
Abstract
Purpose of Review
The evolving information of the initiation, tumor cell heterogeneity, and plasticity of childhood neuroblastoma has opened up new perspectives for developing therapies based on detailed knowledge of the disease.
Recent Findings
The cellular origin of neuroblastoma has begun to unravel and there have been several reports on tumor cell heterogeneity based on transcriptional core regulatory circuitries that have given us important information on the biology of neuroblastoma as a developmental disease. This together with new insight of the tumor microenvironment which acts as a support for neuroblastoma growth has given us the prospect for designing better treatment approaches for patients with high-risk neuroblastoma. Here, we discuss these new discoveries and highlight some emerging therapeutic options.
Summary
Neuroblastoma is a disease with multiple facets. Detailed biological and molecular knowledge on neuroblastoma initiation, heterogeneity, and the communications between cells in the tumor microenvironment holds promise for better therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuroblastoma is the most common and deadly tumor of infancy accounting for 15% of all childhood cancer-related deaths [1]. Neuroblastomas almost exclusively occur in young children and the median age for diagnosis is 18 months. Approximately 40% of the patients are younger than 1 year at diagnosis whereas less than 5% are older than 10 years [2, 3]. Neuroblastoma is characterized histopathologically as a “small round blue cell” tumor, an entity of neoplasms consisting mainly of pediatric solid cancers. Clinically, neuroblastoma manifests as a primary tumor anywhere along with the sympathetic nervous system, with > 50% occurring in the adrenal medulla [1]. Neuroblastomas are heterogeneous diseases, which also is reflected in patient survival. Low-intermediate risk patients have an overall survival rate of > 95%, whereas high-risk patients have < 50% long-term survival [4].
The clinical heterogeneity observed in neuroblastoma can be reflected through chromosomal aberrations. Low-risk patients commonly present with whole chromosomal gains and the tumor cells are frequently hyperdiploid, whereas high-risk patients have a chromosomal makeup consisting of segmental chromosomal gains or losses [5,6,7,8,9]. The most frequent chromosomal aberrations associated with high-risk patients and poor prognosis in neuroblastoma are segmental gain of chromosome 17q, hemizygous loss of chromosome 1p and 11q, and somatically acquired amplification of the oncogene MYCN [3]. Additionally, high-risk neuroblastoma can also present with rearrangements at chromosomal region 5p15.33 that is located close to the telomerase reverse transcriptase gene (TERT) [10, 11]. Similar to the majority of childhood cancers, neuroblastomas show low somatic mutation counts (12–18, median 15) and there is no single mutation that acts as a driver for the development of all neuroblastomas [12,13,14]. The ALK (anaplastic lymphoma kinase) gene harbors the most frequently detected somatic mutations in neuroblastoma, found in 8–10% of neuroblastoma cases. Mutations of ALK are also present in familial neuroblastomas which encompass 1–2% of the neuroblastoma patients [15]. Additionally, germline loss-of-function mutations of the paired-like homeobox 2B (PHOX2B) gene have been found in familial neuroblastoma as well as in approximately 4% of spontaneous neuroblastomas [12, 14, 15]. Recurrent genetic alterations have also been observed for LIN28B, ATRX, ARID1A/1B, BARD1, LMO1, and TP53 [12,13,14, 16,17,18] (Table 1).
Further adding to the complexity of the disease is the presence of frequent inter- and intra-tumorigenic heterogeneity in individual patients and the accumulation of gene mutations observed in recurrent and relapsed tumor tissues [17,18,19,20]. The heterogeneity observed in neuroblastoma is a clinical challenge since tumors that are phenotypically and morphologically alike may respond fundamentally differently to therapy, depending on their molecular makeup. This proposes that neuroblastoma patients in general and specifically those diagnosed with high-risk disease should be carefully examined with regard to their molecular landscape in order to design individual therapies targeting the observed molecular aberrations in addition to current conventional therapies [2]. Here we discuss some recent developments in the understanding of neuroblastoma initiation, heterogeneity, plasticity, and communication between cells in the tumor supporting microenvironment that will have an impact when designing new therapies.
The Neural Crest and Neuroblastoma Cell of Origin
Neuroblastoma is an embryonic cancer originating from cells in the neural crest, a transient structure that appears at the margins of the closing neural tube, consisting of multipotent stem cells active during early embryonic development. Neural crest cells give rise to a number of different cell types including neurons and glial cells within the peripheral nervous system, mesenchymal, pigment and secretory cells, and bone and cartilage cells of the face [21]. During embryonal development, neural crest cells need to undergo epithelial-to-mesenchymal transition and migrate extensively from the neuroepithelium to more distant locations in the embryo where the cells finally maturate and differentiate into established structural and functional networks [22]. The fact that the neural crest is a transient structure has complicated the search for the neuroblastoma cell of origin.
The combination of timing of the onset of disease and its clinical presentation brought about the consensus that neuroblastoma most likely originates from symphatoadrenal progenitor cells within the neural crest that normally differentiate to sympathetic ganglion cells and adrenal catecholamine-secreting cells [23]. The adrenal gland medulla contains secretory cells named chromaffin cells that synthesize and store hormones, including catecholamines [24]. Morphologically, these cells lack neurites and resemble endocrine cells. Earlier studies established the consensus that both adrenal chromaffin cells and sympathetic neurons originate from sympathoadrenal progenitor cells [25].
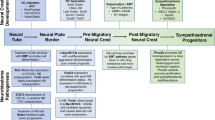
However, this was recently challenged by data demonstrating that Schwann cell precursors deriving from migrating neural crest cells are the ancestors of adrenal medullar chromaffin cells and fetal adrenal neuroblasts (Fig. 1) [26, 27•]. Recent data derived from single-cell RNA sequencing analyses comparing malignant neuroblastoma with neural crest cell signatures resulted in the identification of two different immature cell populations within the neural crest as potential cells of origin for neuroblastoma [28•, 29•, 30•]. One study indicated that neuroblastoma has a predominant chromaffin-cell-like phenotype and that these cells are the cell of origin [28•], whereas two other studies suggested that neuroblastomas transcriptionally resemble normal fetal adrenal neuroblasts (also named sympathoblasts) [29•, 30•].

Normal development of the sympathoadrenal lineage in humans. During neural crest development, Schwann cell precursor (SCPs) differentiate into both chromaffin cells and sympathoblasts [27•, 29•]. The cell annotations in capital letters are genes that were expressed by the corresponding cells. SCPs, sympathoblasts, and chromaffin cells expressed these genes in both studies
Historically, Schwann cell precursors were considered to be primed toward Schwann cell differentiation. However, recent studies suggest that these precursors may give rise to several different cell types including melanocytes, odontoblasts, parasympathetic and enteric neurons, and endoneural fibroblasts in addition to chromaffin cells and neuroblasts (sympathoblasts) [27•, 31,32,33,34,35]. These studies suggest that the plasticity of Schwann cell precursors is more extensively involved in tissue establishment and regeneration than previously assumed [25]. Although these findings have increased our understanding of human sympathoadrenal development and given further insight into the heterogeneity of neuroblastoma, the trajectory from developing normal neural crest cells to malignant neuroblastoma is still incomplete [36].
Neuroblastoma Heterogeneity and Plasticity
Neuroblastomas are characterized by a high degree of heterogeneity which is reflected in clinical presentations spanning from spontaneous regression or differentiation to treatment-refractory progression regardless of intensive multimodal therapies. The spontaneous regression of tumor cells observed in a subgroup, named 4S, of neuroblastoma patients is exceptional among human cancers. Stage 4S neuroblastomas are found in young children up to 18 months of age and usually present with small abdominal tumors with several metastases to the liver, skin, and bone marrow [37]. The majority of stage 4S patients undergo spontaneous regression of their tumor cells with limited or no treatment. The mechanisms for this spontaneous regression are not fully known but epigenetic regulation, neurotrophin deprivation, loss of telomerase activity, or immune responses have been proposed factors for induction of regression [38]. Of note, the spontaneous regression seen in stage 4S neuroblastomas resembles events observed within the neural crest where excess precursor cells undergo apoptosis during the final stages of maturation [39].
Although spontaneous regression occurs in some patients, the majority of neuroblastomas are diagnosed as high risk and most of these patients have metastatic disease already at diagnosis suggesting that induction of metastasis is an early event [3].
Although neuroblastomas have a low mutational burden compared to most adult cancers, recent data have given evidence for the presence of intra-tumor heterogeneity and different evolutionary trajectories with discrete genotypes in different locations of the same tumor [40•]. Also, a comparison of treatment-naïve primary neuroblastomas with matched relapsed tumors demonstrated increased mutational burden at relapse including genes within the RAS-MAPK signaling pathway, Hippo-Yap pathway, and epithelial-mesenchymal transition processes that were not present at initial diagnosis [17, 18]. Adding to this complexity is the recent demonstration of at least two distinct tumor cell types deriving from different cell lineages, adrenergic (ADRN) and mesenchymal (MES) which differ in transcriptomic, phenotypic, and super-enhancer expression [41, 42]. Similarly, a more recent study detected the same cell types in primary neuroblastomas but also discerned additional subtypes in the ADRN group [43•]. The ADRN tumor cells could be further divided into MYCN-amplified, MYCN non-amplified high-risk, and MYCN non-amplified low-risk. These subtypes correlated to different clinical outcomes, whereas the MYCN-amplified subtype correlated with the worst outcome.
MES cells have been shown to resemble neural crest cell precursors, have an active NOTCH signaling pathway, and seem to be more resistant to chemotherapy [41]. They also occur more frequently in relapsed tumors and can be induced in vitro by activation of RAS [43•]. In MYCN-amplified cell lines, six members of the transcriptional core regulatory circuitry that are controlled by super-enhancers have been identified: HAND2, ISL1, PHOX2B, GATA3, ASCL1, and TBX2 [41, 42, 44,45,46]. MYCN regulates the expression of all these genes and acts as a general amplifier of the transcriptional circuitry. These six-core regulatory circuitry transcription factors are expressed in both ADRN and MES cells, although somewhat less in the MES subtype [44]. ADRN and MES cells have been shown to bi-directionally interconvert between the different cell states in culture, suggesting plasticity of neuroblastoma cells. This process, reminiscent of epithelial-to-mesenchymal transition, has been named noradrenergic-to-mesenchymal transition and probably relies on epigenetic reprogramming [47].
All these studies on intra-tumor heterogeneity and different neuroblastoma cell states have focused on the neoplastic cells, whereas the biological and molecular landscape of the non-tumorigenic cells residing in the tumor microenvironment and their molecular communication with the tumor cells has not been investigated until recently. The importance of the non-tumorigenic cellular niche can be illustrated by the demonstration that neuroblastoma cells from the same batch exhibit highly dissimilar growth depending on where they grow. Subcutaneous injection of neuroblastoma cells gives rise to non-invasive cell expansions, whereas identical cells injected orthotopically result in tumor cell expansion with metastatic spread [48]. Similarly, implanting of tumor cells deriving from one single neuroblastoma sample as patient-derived orthotopic xenografts resulted in differences in transcriptional profiles and divergent tumor growth despite the fact that the injected cells should be genetically identical and contain a similar population of ADRN and MES cells [19]. These data point to the assumption that the composition of stromal and immune cells present in the tumor microenvironment has important functions in regulating the tumor cell behavior, cell-state composition, and possibly intra-tumor heterogeneity.
The Tumor Microenvironment in Neuroblastoma
The progress in designing more precise targeted therapies including the striking developments in immunotherapies for certain cancers has spawned the focus on the nature and function of the tumor microenvironment (TME). The paucity of recurrent mutations and oncogene activation in pediatric cancers, including neuroblastoma, poses a major challenge for developing personalized medicine and also limits the effects of checkpoint inhibitors as a treatment option for these cancers. Moreover, since neuroblastoma is an embryonic tumor, an immature or impaired immune system can further complicate the design of successful immunotherapies [49•]. Given the enhanced recent focus on immunotherapy, the innate and adaptive immune cells including T and B lymphocytes, tumor-associated macrophages, dendritic cells, natural killer cells, and natural killer T cells have been given much attention when studying cells within the microenvironment of neuroblastoma. However, non-immune cells like endothelial cells, cancer-associated fibroblasts, pericytes, and mesenchymal stem cells have also recently been shown to have important functions in tumor progression and induction of therapy resistance [50]. Moreover, alterations of the extracellular matrix (ECM) have been reported to be mediators of tumor progression in neuroblastoma by increased collagen cross-linking influencing morphological changes [51]. Also, recent data suggest that extracellular vesicles released by cells in the tumor microenvironment carry oncogenic microRNAs as well as proteins involved in tumor progression such as CD147, a transmembrane protein involved in metastasis, and CD276/B7-H3, an immune checkpoint protein enabling neuroblastoma cells to evade NK cells [50, 52, 53].
The different technological approaches used to study the immune cell landscape in neuroblastoma have sometimes generated conflicting data, and therefore, the pros and cons of the different populations of immune cells detected in neuroblastoma tissues have been difficult to interpret. Nevertheless, a number of studies in neuroblastoma have shown that the immune system plays a critical role in both prognosis and response to treatment.
MYCN Influences the Immune Microenvironment in Neuroblastoma
A general picture of the tumor microenvironment in neuroblastoma is the differences in the cellular landscape of MYCN-amplified vs MYCN non-amplified neuroblastomas [50]. MYCN has been shown to inhibit the expression of MHC class I antigens that contribute to tumor antigen presentation necessary for immune cells to recognize and attack tumor cells [54]. This lack of antigen presentation in MYCN-amplified neuroblastomas is responsible for the escape from cytotoxic T cell and interferon-mediated immune responses [55]. MYCN-amplified neuroblastomas have also been reported to contain less CD4 + and CD8 + T cells, dendritic cells, NK cells, and macrophages compared to non-MYCN-amplified neuroblastomas [56, 57]. Also, T cell receptor analysis showed that MYCN-amplified neuroblastomas had decreased levels of infiltrating lymphocyte signatures compared to non-MYCN-amplified tumors [58]. Furthermore, in metastatic non-MYCN-amplified tumors, a high level of M2 macrophages was present suggesting an increase towards M2 polarization in absence of MYCN expression [59]. Hence, MYCN-amplified tumors are poorer in immune cell infiltration compared to non-MYCN-amplified tumors which contain more infiltrating adaptive and innate immune cells partly through the higher expression of chemokines which are downregulated by MYCN [50].
Targeting the Neuroblastoma Tumor Microenvironment
Cancer-associated fibroblasts (CAFs) are characterized by expression of fibroblast activating protein (FAP), fibroblast-specific protein (FSP-1), platelet-derived growth factor receptor α and β (PDGFR α and PDGFR β), α-smooth-muscle actin (αSMA), and vimentin [60]. However, the heterogeneity in expression of these markers indicates that CAFs are polarized into distinct subpopulations depending on their origin and the local tumor environment [61, 62]. In neuroblastoma, high numbers of CAFs presented within the tumor tissue have been shown to correlate with more aggressive Schwannian stroma-poor tumors [63], and CAFs have been detected in close proximity to tumor-associated macrophages (TAMs) [63, 64]. Secretion of IL-6 from TAMs enhances the activation of STAT3 in CAFs leading to increased proliferation [64, 65]. Our laboratory has shown that prostaglandin E2 secreted by CAFs enhances the M2 polarization of TAMs and increases the proliferation of neuroblastoma cells [66,67,68]. Targeting microsomal prostaglandin E synthase-1 (mPGES-1), one of the important enzymes in the synthesis of prostaglandin E2, using a small molecule inhibitor, blocked the production of prostaglandin E2 from CAFs. This resulted in reduced tumor growth, inhibition of CAF migration and infiltration, and a favorable shift in the macrophage M1/M2 ratio [67]. CAFs located in the tumor microenvironment in neuroblastoma have also been reported to share functional and phenotypical characteristics with bone marrow-derived mesenchymal stem cells. These cells called CAFs-MSCs secrete several inflammatory cytokines and chemokines such as IL-6, IL-8, CCL-2, CxCL-12, and VEGF-A that promote neuroblastoma growth and enhance resistance toward chemotherapeutic drugs [64, 69].
Taken together, neuroblastomas encompass an immunosuppressive microenvironment that contains abundant CD163+ macrophages and C11b+ myeloid-derived cells [70, 71]. High-risk neuroblastoma also presents with a tumor-associated inflammatory signature that facilitates tumor growth, drug resistance, and immune evasion [59, 66, 72]. Hence, combining new modes of immunotherapies with anti-inflammatory drugs may be beneficial for this group of patients.
Except for anti-GD2 (dinutuximab) that has been included in the treatment of high-risk neuroblastoma patients for the last 20 years, no other immune-therapy approaches have proven successful so far. One important reason for this is the lack of neo-antigens present on the surface of neuroblastoma cells. An intriguing recent study may offer a solution to this. By studying the immunopeptidome in neuroblastoma, an enrichment of peptides from proteins essential for tumorigenesis was detected. This included a peptide, QYNPIRTTF, deriving from the master transcription regulator PHOX2B. Development of peptide-centric chimeric antigen receptors targeting this peptide resulted in selective killing of neuroblastoma cells and complete regression of neuroblastoma PDX tumors positive for PHOX2B [73••]. This promising data suggest that a peptide-centric CAR therapy can be applied on tumors with low expression of surface neo-antigens and expand the options of immunotherapeutic targets [73••].
Differentiation of Neuroblastoma as a Treatment Option
Neuroblastoma is an embryonal tumor and can for this reason be characterized as a developmental disease where lack of differentiation or apoptosis during the development and maturation of the peripheral nervous system result in neoplastic transformation and malignant tumor growth. In fact, 13-cis retinoic acid (isotretinoin), which induces differentiation in neuroblastoma, is currently used as maintenance therapy to avoid residual disease in patients with high-risk disease. However, the effects of this treatment are limited in patients compared to the effects seen on neuroblastoma cells growing in culture. Therefore, clinical trials investigating the effects of retinoic acid in combination with other drugs or immunotherapies as well as studies to improve the retinoic acid drug delivery have been initiated [74]. The induction of tumor cell differentiation as a treatment strategy was highlighted by the recent identification of MYCN and TFAP (transcription factor AP-2) as important players in differentiation control [29•].
In MYCN-amplified neuroblastomas, the ADRN core regulatory circuitry of transcription factors is controlled by MYCN. This favors an immature neuroblast cell state and suppresses signals that normally induce differentiation [75]. Recent data suggest that retinoid treatment can reprogram the enhancer landscape, establishing a new retino-sympathetic core regulatory circuitry consisting of RARA, HAND2, TBX2, SOX4, MEIS1, and ISL1, causing downregulation of MYCN expression, proliferative arrest, and sympathetic differentiation [76]. These data provide mechanisms for the beneficial effects of retinoid treatment in neuroblastoma and explain the observed massive downregulation of MYCN expression seen in some neuroblastoma cells when subjected to retinoids.
During embryonal development, neural crest cells must acquire motility through the epithelial-to-mesenchymal transition. Fundamental for the motility is the Wnt-planar cell polarity (PCP) signaling cascade which guides contact inhibition of locomotion inducing polarity of cells within the neural crest. PCP proteins regulate the activity of Rho GTPases locally by stimulating or suppressing the activity of RhoA and Rac1, resulting in cells migrating away from each other upon collision [77]. Activation of Rho signaling by PCP activates rho-associated coiled-coil-containing protein kinases, ROCK1 and ROCK2 [78]. ROCK1 and ROCK2 phosphorylate downstream substrates such as myosin light chain (MLC) and LIMK1/2, which control a number of cellular functions through rearrangement of the actin cytoskeleton [79, 80]. We and others have shown that approximately 30% of neuroblastomas contain mutations in genes involved in Rho/Rac1 signaling and that high expression of ROCK2 corresponds to poor prognosis [13, 81]. Inhibition of ROCK activity induced neuroblastoma cell differentiation and suppressed cell migration, invasion, and xenograft tumor growth in mice [81]. Several ROCK inhibitors are currently undergoing clinical trials or are in clinical use for non-cancerous indications. Hence, further investigations of ROCK inhibitors in combination with other drugs should be performed. Since neuroblastomas can be regarded as a disease caused by obstructed cell differentiation during embryonal development, agents that release this differentiation block are attractive as potential new therapies for high-risk patients.
Targeted Therapy for Neuroblastoma—ALK, a Success Story
ALK, the second most common driver in neuroblastoma after MYCN, is a receptor tyrosine kinase which in the neuroblastoma context first attracted attention in conjunction with the rare hereditary neuroblastomas [15]. The oncogenic mechanism in neuroblastoma operates by activation of RAS-MAPK-ERK and other downstream pathways, and can be initiated by activating mutations of ALK, ALK amplifications, or, possibly, changes to the ALK ligands ALKAL1 and ALKAL2 [82,83,84]. The ALK gene is situated in proximity to MYCN and ALKAL2 on chromosome 2p and it has been argued that co-amplification of these three explains the inferior outcome in neuroblastoma patients with 2p gain [85].
Among targeted therapies proposed for neuroblastoma treatment, ALK inhibitors have come furthest in clinical application. The first clinical trial of the lead substance crizotinib (NCT00939770) started shortly after ALK was discovered as an oncogenic driver in neuroblastoma [86]. While there were some responses in a subset of neuroblastomas, most patients did not respond. Fueled by advances in the field of ALK-positive non-small cell lung cancer (NSCLC), where ALK fusions constitute the predominant driving oncogenic event and where the initial response is frequently followed by the development of acquired resistance, second- and third-generation ALK inhibitors soon became available, including ceritinib, brigatinib, entrectinib, and lorlatinib. Some of these agents also inhibit other tyrosine kinases that may be of importance in neuroblastoma, e.g., ROS1 or TRK. Eventually, it became evident that different ALK hotspot mutations in neuroblastoma confer differential sensitivity toward different ALK inhibitors [87,88,89]. In selected cases, individualized preclinical evaluation of a newly described ALK or ALK ligand mutation has proven beneficial [84, 90]. Secondary resistance toward ALK inhibitors is a frequent observation in NSCLC patients and is also seen in neuroblastoma patients, and sometimes the switch to an alternative ALK inhibitor becomes necessary [91]. In general, ALK inhibitors cause low toxicity, allowing their use even under circumstances when chemotherapy cannot be tolerated [90]. A number of responders have remained on ALK inhibitor treatment well beyond 1 year [84, 86, 88, 90, 92].
Clinical experience with ALK inhibition in neuroblastoma has hitherto mainly been collected in patients with refractory or relapsed tumors. For neuroblastoma with ALK mutations, the TITAN study (transatlantic integration targeting ALK in neuroblastoma) will investigate the frontline addition of lorlatinib, an ALK inhibitor with activity against a wider range of ALK mutations and penetrance to CNS [93,94,95]. In this context, liquid biopsies for ALK mutations might prove useful to monitor lorlatinib treatment response [96].
Conclusions
The rapid advances in Omics techniques and analysis of “big data” have greatly increased our knowledge of the molecular landscape of neuroblastoma. National and international molecular profiling initiatives have provided bioinformatic pipelines to guide treatment options of individual patients with the potential to be implemented as personalized medicines [97].
The new insights concerning the molecular heterogeneity, plasticity, and the involvement of non-tumorigenic cells within the neuroblastoma tumor microenvironment have advanced our understanding of the initiation, progression, and metastatic spread of neuroblastoma. However, these new discoveries also call for new models that more accurately mimic the disease in order to design accurate functional studies and to monitor the effects of novel drug regimens. Patient-derived xenograft models and the establishment of organoids that more accurately resemble the patient tumor may better predict the effects of specific drugs which could then possibly be directly transferred to clinical use if the toxicity profile is acceptable. The advancement in the molecular biology and pathogenesis of neuroblastoma has opened up the possibility to apply more precise treatment to children with this disease. Hopefully, this could lead to the cure of patients whom we currently are unable to cure.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Hematol Oncol Clin North Am. 2010;24(1):65–86.
Johnsen JI, Dyberg C, Fransson S, Wickström M. Molecular mechanisms and therapeutic targets in neuroblastoma. Pharmacol Res. 2018;1(131):164–76.
Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Primers. 2016;10:2.
PDQ Pediatric Treatment Editorial Board. Neuroblastoma Treatment (PDQ®): Health Professional Version. 2022 Feb 17. In: PDQ Cancer Information Summaries [Internet]. Bethesda (MD): National Cancer Institute (US); 2002-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK65747/
Carén H, Kryh H, Nethander M, Sjöberg RM, Träger C, Nilsson S, et al. High-risk neuroblastoma tumors with 11q-deletion display a poor prognostic, chromosome instability phenotype with later onset. Proc Natl Acad Sci U S A. 2010;107(9):4323–8.
Janoueix-Lerosey I, Schleiermacher G, Michels E, Mosseri V, Ribeiro A, Lequin D, et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol. 2009;27(7):1026–33.
Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362(23):2202–11.
Morgenstern DA, London WB, Stephens D, Volchenboum SL, Hero B, di Cataldo A, et al. Metastatic neuroblastoma confined to distant lymph nodes (stage 4N) predicts outcome in patients with stage 4 disease: a study from the International Neuroblastoma Risk Group Database. J Clin Oncol. 2014;32(12):1228–35.
Irwin MS, Park JR. Neuroblastoma. Pediatr Clin North Am. 2015;62(1):225–56.
Valentijn LJ, Koster J, Zwijnenburg DA, Hasselt NE, van Sluis P, Volckmann R, et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet. 2015;47(12):1411.
Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature. 2015;526(7575):700–4.
Cheung NKV, Zhang J, Lu C, Parker M, Bahrami A, Tickoo SK, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA. 2012;307(10):1062.
Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483(7391):589–93.
Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45(3):279–84.
Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455(7215):930–5.
Sweeney SM, Cerami E, Baras A, Pugh TJ, Schultz N, Stricker T, et al. AACR project genie: powering precision medicine through an international consortium. Cancer Discov. 2017;7(8):818–31.
Schramm A, Köster J, Assenov Y, Althoff K, Peifer M, Mahlow E, et al. Mutational dynamics between primary and relapse neuroblastomas. Nat Genet. 2015;47(8):872–7.
Eleveld TF, Oldridge DA, Bernard V, Koster J, Daage LC, Diskin SJ, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet. 2015;47(8):864–71.
Braekeveldt N, von Stedingk K, Fransson S, Martinez-Monleon A, Lindgren D, Axelson H, et al. Patient-derived xenograft models reveal intratumor heterogeneity and temporal stability in neuroblastoma. Cancer Res. 2018;78(20):5958–5969.
Schleiermacher G, Javanmardi N, Bernard V, Leroy Q, Cappo J, Rio Frio T, et al. Emergence of New ALK Mutations at Relapse of Neuroblastoma. J Clin Oncol. 2014;32(25):2727–34.
Groves AK, LaBonne C. Setting appropriate boundaries: fate, patterning and competence at the neural plate border. Dev Biol. 2014;389(1):2–12.
Coelho-Aguiar JM, Le Douarin NM, Dupin E. Environmental factors unveil dormant developmental capacities in multipotent progenitors of the trunk neural crest. Dev Biol. 2013;384(1):13–25. https://doi.org/10.1016/j.ydbio.2013.09.030.
Tomolonis JA, Agarwal S, Shohet JM. Neuroblastoma pathogenesis: deregulation of embryonic neural crest development. Cell Tissue Res. 2018;372(2):245–62. https://doi.org/10.1007/s00441-017-2747-0.
Bornstein SR, Ehrhart-Bornstein M, Androutsellis-Theotokis A, Eisenhofer G, Vukicevic V, Licinio J, et al. Chromaffin cells: the peripheral brain. Mol Psychiatry. 2012;17(4):354–8. https://doi.org/10.1038/mp.2011.176.
Lousado L, Prazeres PHDM, Andreotti JP, Paiva AE, Azevedo PO, Santos GSP, et al. Schwann cell precursors as a source for adrenal gland chromaffin cells. Cell Death Dis. 2017;8(10):e3072.
Furlan A, Dyachuk V, Kastriti ME, Calvo-Enrique L, Abdo H, Hadjab S, et al. Multipotent peripheral glial cells generate neuroendocrine cells of the adrenal medulla. Science. 2017;357(6346):eaal3753. https://doi.org/10.1126/science.aal3753.
•Kameneva P, Artemov AV, Kastriti ME, Faure L, Olsen TK, Otte J, et al. Single-cell transcriptomics of human embryos identifies multiple sympathoblast lineages with potential implications for neuroblastoma origin. Nat Genet. 2021;53(5):694–706. This study describes sympathoblast lineages in human neural crest which may explain neuroblastoma origin.
•Dong R, Yang R, Zhan Y, Lai HD, Ye CJ, Yao XY, et al. Single-cell characterization of malignant phenotypes and developmental trajectories of adrenal neuroblastoma. Cancer Cell. 2020;38(5):716-733.e6. The first description of the cellular landscape of neuroblastoma. Indicating that the cell of origin is chromaffin cells.
•Jansky S, Sharma AK, Körber V, Quintero A, Toprak UH, Wecht EM, et al. Single-cell transcriptomic analyses provide insights into the developmental origins of neuroblastoma. Nat Genet. 2021;53(5):683–93. Describe developmental origin of neuroblastoma. Indicating that the cell of origin is neuroblasts.
•Kildisiute G, Kholosy WM, Young MD, Roberts K, Elmentaite R, van Hooff SR, et al. Tumor to normal single-cell mRNA comparisons reveal a pan-neuroblastoma cancer cell. Sci Adv. 2021;7(6):eabd3311. https://doi.org/10.1126/sciadv.abd3311. A comprehensive analysis of the neuroblastoma cellular landscape indicating that neuroblastoma originates from sympathoblasts.
Adameyko I, Lallemend F, Aquino JB, Pereira JA, Topilko P, Müller T, et al. Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell. 2009;139(2):366–79. https://doi.org/10.1016/j.cell.2009.07.049.
Kaukua N, Shahidi MK, Konstantinidou C, Dyachuk V, Kaucka M, Furlan A, et al. Glial origin of mesenchymal stem cells in a tooth model system. Nature. 2014;513(7519):551–4. https://doi.org/10.1038/nature13536.
Dyachuk V, Furlan A, Shahidi MK, Giovenco M, Kaukua N, Konstantinidou C, et al. Parasympathetic neurons originate from nerve-associated peripheral glial progenitors. Science. 2014;345(6192):82–7. https://doi.org/10.1126/science.1253281.
Espinosa-Medina I, Outin E, Picard CA, Chettouh Z, Dymecki S, Consalez GG, et al. Parasympathetic ganglia derive from Schwann cell precursors. Science. 2014;345(6192):87–90. https://doi.org/10.1126/science.1253286.
Uesaka T, Nagashimada M, Enomoto H. Neuronal differentiation in schwann cell lineage underlies postnatal neurogenesis in the enteric nervous system. J Neurosci. 2015;35(27):9879–88. https://doi.org/10.1523/JNEUROSCI.1239-15.2015.
Rohrer H. Linking human sympathoadrenal development and neuroblastoma. Nat Genet. Nature Research. 2021;53:593–4.
Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol. 2009;27(2):298–303. https://doi.org/10.1200/JCO.2008.16.6876.
Brodeur GM, Bagatell R. Mechanisms of neuroblastoma regression. Nat Rev Clin Oncol. 2014;11(12):704–13. https://doi.org/10.1038/nrclinonc.2014.168.
Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407(6805):802–9. https://doi.org/10.1038/35037739.
•Karlsson J, Valind A, HolmquistMengelbier L, Bredin S, Cornmark L, Jansson C, et al. Four evolutionary trajectories underlie genetic intratumoral variation in childhood cancer. Nat Genet. 2018;50(7):944–50. This study describes the intratumoral variation of pediatric tumors and how these variations occur within a single patient.
van Groningen T, Koster J, Valentijn LJ, Zwijnenburg DA, Akogul N, Hasselt NE, et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat Genet. 2017;49(8):1261–6.
Boeva V, Louis-Brennetot C, Peltier A, Durand S, Pierre-Eugène C, Raynal V, et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat Genet. 2017;49(9):1408–13.
•Gartlgruber M, Sharma AK, Quintero A, Dreidax D, Jansky S, Park YG, et al. Super enhancers define regulatory subtypes and cell identity in neuroblastoma. Nat Cancer. 2021;2(1):114–28. This study defines the super enhancers in neuroblastoma within different cell identities in neuroblastoma primary tumors.
Durbin AD, Zimmerman MW, Dharia NV, Abraham BJ, Iniguez AB, Weichert-Leahey N, et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat Genet. Nature Publishing Group. 2018;50:1240–6.
Decaesteker B, Denecker G, van Neste C, Dolman EM, van Loocke W, Gartlgruber M, et al. TBX2 is a neuroblastoma core regulatory circuitry component enhancing MYCN/FOXM1 reactivation of DREAM targets. Nat Commun. 2018;9(1):4866. https://doi.org/10.1038/s41467-018-06699-9.
Wang L, Tan TK, Durbin AD, Zimmerman MW, Abraham BJ, Tan SH, et al. ASCL1 is a MYCN- and LMO1-dependent member of the adrenergic neuroblastoma core regulatory circuitry. Nat Commun. 2019;10(1):5622. https://doi.org/10.1038/s41467-019-13515-5.
Gautier M, Thirant C, Delattre O, Janoueix-Lerosey I. Plasticity in neuroblastoma cell identity defines a noradrenergic-to-mesenchymal transition (Nmt). Cancers. 2021;13(12):2904. https://doi.org/10.3390/cancers13122904.
Khanna C, Jaboin JJ, Drakos E, Tsokos M, Thiele CJ. Biologically relevant orthotopic neuroblastoma xenograft models: primary adrenal tumor growth and spontaneous distant metastasis. In Vivo. 16(2):77–85.
•Park JA, Cheung N-KV. Targets and antibody formats for immunotherapy of neuroblastoma. J Clin Oncol. 2020;38:1836–48. Available from. This review gives a comprehensive overview of the possibilities using immunotherapy for the treatment of neuroblastoma.
Blavier L, Yang RM, Declerck YA. The tumor microenvironment in neuroblastoma: new players, new mechanisms of interaction and new perspectives. Cancers. MDPI AG. 2020;12:1–18.
Joshi S. Targeting the tumor microenvironment in neuroblastoma: recent advances and future directions. Cancers. MDPI AG. 2020;12:1–22.
Marimpietri D, Petretto A, Raffaghello L, Pezzolo A, Gagliani C, Tacchetti C, et al. Proteome profiling of neuroblastoma-derived exosomes reveal the expression of proteins potentially involved in tumor progression. PLoS One. 2013;8(9):e75054. https://doi.org/10.1371/journal.pone.0075054.
Bottino C, Dondero A, Bellora F, Moretta L, Locatelli F, Pistoia V, et al. Natural killer cells and neuroblastoma: tumor recognition, escape mechanisms, and possible novel immunotherapeutic approaches. Front Immunol. 2014;5:56. https://doi.org/10.3389/fimmu.2014.00056.
Bernards R, Dessain SK, Weinberg RA. N-myc amplification causes down-modulation of MHC class I antigen expression in neuroblastoma. Cell. 1986;47(5):667–74. https://doi.org/10.1016/0092-8674(86)90509-X.
Layer JP, Kronmüller MT, Quast T, Boorn-Konijnenberg D van den, Effern M, Hinze D, et al. Amplification of N-Myc is associated with a T-cell-poor microenvironment in metastatic neuroblastoma restraining interferon pathway activity and chemokine expression. OncoImmunology. 2017;6(6).
Zhang P, Wu X, Basu M, Dong C, Zheng P, Liu Y, et al. MYCN amplification is associated with repressed cellular immunity in neuroblastoma: an in silico immunological analysis of TARGET database. Front Immunol. 2017;88:1473. https://doi.org/10.3389/fimmu.2017.01473.
Mina M, Boldrini R, Citti A, Romania P, D’Alicandro V, de Ioris M, et al. Tumor-infiltrating T lymphocytes improve clinical outcome of therapy-resistant neuroblastoma. OncoImmunology. 2015;4(9):e1019981. https://doi.org/10.1080/2162402X.2015.1019981.
Wei JS, Kuznetsov IB, Zhang S, Song YK, Asgharzadeh S, Sindiri S, et al. Clinically Relevant Cytotoxic Immune Cell Signatures and Clonal Expansion of T-Cell Receptors in High-Risk MYCN-Not-Amplified Human Neuroblastoma. Clin Cancer Res. 2018;24(22):5673–84. https://doi.org/10.1158/1078-0432.CCR-18-0599.
Asgharzadeh S, Salo JA, Ji L, Oberthuer A, Fischer M, Berthold F, et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J Clin Oncol. 2012;30(28):3525–32.
Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H. Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers. 2015;7(4):2443–58. https://doi.org/10.3390/cancers7040902.
Augsten M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol. 2014;27(4):62. https://doi.org/10.3389/fonc.2014.00062.
Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. 2006;5(12):1640–6.
Zeine R, Salwen HR, Peddinti R, Tian Y, Guerrero L, Yang Q, et al. Presence of cancer-associated fibroblasts inversely correlates with Schwannian stroma in neuroblastoma tumors. Mod Pathol. 2009;22(7):950–8. https://doi.org/10.1038/modpathol.2009.52.
Borriello L, Nakata R, Sheard MA, Esteban Fernandez G, Sposto R, Malvar J, et al. Cancer-associated fibroblasts share characteristics and protumorigenic activity with mesenchymal stromal cells. Cancer Res. 2017;77(18):5142–57. https://doi.org/10.1158/0008-5472.CAN-16-2586.
Hashimoto O, Yoshida M, Koma Y-I, Yanai T, Hasegawa D, Kosaka Y. Collaboration of cancer‐associated fibroblasts and tumour‐associated macrophages for neuroblastoma development. J Pathol. 2016;240(2):211–23. https://doi.org/10.1002/path.4769.
Larsson K, Kock A, Idborg H, Henriksson MA, Martinsson T, Johnsen JI, et al. COX/mPGES-1/PGE2 pathway depicts an inflammatory-dependent high-risk neuroblastoma subset. Proc Natl Acad Sci U S A. 2015;112(26):8070–5.
Kock A, Larsson K, Bergqvist F, Eissler N, Elfman LHM, Raouf J, et al. Inhibition of microsomal prostaglandin E synthase-1 in cancer-associated fibroblasts suppresses neuroblastoma tumor growth. EBioMedicine. 2018;1(32):84–92.
Kock A, Bergqvist F, Steinmetz J, Elfman LHM, Korotkova M, Johnsen JI, et al. Establishment of an in vitro 3D model for neuroblastoma enables preclinical investigation of combined tumor-stroma drug targeting. FASEB J. 2020;34(8):11101–14.
Ma M, Ye JY, Deng R, Dee CM, Chan GC-F. Mesenchymal stromal cells may enhance metastasis of neuroblastoma via SDF-1/CXCR4 and SDF-1/CXCR7 signaling. Cancer Lett. 2011;312(1):1–10.
Mao Y, Eissler N, le Blanc K, Johnsen JI, Kogner P, Kiessling R. Targeting suppressive myeloid cells potentiates checkpoint inhibitors to control spontaneous neuroblastoma. Clin Cancer Res. 2016;22(15):3849–59.
Eissler N, Mao Y, Brodin D, Reuterswärd P, AnderssonSvahn H, Johnsen JI, et al. Regulation of myeloid cells by activated T cells determines the efficacy of PD-1 blockade. OncoImmunology. 2016;5(12):e1232222.
Ara T, Nakata R, Sheard MA, Shimada H, Buettner R, Groshen SG, et al. Critical role of STAT3 in IL-6-mediated drug resistance in human neuroblastoma. Cancer Res. 2013;73(13):3852–64.
••Yarmarkovich M, Marshall QF, Warrington JM, Premaratne R, Farrel A, Groff D, et al. Cross-HLA targeting of intracellular oncoproteins with peptide-centric CARs. Nature. 2021;599(7885):477–84. This study describes the immunopeptidome of neuroblastoma. This information was used to develop peptide-centric chimeric antigen receptors that can offer new immunotherapeutic options for the treatment of cancers with low neo-antigen expression.
Bayeva N, Coll E, Piskareva O. Differentiating neuroblastoma: a systematic review of the retinoic acid, its derivatives, and synergistic interactions. J Pers Med. 2021;11(3):211.
Zeid R, Lawlor MA, Poon E, Reyes JM, Fulciniti M, Lopez MA, et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat Genet. 2018;50(4):515–23.
Zimmerman MW, Durbin AD, He S, Oppel F, Shi H, Tao T, et al. Retinoic acid rewires the adrenergic core regulatory circuitry of childhood neuroblastoma. Sci Adv. 2021;7(43):eabe0834. https://doi.org/10.1126/sciadv.abe0834.
Sebbagh M, Borg JP. Insight into planar cell polarity. Exp Cell Res. 2014;328(2):284.
Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13(1):11–26.
Riento K, Ridley AJ. Inhibition of ROCK by RhoE. Methods Enzymol. 2006;406:533–41. https://doi.org/10.1016/S0076-6879(06)06041-1.
Hahmann C, Schroeter T. Rho-kinase inhibitors as therapeutics: from pan inhibition to isoform selectivity. Cell Mol Life Sci. 2010;67(2):171–7. https://doi.org/10.1007/s00018-009-0189-x.
Dyberg C, Fransson S, Andonova T, Sveinbjörnsson B, Lännerholm-Palm J, Olsen TK, et al. Rho-associated kinase is a therapeutic target in neuroblastoma. Proc Natl Acad Sci U S A. 2017;114(32):E6603–E6612. https://doi.org/10.1073/pnas.1706011114.
Borenäs M, Umapathy G, Lai W, Lind DE, Witek B, Guan J, et al. ALK ligand ALKAL2 potentiates MYCN-driven neuroblastoma in the absence of ALK mutation. EMBO J. 2021;40(3):e105784. https://doi.org/10.15252/embj.2020105784.
Fadeev A, Mendoza-Garcia P, Irion U, Guan J, Pfeifer K, Wiessner S, et al. ALKALs are in vivo ligands for ALK family receptor tyrosine kinases in the neural crest and derived cells. Proc Natl Acad Sci U S A. 2018;115(4):E630–E638. https://doi.org/10.1073/pnas.1719137115.
Treis D, Umapathy G, Fransson S, Guan J, Mendoza-García P, Siaw JT, et al. Sustained response to entrectinib in an infant with a germline ALKAL2 variant and refractory metastatic neuroblastoma with chromosomal 2p gain and anaplastic lymphoma kinase and tropomyosin receptor kinase activation. JCO Precis Oncol. 2022;6:e2100271.
Javanmardi N, Fransson S, Djos A, Umapathy G, Östensson M, Milosevic J, et al. Analysis of ALK, MYCN, and the ALK ligand ALKAL2 (FAM150B/AUGα) in neuroblastoma patient samples with chromosome arm 2p rearrangements. Genes Chromosomes Cancer. 2020;59(1):50–7.
Mossé YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14(6):472–80.
Foster JH, Voss SD, Hall DC, Minard CG, Balis FM, Wilner K, et al. Activity of crizotinib in patients with ALK-aberrant relapsed/refractory neuroblastoma: a Children’s Oncology Group Study (ADVL0912). Clin Cancer Res. 2021;27(13):3543–8.
Liu T, Merguerian MD, Rowe SP, Pratilas CA, Chen AR, Ladle BH. Exceptional response to the ALK and ROS1 inhibitor lorlatinib and subsequent mechanism of resistance in relapsed ALK F1174L-mutated neuroblastoma. Cold Spring Harb Mol Case Stud. 2021;7(4):a006064. https://doi.org/10.1101/mcs.a006064.
Umapathy G, Mendoza-Garcia P, Hallberg B, Palmer RH. Targeting anaplastic lymphoma kinase in neuroblastoma. APMIS. 2019;127(5):288–302. https://doi.org/10.1111/apm.12940.
Guan J, Fransson S, Siaw JT, Treis D, van den Eynden J, Chand D, et al. Clinical response of the novel activating ALK-I1171T mutation in neuroblastoma to the ALK inhibitor ceritinib. Cold Spring Harb Mol Case Stud. 2018;4(4):a002550. https://doi.org/10.1101/mcs.a002550.
Heath JA, Campbell MA, Thomas A, Solomon B. Good clinical response to alectinib, a second generation ALK inhibitor, in refractory neuroblastoma. Pediatr Blood Cancer. 2018;65(7):e27055.
Fischer M, Moreno L, Ziegler DS, Marshall LV, Zwaan CM, Irwin MS, et al. Ceritinib in paediatric patients with anaplastic lymphoma kinase-positive malignancies: an open-label, multicentre, phase 1, dose-escalation and dose-expansion study. Lancet Oncol. 2021;22(12):1764–76.
Guan J, Tucker ER, Wan H, Chand D, Danielson LS, Ruuth K, et al. The ALK inhibitor PF-06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. Dis Model Mech 2016;(9):941-52. https://doi.org/10.1242/dmm.024448.
Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, Szigety KM, et al. The ALK/ROS1 inhibitor PF-06463922 overcomes primary resistance to crizotinib in ALK-driven neuroblastoma. Cancer Discov. 2016;6(1):96–107. https://doi.org/10.1158/2159-8290.CD-15-1056.
Pearson ADJ, Barry E, Mossé YP, Ligas F, Bird N, de Rojas T, et al. Second Paediatric Strategy Forum for anaplastic lymphoma kinase (ALK) inhibition in paediatric malignancies. Eur J Cancer. 2021;157:198–213. https://doi.org/10.1016/j.ejca.2021.08.022.
Kahana-Edwin S, Cain LE, McCowage G, Darmanian A, Wright D, Mullins A, et al. Neuroblastoma molecular risk-stratification of DNA copy number and ALK genotyping via cell-free circulating tumor DNA profiling. Cancers. 2021;13(13):3365. https://doi.org/10.3390/cancers13133365.
Segura MF, Soriano A, Roma J, Piskareva O, Jiménez C, Boloix A, et al. Methodological advances in the discovery of novel neuroblastoma therapeutics. Expert Opin Drug Discov. 2021;22:1–13.
Acknowledgements
This work was supported with grants from the Swedish Childhood Cancer Foundation, the Swedish Cancer Foundation and The Cancer Research Foundations of Radiumhemmet.
Funding
Open access funding provided by Karolinska Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Kristina Ihrmark Lundberg, Diana Treis, and John Inge Johnsen declare they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Pediatric Oncology
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lundberg, K.I., Treis, D. & Johnsen, J.I. Neuroblastoma Heterogeneity, Plasticity, and Emerging Therapies. Curr Oncol Rep 24, 1053–1062 (2022). https://doi.org/10.1007/s11912-022-01270-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11912-022-01270-8