi.nlm.nih.gov/35170238
Hain BA, Xu H, Waning DL. Loss of REDD1 prevents chemotherapy-induced muscle atrophy and weakness in mice. J Cachexia Sarcopenia Muscle [Internet]. Springer nature; 2021;12:1597–612. Available from. https://doi.org/10.1002/jcsm.12795.
Dasgupta A, Arneson-Wissink PC, Schmitt RE, Cho DS, Ducharme AM, Hogenson TL, et al. Anticachectic regulator analysis reveals Perp-dependent antitumorigenic properties of 3-methyladenine in pancreatic cancer. JCI insight [Internet]. Am Soc Clin Invest; 2022;7:e153842. Available from: https://pubmed.ncbi.nlm.nih.gov/34874916
Chen R, Lei S, Jiang T, She Y, Shi H. Regulation of skeletal muscle atrophy in cachexia by MicroRNAs and long non-coding RNAs. Front cell Dev Biol [Internet]. Frontiers Media S.A.; 2020;8:577010. Available from: https://pubmed.ncbi.nlm.nih.gov/33043011
Xie K, Xiong H, Xiao W, Xiong Z, Hu W, Ye J, et al. Downregulation of miR-29c promotes muscle wasting by modulating the activity of leukemia inhibitory factor in lung cancer cachexia. Cancer Cell Int [Internet]. 2021;21:627. Available from. https://doi.org/10.1186/s12935-021-02332-w.
Qiu L, Chen W, Wu C, Yuan Y, Li Y. Exosomes of oral squamous cell carcinoma cells containing miR-181a-3p induce muscle cell atrophy and apoptosis by transmissible endoplasmic reticulum stress signaling. Biochem Biophys Res Commun [Internet]. 2020;533:831–7 Available from: https://www.sciencedirect.com/science/article/pii/S0006291X20318118.
Miao C, Zhang W, Feng L, Gu X, Shen Q, Lu S, et al. Cancer-derived exosome miRNAs induce skeletal muscle wasting by Bcl-2-mediated apoptosis in colon cancer cachexia. Mol Ther - Nucleic Acids [Internet]. 2021;24:923–38. Available from: https://www.sciencedirect.com/science/article/pii/S2162253121001098
Freire PP, Cury SS, Lopes LO, Fernandez GJ, Liu J, de Moraes LN, et al. Decreased miR-497-5p suppresses IL-6 induced atrophy in muscle cells. Cells [Internet]. MDPI; 2021;10:3527. Available from: https://pubmed.ncbi.nlm.nih.gov/34944037
van de Worp WRPH, Schols AMWJ, Dingemans A-MC, Op den Kamp CMH, Degens JHRJ, Kelders MCJM, et al. Identification of microRNAs in skeletal muscle associated with lung cancer cachexia. J Cachexia Sarcopenia Muscle [Internet]. 2019/12/11. John Wiley and Sons Inc.; 2020;11:452–63. Available from: https://pubmed.ncbi.nlm.nih.gov/31828982
Lee AQ, Li Y, Gong Z. Inducible liver cancer models in transgenic zebrafish to investigate cancer biology. Cancers (Basel) [Internet]. MDPI; 2021;13:5148. Available from: https://pubmed.ncbi.nlm.nih.gov/34680297
Olson B, Norgard MA, Levasseur PR, Zhu X, Marks DL. Physiologic and molecular characterization of a novel murine model of metastatic head and neck cancer cachexia. J Cachexia Sarcopenia Muscle. 2021;12:1312–32.
Kim H-J, Lee J-H, Kim S-W, Lee S-H, Jung D-W, Williams DR. Investigation of niclosamide as a repurposing agent for skeletal muscle atrophy. PLoS One [Internet]. Public Library of Science; 2021;16:e0252135–e0252135. Available from: https://pubmed.ncbi.nlm.nih.gov/34038481
Shen Q, Kuang J-X, Miao C-X, Zhang W-L, Li Y-W, Zhang X-W, et al. Alantolactone ameliorates cancer cachexia-associated muscle atrophy mainly by inhibiting the STAT3 signaling pathway. Phytomedicine [Internet]. 2022;95:153858. Available from: https://www.sciencedirect.com/science/article/pii/S0944711321003998
Bae T, Jang J, Lee H, Song J, Chae S, Park M, et al. Paeonia lactiflora root extract suppresses cancer cachexia by down-regulating muscular NF-κB signalling and muscle-specific E3 ubiquitin ligases in cancer-bearing mice. J Ethnopharmacol [Internet]. 2020;246:112222. Available from: https://www.sciencedirect.com/science/article/pii/S0378874119324900
Lee H, Heo J-W, Kim A-R, Kweon M, Nam S, Lim J-S, et al. Z-ajoene from crushed garlic alleviates cancer-induced skeletal muscle atrophy. Nutrients [Internet]. MDPI. 2019;11:2724 Available from: https://pubmed.ncbi.nlm.nih.gov/31717643.
Chiappalupi S, Sorci G, Vukasinovic A, Salvadori L, Sagheddu R, Coletti D, et al. Targeting RAGE prevents muscle wasting and prolongs survival in cancer cachexia. J Cachexia Sarcopenia Muscle [Internet]. 2020/03/11. John Wiley and Sons Inc.; 2020;11:929–46. Available from: https://pubmed.ncbi.nlm.nih.gov/32159297
Chen L, Yang Q, Zhang H, Wan L, Xin B, Cao Y, et al. Cryptotanshinone prevents muscle wasting in CT26-induced cancer cachexia through inhibiting STAT3 signaling pathway. J Ethnopharmacol [Internet]. 2020;260:113066. Available from: https://www.sciencedirect.com/science/article/pii/S0378874119346847
Liu D, Qiao X, Ge Z, Shang Y, Li Y, Wang W, et al. IMB0901 inhibits muscle atrophy induced by cancer cachexia through MSTN signaling pathway. Skelet Muscle [Internet]. BioMed Central; 2019;9:8. Available from: https://pubmed.ncbi.nlm.nih.gov/30922397
Huot JR, Pin F, Narasimhan A, Novinger LJ, Keith AS, Zimmers TA, et al. ACVR2B antagonism as a countermeasure to multi-organ perturbations in metastatic colorectal cancer cachexia. J Cachexia Sarcopenia Muscle [Internet]. 2020/11/16. John Wiley and Sons Inc.; 2020;11:1779–98. Available from: https://pubmed.ncbi.nlm.nih.gov/33200567
Pettersen K, Andersen S, van der Veen A, Nonstad U, Hatakeyama S, Lambert C, et al. Autocrine activin a signalling in ovarian cancer cells regulates secretion of interleukin 6, autophagy, and cachexia. J cachexia sarcopenia muscle [internet]. Springer nature; 2020;11:195–207. Available from. https://doi.org/10.1002/jcsm.12489.
Levolger S, Wiemer EAC, van Vugt JLA, Huisman SA, van Vledder MG, van Damme-van Engel S, et al. Inhibition of activin-like kinase 4/5 attenuates cancer cachexia associated muscle wasting. Sci Rep [Internet]. 2019;9:9826. Available from. https://doi.org/10.1038/s41598-019-46178-9.
Ozawa T, Morikawa M, Morishita Y, Ogikubo K, Itoh F, Koinuma D, et al. Systemic administration of monovalent follistatin-like 3-Fc-fusion protein increases muscle mass in mice. iScience [Internet]. 2021;24:102488. Available from: https://www.sciencedirect.com/science/article/pii/S2589004221004569
Loumaye A, Lause P, Zhong X, Zimmers TA, Bindels LB, Thissen J-P. Activin A causes muscle atrophy through MEF2C-dependent impaired myogenesis. Cells [Internet]. MDPI; 2022;11:1119. Available from: https://pubmed.ncbi.nlm.nih.gov/35406681
Suriben R, Chen M, Higbee J, Oeffinger J, Ventura R, Li B, et al. Antibody-mediated inhibition of GDF15–GFRAL activity reverses cancer cachexia in mice. Nat med [internet]. 2020;26:1264–70. Available from. https://doi.org/10.1038/s41591-020-0945-x.
Murphy KT, Hossain MI, Swiderski K, Chee A, Naim T, Trieu J, et al. Mas receptor activation slows tumor growth and attenuates muscle wasting in cancer. Cancer Res [Internet]. 2018/11/12. 2019;79:706–19. Available from: https://pubmed.ncbi.nlm.nih.gov/30420474
• Pin F, Huot JR, Bonetto A. The mitochondria-targeting agent MitoQ Improves muscle atrophy, weakness and oxidative metabolism in C26 tumor-bearing mice. Front Cell Dev Biol. 2022;10:1–13 In this study, authors reported anti-cachectic properties of mitochondria-targeting antioxidant indicating that prevention of mitochondrial alterations could be a promising tool for the preservation of skeletal mass.
Ballarò R, Lopalco P, Audrito V, Beltrà M, Pin F, Angelini R, et al. Targeting mitochondria by SS-31 ameliorates the whole body energy status in cancer- and chemotherapy-induced cachexia. Cancers. 2021.
Wang M, Xia F, Wei Y, Wei X. Molecular mechanisms and clinical management of cancer bone metastasis. Bone Res [Internet]. Nature Publishing Group UK; 2020;8:30. Available from: https://pubmed.ncbi.nlm.nih.gov/32793401
Ribatti D, Tamma R, Annese T. Epithelial-mesenchymal transition in cancer: a historical overview. Transl Oncol [Internet]. 2020/04/22. Neoplasia Press; 2020;13:100773. Available from: https://pubmed.ncbi.nlm.nih.gov/32334405
Romeo E, Caserta CA, Rumio C, Marcucci F. The vicious cross-talk between tumor cells with an EMT phenotype and cells of the immune system. Cells [Internet]. MDPI; 2019;8:460. Available from: https://pubmed.ncbi.nlm.nih.gov/31096701
Weng Y-S, Tseng H-Y, Chen Y-A, Shen P-C, Al Haq AT, Chen L-M, et al. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol Cancer [Internet]. BioMed Central; 2019;18:42. Available from: https://pubmed.ncbi.nlm.nih.gov/30885232
Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity [internet]. Front. Immunol. 2020. Available from: https://www.frontiersin.org/article/10.3389/fimmu.2020.583084
Huang R, Wang S, Wang N, Zheng Y, Zhou J, Yang B, et al. CCL5 derived from tumor-associated macrophages promotes prostate cancer stem cells and metastasis via activating β-catenin/STAT3 signaling. Cell Death Dis [Internet]. Nature Publishing Group UK; 2020;11:234. Available from: https://pubmed.ncbi.nlm.nih.gov/32300100
Waning DL, Guise TA, Mohammad KS. A “Connexin” responsible for the fatal attraction of cancer to bone. Cell Metab [Internet]. 2019;29:6–8 Available from: https://pubmed.ncbi.nlm.nih.gov/30625309.
Loftus A, Cappariello A, George C, Ucci A, Shefferd K, Green A, et al. Extracellular vesicles from osteotropic breast cancer cells affect bone resident cells. J Bone Miner Res [Internet]. John Wiley & Sons, Ltd; 2020;35:396–412. Available from. https://doi.org/10.1002/jbmr.3891.
• Yuan X, Qian N, Ling S, Li Y, Sun W, Li J, et al. Breast cancer exosomes contribute to pre-metastatic niche formation and promote bone metastasis of tumor cells. Theranostics [Internet]. Ivyspring International Publisher; 2021;11:1429–45. Available from: https://pubmed.ncbi.nlm.nih.gov/33391543. Authors describe an essential role of breast cancer cell-derived exosomes in promoting bone metastasis by inducing osteoclastogenesis and pre-metastatic niche formation. These exosomes could be a potential target for treatment of breast cancer bone metastasis.
Xu Y, Zhang S, Liao X, Li M, Chen S, Li X, et al. Circular RNA circIKBKB promotes breast cancer bone metastasis through sustaining NF-κB/bone remodeling factors signaling. Mol Cancer [Internet]. BioMed Central; 2021;20:98. Available from: https://pubmed.ncbi.nlm.nih.gov/34325714
Chuang L, Inés M-J, Tengteng T, Wolfgang W, N. DM, et al. Breast cancer–secreted factors perturb murine bone growth in regions prone to metastasis. Sci Adv [Internet]. Am Assoc Advanc Sci; 2022;7:eabf2283. Available from. https://doi.org/10.1126/sciadv.abf2283.
Yue Z, Niu X, Yuan Z, Qin Q, Jiang W, He L, et al. RSPO2 and RANKL signal through LGR4 to regulate osteoclastic premetastatic niche formation and bone metastasis. J Clin Invest [Internet]. Am Soc Clin Invest; 2022;132:e144579–e144579. Available from: https://pubmed.ncbi.nlm.nih.gov/34847079
Monteiro AC, Bonomo A. Dendritic cells development into osteoclast-type APCs by 4T1 breast tumor T cells milieu boost bone consumption. Bone [Internet]. 2021;143:115755 Available from: https://www.sciencedirect.com/science/article/pii/S8756328220305433.
Yadav AS, Pandey PR, Butti R, Radharani NN V, Roy S, Bhalara SR, et al. The biology and therapeutic implications of tumor dormancy and reactivation [internet]. Front. Oncol. 2018. Available from: https://www.frontiersin.org/article/10.3389/fonc.2018.00072
Owen KL, Gearing LJ, Zanker DJ, Brockwell NK, Khoo WH, Roden DL, et al. Prostate cancer cell-intrinsic interferon signaling regulates dormancy and metastatic outgrowth in bone. EMBO Rep [Internet]. 2020/04/21. John Wiley and Sons Inc.; 2020;21:e50162–e50162. Available from: https://pubmed.ncbi.nlm.nih.gov/32314873
Ren D, Dai Y, Yang Q, Zhang X, Guo W, Ye L, et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J Exp med [internet]. 2018/12/28. Rockefeller University Press; 2019;216:428–449. Available from: https://pubmed.ncbi.nlm.nih.gov/30593464
Capulli M, Hristova D, Valbret Z, Carys K, Arjan R, Maurizi A, et al. Notch2 pathway mediates breast cancer cellular dormancy and mobilisation in bone and contributes to haematopoietic stem cell mimicry. Br J cancer [internet]. 2019/06/26. Nature Publishing Group UK; 2019;121:157–171. Available from: https://pubmed.ncbi.nlm.nih.gov/31239543
Maurizi A, Ciocca M, Giuliani C, Di Carlo I, Teti A. Role of neural (N)-cadherin in breast cancer cell stemness and dormancy in the bone microenvironment. Cancers (Basel) [Internet]. MDPI; 2022;14:1317. Available from: https://pubmed.ncbi.nlm.nih.gov/35267624
Ren Q, Khoo WH, Corr AP, Phan TG, Croucher PI, Stewart SA. Gene expression predicts dormant metastatic breast cancer cell phenotype. Breast Cancer Res [Internet]. BioMed Central; 2022;24:10. Available from: https://pubmed.ncbi.nlm.nih.gov/35093137
Carlson P, Dasgupta A, Grzelak CA, Kim J, Barrett A, Coleman IM, et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat Cell Biol [Internet]. 2019/01/21. 2019;21:238–50. Available from: https://pubmed.ncbi.nlm.nih.gov/30664790
Fox GC, Su X, Davis JL, Xu Y, Kwakwa KA, Ross MH, et al. Targeted therapy to β3 integrin reduces chemoresistance in breast cancer bone metastases. Mol Cancer Ther [Internet]. 2021/03/30. 2021;20:1183–98. Available from: https://pubmed.ncbi.nlm.nih.gov/33785647
Al-Bari AA, Al Mamun A. Current advances in regulation of bone homeostasis. FASEB bioAdvances [Internet]. John Wiley and Sons Inc.; 2020;2:668–79. Available from: https://pubmed.ncbi.nlm.nih.gov/33205007
Zhang X. Interactions between cancer cells and bone microenvironment promote bone metastasis in prostate cancer. Cancer Commun [Internet]. 2019;39:76. Available from. https://doi.org/10.1186/s40880-019-0425-1.
Allocca G, Hughes R, Wang N, Brown HK, Ottewell PD, Brown NJ, et al. The bone metastasis niche in breast cancer-potential overlap with the haematopoietic stem cell niche in vivo. J bone Oncol [Internet]. Elsevier; 2019;17:100244. Available from: https://pubmed.ncbi.nlm.nih.gov/31236323
Hughes R, Chen X, Cowley N, Ottewell PD, Hawkins RJ, Hunter KD, et al. Osteoblast-derived paracrine and juxtacrine signals protect disseminated breast cancer cells from stress. Cancers (Basel) [Internet]. MDPI; 2021;13:1366. Available from: https://pubmed.ncbi.nlm.nih.gov/33803526
Kolb AD, Shupp AB, Mukhopadhyay D, Marini FC, Bussard KM. Osteoblasts are “educated” by crosstalk with metastatic breast cancer cells in the bone tumor microenvironment. Breast Cancer Res [Internet]. BioMed Central; 2019;21:31. Available from: https://pubmed.ncbi.nlm.nih.gov/30813947
Shupp AB, Neupane M, Agostini LC, Ning G, Brody JR, Bussard KM. Stromal-derived extracellular vesicles suppress proliferation of bone metastatic cancer cells mediated by ERK2. Mol Cancer Res [Internet]. 2021/05/21. 2021;19:1763–77. Available from: https://pubmed.ncbi.nlm.nih.gov/34021072
Haider M-T, Saito H, Zarrer J, Uzhunnumpuram K, Nagarajan S, Kari V, et al. Breast cancer bone metastases are attenuated in a Tgif1-deficient bone microenvironment. Breast Cancer Res [Internet]. BioMed Central; 2020;22:34. Available from: https://pubmed.ncbi.nlm.nih.gov/32272947
Yamagata AS, Freire PP, Jones Villarinho N, Teles RHG, Francisco KJM, Jaeger RG, et al. Transcriptomic response to acidosis reveals its contribution to bone metastasis in breast cancer cells. Cells [Internet]. MDPI. 2022;11:544 Available from: https://pubmed.ncbi.nlm.nih.gov/35159353.
Di Pompo G, Errani C, Gillies R, Mercatali L, Ibrahim T, Tamanti J, et al. Acid-induced inflammatory cytokines in osteoblasts: a guided path to osteolysis in bone metastasis [Internet]. Front. Cell Dev. Biol. 2021. Available from: https://www.frontiersin.org/article/10.3389/fcell.2021.678532
Back J, Nguyen MN, Li L, Lee S, Lee I, Chen F, et al. Inflammatory conversion of quiescent osteoblasts by metastatic breast cancer cells through pERK1/2 aggravates cancer-induced bone destruction. Bone Res [Internet]. Nature Publishing Group UK. 2021;9:43 Available from: https://pubmed.ncbi.nlm.nih.gov/34588427.
Cheng JN, Frye JB, Whitman SA, Kunihiro AG, Pandey R, Funk JL. A Role for TGFβ signaling in preclinical osteolytic estrogen receptor-positive breast cancer bone metastases progression. Int J Mol Sci [Internet]. MDPI; 2021;22:4463. Available from: https://pubmed.ncbi.nlm.nih.gov/33923316
Esposito M, Fang C, Cook KC, Park N, Wei Y, Spadazzi C, et al. TGF-β-induced DACT1 biomolecular condensates repress Wnt signalling to promote bone metastasis. Nat Cell Biol [Internet]. 2021/03/09. 2021;23:257–67. Available from: https://pubmed.ncbi.nlm.nih.gov/33723425
Noh J, Yu J, Kim W, Park A, Park K-S. Bone marrow-derived mesenchymal stem cells migrate toward hormone-insensitive prostate tumor cells expressing TGF-β via N-cadherin. Biomedicines [Internet]. MDPI; 2021;9:1572. Available from: https://pubmed.ncbi.nlm.nih.gov/34829800
Choi S, Yu J, Kim W, Park K-S. N-cadherin mediates the migration of bone marrow-derived mesenchymal stem cells toward breast tumor cells. Theranostics [internet]. Ivyspring Int Publisher. 2021;11:6786–99 Available from: https://pubmed.ncbi.nlm.nih.gov/34093853.
Siddiqui JA, Seshacharyulu P, Muniyan S, Pothuraju R, Khan P, Vengoji R, et al. GDF15 promotes prostate cancer bone metastasis and colonization through osteoblastic CCL2 and RANKL activation. Bone Res [Internet]. 2022;10:6. Available from. https://doi.org/10.1038/s41413-021-00178-6.
Wang W, Yang X, Dai J, Lu Y, Zhang J, Keller ET. Prostate cancer promotes a vicious cycle of bone metastasis progression through inducing osteocytes to secrete GDF15 that stimulates prostate cancer growth and invasion. Oncogene [Internet]. 2019;38:4540–59. Available from. https://doi.org/10.1038/s41388-019-0736-3.
Ye L, Jiang WG. Bone morphogenetic proteins in tumour associated angiogenesis and implication in cancer therapies. Cancer Lett [Internet]. 2016;380:586–97. Available from: https://www.sciencedirect.com/science/article/pii/S0304383515007041
Sun Z, Cai S, Zabkiewicz C, Liu C, Ye L. Bone morphogenetic proteins mediate crosstalk between cancer cells and the tumour microenvironment at primary tumours and metastases (Review). Int J Oncol [Internet]. VIP-II Division of Medical Department, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education, Beijing), Peking University Cancer Hospital and Institute, Beijing 100142, P.R. China Cardiff China Medical Research Collaborative; 2020;56:1335–51. Available from: https://doi.org/10.3892/ijo.2020.5030
Ihle CL, Straign DM, Provera MD, Novitskiy S V, Owens P. Loss of myeloid BMPR1a alters differentiation and reduces mouse prostate cancer growth. Front Oncol [Internet]. Frontiers Media S.A.; 2020;10:357. Available from: https://pubmed.ncbi.nlm.nih.gov/32318332
Gooding S, Olechnowicz SWZ, Morris E V, Armitage AE, Arezes J, Frost J, et al. Transcriptomic profiling of the myeloma bone-lining niche reveals BMP signalling inhibition to improve bone disease. Nat Commun [Internet]. Nature Publishing Group UK; 2019;10:4533. Available from: https://pubmed.ncbi.nlm.nih.gov/31586071
Straign DM, Ihle CL, Provera MD, Owens P. Targeting the BMP pathway in prostate cancer induced bone disease [internet]. Front. Endocrinol. . 2021. Available from: https://www.frontiersin.org/article/10.3389/fendo.2021.769316
Bach D-H, Park HJ, Lee SK. The dual role of bone morphogenetic proteins in cancer. Mol Ther oncolytics [Internet]. Am Soc Gene Cell Ther. 2017;8:1–13. Available from: https://pubmed.ncbi.nlm.nih.gov/29234727
Vollaire J, Machuca-Gayet I, Lavaud J, Bellanger A, Bouazza L, El Moghrabi S, et al. The bone morphogenetic protein signaling inhibitor LDN-193189 enhances metastasis development in mice [internet]. Front. Pharmacol. . 2019. Available from: https://www.frontiersin.org/article/10.3389/fphar.2019.00667
Monteran L, Ershaid N, Sabah I, Fahoum I, Zait Y, Shani O, et al. Bone metastasis is associated with acquisition of mesenchymal phenotype and immune suppression in a model of spontaneous breast cancer metastasis. Sci Rep [Internet]. Nature Publishing Group UK; 2020;10:13838. Available from: https://pubmed.ncbi.nlm.nih.gov/32796899
Kfoury Y, Baryawno N, Severe N, Mei S, Gustafsson K, Hirz T, et al. Human prostate cancer bone metastases have an actionable immunosuppressive microenvironment. Cancer Cell [Internet]. 2021;39:1464-1478.e8. Available from: https://www.sciencedirect.com/science/article/pii/S1535610821004943
Ma R-Y, Zhang H, Li X-F, Zhang C-B, Selli C, Tagliavini G, et al. Monocyte-derived macrophages promote breast cancer bone metastasis outgrowth. J Exp Med [Internet]. 2020;217:e20191820. Available from. https://doi.org/10.1084/jem.20191820.
Lin Q, Fang X, Liang G, Luo Q, Cen Y, Shi Y, et al. Silencing CTNND1 mediates triple-negative breast cancer bone metastasis via upregulating CXCR4/CXCL12 axis and neutrophils infiltration in bone. Cancers (Basel) [Internet]. MDPI; 2021;13:5703. Available from: https://pubmed.ncbi.nlm.nih.gov/34830862
Costanzo-Garvey DL, Keeley T, Case AJ, Watson GF, Alsamraae M, Yu Y, et al. Neutrophils are mediators of metastatic prostate cancer progression in bone. Cancer Immunol Immunother [internet]. 2020/02/29. Springer Berlin Heidelberg; 2020;69:1113–1130. Available from: https://pubmed.ncbi.nlm.nih.gov/32114681
Bouchet M, Lainé A, Boyault C, Proponnet-Guerault M, Meugnier E, Bouazza L, et al. ERRα expression in bone metastases leads to an exacerbated antitumor immune response. Cancer Res [Internet]. 2020;80:2914–26. Available from. https://doi.org/10.1158/0008-5472.CAN-19-3584.
Li F, Du X, Lan F, Li N, Zhang C, Zhu C, et al. Eosinophilic inflammation promotes CCL6-dependent metastatic tumor growth. Sci Adv [Internet]. American Association for the Advancement of Science; 2021;7:eabb5943–eabb5943. Available from: https://pubmed.ncbi.nlm.nih.gov/34039594
Haider M-T, Ridlmaier N, Smit DJ, Taipaleenmäki H. Interleukins as mediators of the tumor cell-bone cell crosstalk during the initiation of breast cancer bone metastasis. Int J Mol Sci [Internet]. MDPI; 2021;22:2898. Available from: https://pubmed.ncbi.nlm.nih.gov/33809315
Liang M, Ma Q, Ding N, Luo F, Bai Y, Kang F, et al. IL-11 is essential in promoting osteolysis in breast cancer bone metastasis via RANKL-independent activation of osteoclastogenesis. Cell Death Dis [Internet]. Nature Publishing Group UK; 2019;10:353. Available from: https://pubmed.ncbi.nlm.nih.gov/31040267
Jin Q, Yang H, Jing Z, Hong-Hua W, Ben-Jing S, Li-Ting W, et al. IL4/IL4R signaling promotes the osteolysis in metastatic bone of CRC through regulating the proliferation of osteoclast precursors. Mol Med [Internet]. BioMed Central; 2021;27:152. Available from: https://pubmed.ncbi.nlm.nih.gov/34863091
Eyre R, Alférez DG, Santiago-Gómez A, Spence K, McConnell JC, Hart C, et al. Microenvironmental IL1β promotes breast cancer metastatic colonisation in the bone via activation of Wnt signalling. Nat Commun [Internet]. 2019;10:5016. Available from. https://doi.org/10.1038/s41467-019-12807-0.
Tulotta C, Lefley DV, Freeman K, Gregory WM, Hanby AM, Heath PR, et al. Endogenous production of IL1B by breast cancer cells drives metastasis and colonization of the bone microenvironment. Clin Cancer Res [Internet]. 2019;25:2769–2782. Available from. https://doi.org/10.1158/1078-0432.CCR-18-2202.
Zhao C, Cai X, Wang Y, Wang D, Wang T, Gong H, et al. NAT1 promotes osteolytic metastasis in luminal breast cancer by regulating the bone metastatic niche via NF-κB/IL-1B signaling pathway. Am J Cancer Res [Internet]. e-Century Publishing Corporation; 2020;10:2464–79. Available from: https://pubmed.ncbi.nlm.nih.gov/32905535
Tulotta C, Lefley D V, Moore CK, Amariutei AE, Spicer-Hadlington AR, Quayle LA, et al. IL-1B drives opposing responses in primary tumours and bone metastases; harnessing combination therapies to improve outcome in breast cancer. NPJ breast cancer [Internet]. Nature Publishing Group UK; 2021;7:95. Available from: https://pubmed.ncbi.nlm.nih.gov/34290237
McGuire JJ, Frieling JS, Lo CH, Li T, Muhammad A, Lawrence HR, et al. Mesenchymal stem cell-derived interleukin-28 drives the selection of apoptosis resistant bone metastatic prostate cancer. Nat Commun [Internet]. Nature Publishing Group UK; 2021;12:723. Available from: https://pubmed.ncbi.nlm.nih.gov/33526787
Romero-Moreno R, Curtis KJ, Coughlin TR, Miranda-Vergara MC, Dutta S, Natarajan A, et al. The CXCL5/CXCR2 axis is sufficient to promote breast cancer colonization during bone metastasis. Nat Commun [Internet]. Nature Publishing Group UK; 2019;10:4404. Available from: https://pubmed.ncbi.nlm.nih.gov/31562303
Furesi G, Rauner M, Hofbauer LC. Emerging players in prostate cancer–Bone Niche Communication. Trends in Cancer [Internet]. Elsevier; 2021;7:112–21. Available from: https://doi.org/10.1016/j.trecan.2020.09.006
Probert C, Dottorini T, Speakman A, Hunt S, Nafee T, Fazeli A, et al. Communication of prostate cancer cells with bone cells via extracellular vesicle RNA; a potential mechanism of metastasis. Oncogene [internet]. 2018/10/23. Nature Publishing Group UK; 2019;38:1751–1763. Available from: https://pubmed.ncbi.nlm.nih.gov/30353168
Giannandrea D, Citro V, Lesma E, Bignotto M, Platonova N, Chiaramonte R. Restoring tissue homeostasis at metastatic sites: a focus on extracellular vesicles in bone metastasis. Front Oncol [Internet]. Frontiers Media S.A.; 2021;11:644109. Available from: https://pubmed.ncbi.nlm.nih.gov/33869035
Henrich SE, McMahon KM, Plebanek MP, Calvert AE, Feliciano TJ, Parrish S, et al. Prostate cancer extracellular vesicles mediate intercellular communication with bone marrow cells and promote metastasis in a cholesterol-dependent manner. J Extracell vesicles [Internet]. 2020/12/31. John Wiley and Sons Inc.; 2020;10:e12042–e12042. Available from: https://pubmed.ncbi.nlm.nih.gov/33408816
Hu C-Y, Chen J, Qin X-H, You P, Ma J, Zhang J, et al. Long non-coding RNA NORAD promotes the prostate cancer cell extracellular vesicle release via microRNA-541-3p-regulated PKM2 to induce bone metastasis of prostate cancer. J Exp Clin Cancer Res [Internet]. BioMed Central; 2021;40:98. Available from: https://pubmed.ncbi.nlm.nih.gov/33722248
Dai J, Escara-Wilke J, Keller JM, Jung Y, Taichman RS, Pienta KJ, et al. Primary prostate cancer educates bone stroma through exosomal pyruvate kinase M2 to promote bone metastasis. J Exp Med [Internet]. 2019;216:2883–99. Available from. https://doi.org/10.1084/jem.20190158.
Raimondi L, De Luca A, Fontana S, Amodio N, Costa V, Carina V, et al. Multiple myeloma-derived extracellular vesicles induce osteoclastogenesis through the activation of the XBP1/IRE1α Axis. Cancers (Basel) [Internet]. MDPI; 2020;12:2167. Available from: https://pubmed.ncbi.nlm.nih.gov/32759820
Raimondo S, Saieva L, Vicario E, Pucci M, Toscani D, Manno M, et al. Multiple myeloma-derived exosomes are enriched of amphiregulin (AREG) and activate the epidermal growth factor pathway in the bone microenvironment leading to osteoclastogenesis. J Hematol Oncol [Internet]. BioMed Central; 2019;12:2. Available from: https://pubmed.ncbi.nlm.nih.gov/30621731
Bravo Vázquez LA, Moreno Becerril MY, Mora Hernández EO, León Carmona GG de, Aguirre Padilla ME, Chakraborty S, et al. The emerging role of MicroRNAs in bone diseases and their therapeutic potential. Molecules [Internet]. MDPI; 2021;27:211. Available from: https://pubmed.ncbi.nlm.nih.gov/35011442
Puppo M, Taipaleenmäki H, Hesse E, Clézardin P. Non-coding RNAs in bone remodelling and bone metastasis: mechanisms of action and translational relevance. Br J Pharmacol [internet]. John Wiley & Sons, ltd; 2021;178:1936–54. Available from. https://doi.org/10.1111/bph.14836.
Guo L, Zhu Y, Li L, Zhou S, Yin G, Yu G, et al. Breast cancer cell-derived exosomal miR-20a-5p promotes the proliferation and differentiation of osteoclasts by targeting SRCIN1. Cancer Med [Internet]. 2019/08/06. John Wiley and Sons Inc.; 2019;8:5687–701. Available from: https://pubmed.ncbi.nlm.nih.gov/31385464
Wang S, Liu Z, Wang J, Ji X, Yao Z, Wang X. miR-21 promotes osteoclastogenesis through activation of PI3K/Akt signaling by targeting Pten in RAW264.7 cells. Mol Med Rep [Internet]. 2020/01/13. D.A. Spandidos; 2020;21:1125–32. Available from: https://pubmed.ncbi.nlm.nih.gov/32016444
Kitayama K, Kawamoto T, Kawakami Y, Hara H, Takemori T, Fujiwara S, et al. Regulatory roles of miRNAs 16, 133a, and 223 on osteoclastic bone destruction caused by breast cancer metastasis. Int J Oncol [internet]. Department of Orthopaedic Surgery, Kobe University graduate School of Medicine, Kobe, Hyogo 650-0017, Japan; 2021;59:97. Available from. https://doi.org/10.3892/ijo.2021.5277.
Furesi G, de Jesus Domingues AM, Alexopoulou D, Dahl A, Hackl M, Schmidt JR, et al. Exosomal miRNAs from prostate cancer impair osteoblast function in mice. Int J Mol Sci [Internet]. MDPI; 2022;23:1285. Available from: https://pubmed.ncbi.nlm.nih.gov/35163219
Sun Z, Hu J, Ren W, Fang Y, Hu K, Yu H, et al. LncRNA SNHG3 regulates the BMSC osteogenic differentiation in bone metastasis of breast cancer by modulating the miR-1273g-3p/BMP3 axis. Biochem Biophys Res Commun [Internet]. 2022;594:117–23 Available from: https://www.sciencedirect.com/science/article/pii/S0006291X21017162.
Chen L, Wang Y, Lu X, Zhang L, Wang Z. miRNA-7062-5p promoting bone resorption after bone metastasis of colorectal cancer through inhibiting GPR65 [Internet]. Front. Cell Dev. Biol. 2021. Available from: https://www.frontiersin.org/article/10.3389/fcell.2021.681968
Kim B, Jung S, Kim H, Kwon J-O, Song M-K, Kim MK, et al. The role of S100A4 for bone metastasis in prostate cancer cells. BMC Cancer [Internet]. BioMed Central; 2021;21:137. Available from: https://pubmed.ncbi.nlm.nih.gov/33549040
Kim H, Kim B, Il Kim S, Kim HJ, Ryu BY, Chung J, et al. S100A4 released from highly bone-metastatic breast cancer cells plays a critical role in osteolysis. Bone Res [Internet]. Nature Publishing Group UK; 2019;7:30. Available from: https://pubmed.ncbi.nlm.nih.gov/31667000
Chengling L, Yulin Z, Xiaoyu X, Xingchen L, Sen Z, Ziming W, et al. miR-325-3p, a novel regulator of osteoclastogenesis in osteolysis of colorectal cancer through targeting S100A4. Mol med [internet]. 2021;27:23. Available from. https://doi.org/10.1186/s10020-021-00282-7.
Li Y, Liang Y, Ma T, Yang Q. Identification of DGUOK-AS1 as a prognostic factor in breast cancer by bioinformatics analysis. Front Oncol [Internet]. Frontiers Media S.A.; 2020;10:1092. Available from: https://pubmed.ncbi.nlm.nih.gov/32766141
Zhang L, Niu H, Yang P, Ma J, Yuan B-Y, Zeng Z-C, et al. Serum lnc34a is a potential prediction biomarker for bone metastasis in hepatocellular carcinoma patients. BMC Cancer [Internet]. BioMed Central; 2021;21:161. Available from: https://pubmed.ncbi.nlm.nih.gov/33588789
Chen Y, Chen Z, Mo J, Pang M, Chen Z, Feng F, et al. Identification of HCG18 and MCM3AP-AS1 that associate with bone metastasis, poor prognosis and increased abundance of M2 macrophage infiltration in prostate cancer. Technol Cancer Res Treat [Internet]. SAGE Publications; 2021;20:1533033821990064–1533033821990064. Available from: https://pubmed.ncbi.nlm.nih.gov/33596783
Wang Y, Fang Y-X, Dong B, Du X, Wang J, Wang X, et al. Discovery of extracellular vesicles derived miR-181a-5p in patient’s serum as an indicator for bone-metastatic prostate cancer. Theranostics [internet]. Ivyspring International Publisher; 2021;11:878–892. Available from: https://pubmed.ncbi.nlm.nih.gov/33391510
Olivan M, Garcia M, Suárez L, Guiu M, Gros L, Méndez O, et al. Loss of microRNA-135b enhances bone metastasis in prostate cancer and predicts aggressiveness in human prostate samples. Cancers (Basel) [Internet]. MDPI; 2021;13:6202. Available from: https://pubmed.ncbi.nlm.nih.gov/34944822
Liang Y, Ye F, Wang Y, Li Y, Li Y, Song X, et al. DGUOK-AS1 acts as a tumor promoter through regulating miR-204-5p/IL-11 axis in breast cancer. Mol Ther - nucleic acids [internet]. Elsevier; 2021;26:1079–91. Available from. https://doi.org/10.1016/j.omtn.2021.10.018.
Wa Q, Zou C, Lin Z, Huang S, Peng X, Yang C, et al. Ectopic expression of miR-532-3p suppresses bone metastasis of prostate cancer cells via inactivating NF-κB signaling. Mol Ther oncolytics [Internet]. Am Soc Gene Cell Therapy; 2020;17:267–77. Available from: https://pubmed.ncbi.nlm.nih.gov/32368615
Wa Q, Huang S, Pan J, Tang Y, He S, Fu X, et al. miR-204-5p represses bone metastasis via inactivating NF-κB signaling in prostate cancer. Mol Ther Nucleic Acids [Internet]. 2019/09/18. Am Soc Gene Cell Ther; 2019;18:567–79. Available from: https://pubmed.ncbi.nlm.nih.gov/31678733
Huang S, Zou C, Tang Y, Wa Q, Peng X, Chen X, et al. miR-582-3p and miR-582-5p suppress prostate cancer metastasis to bone by repressing TGF-β signaling. Mol Ther Nucleic Acids [Internet]. 2019/01/15. Am Soc Gene Cell Ther; 2019;16:91–104. Available from: https://pubmed.ncbi.nlm.nih.gov/30852380
Dai Y, Wu Z, Lang C, Zhang X, He S, Yang Q, et al. Copy number gain of ZEB1 mediates a double-negative feedback loop with miR-33a-5p that regulates EMT and bone metastasis of prostate cancer dependent on TGF-β signaling. Theranostics [internet]. Ivyspring International Publisher. 2019;9:6063–79 Available from: https://pubmed.ncbi.nlm.nih.gov/31534537.
Wang J, Du X, Wang X, Xiao H, Jing N, Xue W, et al. Tumor-derived miR-378a-3p-containing extracellular vesicles promote osteolysis by activating the Dyrk1a/Nfatc1/Angptl2 axis for bone metastasis. Cancer Lett [Internet]. 2022;526:76–90. Available from: https://www.sciencedirect.com/science/article/pii/S030438352100584X
Ma Q, Liang M, Wu Y, Dou C, Xu J, Dong S, et al. Small extracellular vesicles deliver osteolytic effectors and mediate cancer-induced osteolysis in bone metastatic niche. J Extracell vesicles [internet]. John Wiley & Sons, Ltd; 2021;10:e12068. Available from. https://doi.org/10.1002/jev2.12068.
Zhang L, Niu H, Ma J, Yuan B-Y, Chen Y-H, Zhuang Y, et al. The molecular mechanism of LncRNA34a-mediated regulation of bone metastasis in hepatocellular carcinoma. Mol Cancer [Internet]. BioMed Central; 2019;18:120. Available from: https://pubmed.ncbi.nlm.nih.gov/31349837
Ni J, Zhang X, Li J, Zheng Z, Zhang J, Zhao W, et al. Tumour-derived exosomal lncRNA-SOX2OT promotes bone metastasis of non-small cell lung cancer by targeting the miRNA-194-5p/RAC1 signalling axis in osteoclasts. Cell Death Dis [Internet]. Nature Publishing Group UK; 2021;12:662. Available from: https://pubmed.ncbi.nlm.nih.gov/34215717
Safarulla S, Khillar PS, Kini S, Jaiswal AK. Tissue engineered scaffolds as 3D models for prostate cancer metastasis to bone. Mater Today Commun [Internet]. 2021;28:102641. Available from: https://www.sciencedirect.com/science/article/pii/S2352492821006334
Hughes AM, Kolb AD, Shupp AB, Shine KM, Bussard KM. Printing the pathway forward in bone metastatic cancer research: applications of 3D engineered models and bioprinted scaffolds to recapitulate the bone-tumor niche. Cancers (Basel) [Internet]. MDPI; 2021;13:507. Available from: https://pubmed.ncbi.nlm.nih.gov/33572757
Lee S, Mendoza TR, Burner DN, Muldong MT, Wu CCN, Arreola-Villanueva C, et al. Novel dormancy mechanism of castration resistance in bone metastatic prostate cancer organoids. Int J Mol Sci [Internet]. MDPI; 2022;23:3203. Available from: https://pubmed.ncbi.nlm.nih.gov/35328625
Laranga R, Duchi S, Ibrahim T, Guerrieri AN, Donati DM, Lucarelli E. Trends in bone metastasis modeling. Cancers (Basel) [Internet]. MDPI; 2020;12:2315. Available from: https://pubmed.ncbi.nlm.nih.gov/32824479
Brylka L, Jähn-Rickert K, Baranowsky A, Neven M, Horn M, Yorgan T, et al. Spine metastases in immunocompromised mice after intracardiac injection of MDA-MB-231-SCP2 breast cancer cells. Cancers (Basel) [Internet]. MDPI; 2022;14:556. Available from: https://pubmed.ncbi.nlm.nih.gov/35158823
Rimal R, Desai P, Marquez AB, Sieg K, Marquardt Y, Singh S. 3-D vascularized breast cancer model to study the role of osteoblast in formation of a pre-metastatic niche. Sci Rep [Internet]. Nature Publishing Group UK; 2021;11:21966. Available from: https://pubmed.ncbi.nlm.nih.gov/34754042
Lefley D, Howard F, Arshad F, Bradbury S, Brown H, Tulotta C, et al. Development of clinically relevant in vivo metastasis models using human bone discs and breast cancer patient-derived xenografts. Breast cancer res [internet]. 2019;21:130. Available from. https://doi.org/10.1186/s13058-019-1220-2.
Han Y, Nakayama J, Hayashi Y, Jeong S, Futakuchi M, Ito E, et al. Establishment and characterization of highly osteolytic luminal breast cancer cell lines by intracaudal arterial injection. Genes to cells [internet]. John Wiley & Sons, ltd; 2020;25:111–23. Available from. https://doi.org/10.1111/gtc.12743.
Bock N, Shokoohmand A, Kryza T, Röhl J, Meijer J, Tran PA, et al. Engineering osteoblastic metastases to delineate the adaptive response of androgen-deprived prostate cancer in the bone metastatic microenvironment. Bone res [internet]. 2019;7:13. Available from. https://doi.org/10.1038/s41413-019-0049-8.
Nathalie B, Thomas K, Ali S, Joan R, Akhilandeshwari R, Marie-Luise W, et al. In vitro engineering of a bone metastases model allows for study of the effects of antiandrogen therapies in advanced prostate cancer. Sci Adv [internet]. American Association for the Advancement of Science; 2022;7:eabg2564. Available from. https://doi.org/10.1126/sciadv.abg2564.
Jiang W, Rixiati Y, Huang H, Shi Y, Huang C, Jiao B. Asperolide A prevents bone metastatic breast cancer via the PI3K/AKT/mTOR/c-Fos/NFATc1 signaling pathway. Cancer Med [Internet]. 2020/09/25. John Wiley and Sons Inc.; 2020;9:8173–85. Available from: https://pubmed.ncbi.nlm.nih.gov/32976685
Li T, Jiang G, Hu X, Yang D, Tan T, Gao Z, et al. Punicalin attenuates breast cancer-associated osteolysis by inhibiting the NF-κB signaling pathway of osteoclasts [internet]. Front. Pharmacol. . 2021. Available from: https://www.frontiersin.org/article/10.3389/fphar.2021.789552
Nakai Y, Okamoto K, Terashima A, Ehata S, Nishida J, Imamura T, et al. Efficacy of an orally active small-molecule inhibitor of RANKL in bone metastasis. Bone res [internet]. 2019;7:1. Available from. https://doi.org/10.1038/s41413-018-0036-5.
Bishop RT, Marino S, Carrasco G, Li B, Allen RJ, Sparatore A, et al. Combined administration of a small-molecule inhibitor of TRAF6 and Docetaxel reduces breast cancer skeletal metastasis and osteolysis. Cancer Lett [Internet]. 2020;488:27–39. Available from: https://www.sciencedirect.com/science/article/pii/S0304383520302822
Pantano F, Croset M, Driouch K, Bednarz-Knoll N, Iuliani M, Ribelli G, et al. Integrin alpha5 in human breast cancer is a mediator of bone metastasis and a therapeutic target for the treatment of osteolytic lesions. Oncogene [internet]. 2021/01/08. Nature Publishing Group UK; 2021;40:1284–1299. Available from: https://pubmed.ncbi.nlm.nih.gov/33420367
Kunihiro AG, Brickey JA, Frye JB, Cheng JN, Luis PB, Schneider C, et al. Curcumin inhibition of TGFβ signaling in bone metastatic breast cancer cells and the possible role of oxidative metabolites. J Nutr Biochem [Internet]. 2022;99:108842. Available from: https://www.sciencedirect.com/science/article/pii/S095528632100262X
Kunihiro AG, Brickey JA, Frye JB, Luis PB, Schneider C, Funk JL. Curcumin, but not curcumin-glucuronide, inhibits Smad signaling in TGFβ-dependent bone metastatic breast cancer cells and is enriched in bone compared to other tissues. J Nutr Biochem [Internet]. 2018/10/11. 2019;63:150–6. Available from: https://pubmed.ncbi.nlm.nih.gov/30393127
Zhang N, Su P, Li X, Xi J, Li X, Xu L. Downregulated Krüppel-like factor 4 expression is associated with the aggressiveness of prostate cancer . Oncol Rep [Internet]. Department of Urology, The Affiliated Hospital of Zunyi Medical College, Zunyi, Guizhou 563003, P.R. China; 2019;41:1789–96. Available from: https://doi.org/10.3892/or.2019.6975
Liang H, Chen Q, Hu Z, Zhou L, Meng Q, Zhang T, et al. Siglec15 facilitates the progression of non-small cell lung cancer and is correlated with spinal metastasis. Ann Transl Med [Internet]. AME Publishing Company; 2022;10:281. Available from: https://pubmed.ncbi.nlm.nih.gov/35434017
Huang F, Cao Y, Wang C, Lan R, Wu B, Xie X, et al. PNMA5 Promotes Bone Metastasis of Non-small-Cell Lung Cancer as a Target of BMP2 Signaling. Front cell Dev Biol [Internet]. Frontiers Media S.A.; 2021;9:678931. Available from: https://pubmed.ncbi.nlm.nih.gov/34136487
Qian J, Gong Z-C, Zhang Y-N, Wu H-H, Zhao J, Wang L-T, et al. Lactic acid promotes metastatic niche formation in bone metastasis of colorectal cancer. Cell Commun Signal [Internet]. BioMed Central; 2021;19:9. Available from: https://pubmed.ncbi.nlm.nih.gov/33478523
Kovacheva M, Zepp M, Berger S, Berger MR. Conditional knockdown of integrin beta-3 reveals its involvement in osteolytic and soft tissue lesions of breast cancer skeletal metastasis. J cancer res Clin Oncol [internet]. 2020/10/20. Springer. Berlin Heidelberg. 2021;147:361–71 Available from: https://pubmed.ncbi.nlm.nih.gov/33083904.
Tiedemann K, Sadvakassova G, Mikolajewicz N, Juhas M, Sabirova Z, Tabariès S, et al. Exosomal release of L-plastin by breast cancer cells facilitates metastatic bone osteolysis. Transl Oncol [internet]. 2018/12/21. Neoplasia Press; 2019;12:462–474. Available from: https://pubmed.ncbi.nlm.nih.gov/30583289
Guerrieri AN, Montesi M, Sprio S, Laranga R, Mercatali L, Tampieri A, et al. Innovative options for bone metastasis treatment: an extensive analysis on biomaterials-based strategies for orthopedic surgeons [internet]. Front. Bioeng. Biotechnol. 2020. Available from: https://www.frontiersin.org/article/10.3389/fbioe.2020.589964
Liu S-Z, Sun X, Li K-X, Lin C-C, Na S. Li B-Y, et al. Onco: Tumor cell secretomes in response to anti- and pro-tumorigenic agents; 2021.
Liu S, Wu D, Sun X, Fan Y, Zha R, Jalali A, et al. Overexpression of Lrp5 enhanced the anti-breast cancer effects of osteocytes in bone. Bone Res [Internet]. Nature Publishing Group UK; 2021;9:32. Available from: https://pubmed.ncbi.nlm.nih.gov/34230453
Sano T, Sun X, Feng Y, Liu S, Hase M, Fan Y, et al. Inhibition of the growth of breast cancer-associated brain tumors by the osteocyte-derived conditioned medium. Cancers (Basel) [Internet]. MDPI; 2021;13:1061. Available from: https://pubmed.ncbi.nlm.nih.gov/33802279
Sun X, Li K, Zha R, Liu S, Fan Y, Wu D, et al. Preventing tumor progression to the bone by induced tumor-suppressing MSCs. Theranostics [Internet]. Ivyspring International Publisher. 2021;11:5143–59 Available from: https://pubmed.ncbi.nlm.nih.gov/33859739.
Li K-X, Sun X, Li B-Y, Yokota H. Conversion of osteoclasts into bone-protective, tumor-suppressing cells. Cancers (Basel) [Internet]. MDPI; 2021;13:5593. Available from: https://pubmed.ncbi.nlm.nih.gov/34830748
• Sun X, Li K, Hase M, Zha R, Feng Y, Li B-Y, et al. Suppression of breast cancer-associated bone loss with osteoblast proteomes via Hsp90ab1/moesin-mediated inhibition of TGFβ/FN1/CD44 signaling. Theranostics [Internet]. Ivyspring International Publisher; 2022;12:929–43. Available from: https://pubmed.ncbi.nlm.nih.gov/34976221. In most recent study, authors suggest that activation of WNT signaling generates tumor-suppressive and bone-protective secretomes derived by osteoblasts. This has a potential for developing secretome-based therapy for breast cancer-associated bone metastasis.
Liu S, Sun X, Li K, Zha R, Feng Y, Sano T, et al. Generation of the tumor-suppressive secretome from tumor cells. Theranostics [internet]. Ivyspring Int Publisher. 2021;11:8517–34 Available from: https://pubmed.ncbi.nlm.nih.gov/34373756.
Su Y, Huang H, Luo T, Zheng Y, Fan J, Ren H, et al. Cell-in-cell structure mediates in-cell killing suppressed by CD44. Cell Discov [internet]. 2022;8:35. Available from. https://doi.org/10.1038/s41421-022-00387-1.
Zhang X, Liu Q, Zhang T, Gao P, Wang H, Yao L, et al. Bone-targeted nanoplatform enables efficient modulation of bone tumor microenvironment for prostate cancer bone metastasis treatment. Drug Deliv [Internet]. Taylor Francis; 2022;29:889–905. Available from: https://pubmed.ncbi.nlm.nih.gov/35285760
Bai S, Liu D, Cheng Y, Cui H, Liu M, Cui M, et al. Osteoclasts and tumor cells dual targeting nanoparticle to treat bone metastases of lung cancer. Nanomedicine Nanotechnology, Biol Med [Internet]. 2019;21:102054. Available from: https://www.sciencedirect.com/science/article/pii/S1549963419301388
Huang Y, Xiao Z, Guan Z, Zeng Z, Shen Y, Xu X, et al. Bone-seeking nanoplatform co-delivering cisplatin and zoledronate for synergistic therapy of breast cancer bone metastasis and bone resorption. Acta pharm Sin B [internet]. 2020/06/19. Elsevier; 2020;10:2384–2403. Available from: https://pubmed.ncbi.nlm.nih.gov/33354509
Zhang Z, Yao Y, Yuan Q, Lu C, Zhang X, Yuan J, et al. Gold clusters prevent breast cancer bone metastasis by suppressing tumor-induced osteoclastogenesis. Theranostics [internet]. Ivyspring International Publisher; 2020;10:4042–55. Available from: https://pubmed.ncbi.nlm.nih.gov/32226538
•• Pin F, Jones AJ, Huot JR, Narasimhan A, Zimmers TA, Bonewald LF, et al. RANKL blockade reduces cachexia and bone loss induced by non-metastatic ovarian cancer in mice. J Bone Miner Res [Internet]. John Wiley & Sons, Ltd; 2022;37:381–96. Available from: 10.1002/jbmr.4480. This study identifies tumor-derived circulating RANKL as an important factor contributing to bone loss and skeletal muscle atrophy in cachetic patients and ovarian cancer-bearing mice. Furthermore, anti-RANKL therapy was shown to be effective in improving both muscle and bone deficits.
Essex AL, Pin F, Huot JR, Bonewald LF, Plotkin LI, Bonetto A. Bisphosphonate treatment ameliorates chemotherapy-induced bone and muscle abnormalities in young mice. Front Endocrinol (Lausanne) [Internet]. Frontiers Media S.A.; 2019;10:809. Available from: https://pubmed.ncbi.nlm.nih.gov/31803146
Funding
Open Access funding enabled and organized by Projekt DEAL. This work has been funded by research grants from the German Research Foundation (Deutsche Forschungsgemeinchaft; DFG) SA 4275/1-1 to H.S. HE 5208/5-1 to E.H. and TA 1154/1-2 and TA 1152/2-2 to H.T.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Muscle and Bone
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pauk, M., Saito, H., Hesse, E. et al. Muscle and Bone Defects in Metastatic Disease. Curr Osteoporos Rep 20, 273–289 (2022). https://doi.org/10.1007/s11914-022-00741-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-022-00741-y