
Abstract
Purpose of Review
The field of acute myeloid leukemia (AML) has been revolutionized in recent years by the advent of high-throughput techniques, such as next-generation sequencing. In this review, we will discuss some of the recently identified mutations that have defined a new molecular landscape in this disease, as well as their prognostic, predictive, and therapeutic implications.
Recent Findings
Recent studies have shown how many cases of AML evolve from a premalignant period of latency characterized by the accumulation of several mutations and the emergence of one or multiple dominant clones. The pattern of co-occurring mutations and cytogenetic abnormalities at diagnosis defines risk and can determine therapeutic approaches to induce remission. Besides the genetic landscape at diagnosis, the continued presence of particular gene mutations during or after treatment carries prognostic information that should further influence strategies to maintain remission in the long term.
Summary
The recent progress made in AML research is a seminal example of how basic science can translate into improving clinical practice. Our ability to characterize the genomic landscape of individual patients has not only improved our ability to diagnose and prognosticate but is also bringing the promise of precision medicine to fruition in the field.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute myeloid leukemia is an aggressive and highly heterogeneous disease, with biologically and prognostically different subtypes [1]. Over 40 years have passed since pioneering work by Janet Rowley defined AML as a genetic disease with the description of the t(15;17) as a recurrent event in acute promyelocytic leukemia (APML) [2, 3]. Over the last few decades, we have witnessed a growing application of several high-throughput sequencing technologies, including whole genome, exome, and panel-based capture sequencing, that have helped refine the classification and prognostic scoring of AML. Current state-of-the-art iterations of these tools, the WHO classification (2016) and the European Leukemia Net (ELN) guidelines, have become the “industry standards.”
Well-established poor-risk prognostic variables in AML include older age, secondary disease, and adverse cytogenetics; however, recent analyses have also suggested the value of incorporating gene mutations beyond FLT3, NPM1, and CEBPA (e.g., IDH1and IDH2, ASXL1, MLL, DNMT3A, and TET2) into AML risk classifications [4]. Despite the advances in our understanding of the pathogenesis of AML, the standard of care still remains based around a “one size fits all” approach of age-adapted remission induction with chemotherapy and post-remission consolidation with chemotherapy and/or allogeneic hematopoietic stem cell transplant in younger patients. However, as the mutational landscape of AML is mapped in more detail, our therapeutic expectation in AML must evolve to match our increasing understanding of AML pathogenesis, with potential associated therapeutic vulnerabilities identified.
Cytogenetics in the DNA Era
Diagnostic karyotype remains the most powerful prognostic indicator in AML and forms the basis of current prognostic scores. To give an example of the key role cytogenetics plays in the diagnosis of AML, the WHO classification has established that patients with the clonal, recurring cytogenetic abnormalities t(8;21)(q22;q22), inv. (16)(p13q22) or t(16;16)(p13;q22), and t(15;17)(q22;q12) should be considered to have AML regardless of the blast percentage [5]. By dividing patients into three risk groups, favorable, intermediate, and adverse [6], cytogenetics has been widely adopted to provide the framework for risk-adapted treatment approaches [7]. In certain situations, cytogenetics also allow for prediction of effective therapy; in patients with the t(15;17)(q22;q21)/PML-RARA, for instance, the combination of all-trans retinoic acid (ATRA) and anthracycline-based protocols has resulted in a markedly improved outcome. In contrast patients with complex karyotype (≥ 3 or ≥ 5 abnormalities depending on the classification system), monosomal karyotype (such as monosomy 5/del(5q) or monosomy 7/del(7q)), or abnormalities of 3q have been shown to have inferior complete remission rates and overall survival and are currently considered for allogeneic stem cell transplant in first remission.
However, although cytogenetic analysis remains mandatory in the evaluation of suspected myeloid leukemia, it also presents several limitations. Apart from technical failures, cytogenetics cannot identify cryptic rearrangements, for example, 5% of PML/RARA positive AML lack the classic t (15, 17), with the fusion gene resulting from more complex rearrangements [8]. These patients not only respond to targeted therapy in a similar fashion to patients with the classic translocation but also share the same favorable prognosis and requirement for ATRA to prevent catastrophic coagulopathy, therefore cannot be missed. Moreover, around 40–50% of adult and 25% of pediatric AML patients have a normal karyotype (CN-AML), and these individuals are highly heterogeneous in terms of clinical outcomes [11]. Therefore, improving risk stratification and clinical decision making for this group of patients is a vibrant focus of research. In this effort, the mutational analysis of FLT3, NPM1, and CEBPA has become standard practice to improve their risk stratification [12]. There are however several additional gene mutations that also appear to carry prognostic relevance that include IDH1, IDH2, KIT, WT1, and RUNX1 [13], and their incorporation into risk stratification scoring is a matter of ongoing debate.
The Molecular Landscape of AML
The advent of massive parallel sequencing heralded a new age in molecular diagnosis, prognosis, and prediction. AML was the first cancer genome to be sequenced [14] and remains one of the most highly sequenced tumors, with ease of access of tumor tissue an obvious facilitating factor. However, even prior to this, a number of candidate gene studies had determined point and more complex mutations in critical genes, and we will summarize these below.
The Nucleolar Protein Nucleophosmin 1 (NPM1)
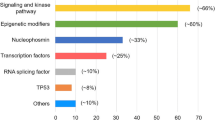
Mutations in the NPM1 gene are among the most common genetic changes in AML (occurring in 25–35% of patients), especially in CN-AML (present in 45–64%) [15]. NPM1 plays a role in numerous cellular functions, including ribosome biogenesis, DNA repair, and regulation of apoptosis. More than 40 different mutations in the C-terminal region of the protein have been described, but these uniformly result in the disruption of an N-terminal nucleolar localization signal and cause the protein to be aberrantly localized to the cytosol [16]. NPM1 mutations appear to be late driver events often occurring after DNMT3A, IDH1, or NRAS mutations [9••]. Interestingly, however, NPM1-mutated AML behaves as an entity on its own and is the largest classification category in a recent 11 component classification [9••]. NPM1 mutations are not normally observed in patients with AML associated recurrent translocations, and murine models of NPM1 mutation are associated with expanded myelopoiesis and the development of AML [17]. The prognostic implications of NPM1 mutations in individual patients are highly dependent on the pattern of co-occurring mutations and confer favorable prognosis only if associated with FLT3-ITD wild-type or low allelic ratio. Growing evidence suggests that carrying an NPM1 mutation confers sensitivity to novel agents such as venetoclax [10].
Mutations in Signaling Pathway Components
Fms-Like Tyrosine Kinase 3 (FLT3)
FLT3 is a tyrosine kinase that acts as a cytokine receptor for the FLT3 ligand. First described in 1991, FLT3 is strongly expressed in hematopoietic stem cells with important roles in cell survival and proliferation [18]. FLT3 mutations are among the most common mutations in AML and occur as either in-frame duplications within the juxtamembrane region (FLT3-ITD, internal tandem duplication) or as point mutations within the tyrosine kinase domain (FLT3-TKD) at a frequency of around 25% and 7% of AML cases, respectively [1]. Both mutations constitutively activate the tyrosine kinase leading to enhanced RAS, MAPK, and STAT5 signaling that results in blast proliferation [19, 20]. The effect on prognosis is modulated by the mutated to wild-type allele ratio. This may reflect a dominant clone and/or uniparental disomy of Ch13 on which FLT3 resides and an increased ratio is associated with an inferior outcome. In addition, FLT3-ITD mutations are associated with increased risk of relapse, whereas the prognostic relevance of FLT3-TKD mutations remains controversial [21]. Recent studies have suggested that inhibitors of FLT3 are effective as single agents in the relapsed refractory setting, as up-front adjuvants to conventional therapy in newly diagnosed patients and possibly in the maintenance setting also (RATIFY, QuANTUM-R and ADMIRAL studies).
With the ability to sequence AML genomes, it has become apparent that a number of other genes encoding for signaling pathways components (RAS, cKIT, NF1, and others) are mutated in AML. The observation that mutations in signaling pathways proteins frequently co-occur with chromosomal rearrangements in hematopoietic transcription factors (PML-RARA, CBFβ-MYH11, RUNX1-RUNX1T1) led to the hypothesis that AML results from the cooperation of mutations that confer a proliferative advantage (class I mutations) with mutations that induce a block in differentiation (class II mutations), the 2-hit model [1]. However, over 40% of AML cases lack mutations in classical signaling pathway genes, suggesting that the evolution of acute leukemia is a more complex and individual phenomenon [22].
Mutations in Epigenetic/Chromatin Modifiers
In recent years, a number of epigenetic and chromatin modifiers have been identified as mutated in AML. These mutations are classically found at the highest variant allele frequencies (VAF) in AML patients [9••] and can also persist in remission, [23] leading to the acceptance that these mutations often represent preleukemic events [24]. In the most recent large classification of AML, this group was also demonstrated to have a poor prognosis, [9••] a finding recapitulated for individual genes [25]. Efforts are therefore ongoing to dissect this group further, as epigenetic and chromatin modifiers represent effective therapeutic targets [26].
DNA Methyltransferase 3A (DNMT3a)
DNMT3A mutations occur in 18–22% of all AML cases and approximately 34% of CN-AML cases [27], and they are mostly heterozygous and commonly affect a hotspot encoding arginine at codon 882 (~ 60% of AML cases). R882 mutations appear to result in a hypomorphic protein that acts in a dominant negative manner, inhibiting the methyltransferase activity of the remaining wild-type DNMT3A [28]. In murine models, when Dnmt3a is conditionally deleted, self-renewal is favored over differentiation [29], but the underlying mechanisms, and their relationship to DNA methylation, remain unexplained. The prognostic significance of DNMT3A mutations remain controversial, but recent evidence suggests that DNMT3A-mutated AML patients may benefit from higher doses of anthracyclines [30].
Ten–Eleven Translocation 2 (TET2)
TET2 is found mutated in about 9%–23% of AML patients [31]. TET2 regulates the initial step in DNA demethylation through the conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC). In general, TET2 mutations are thought to be loss-of-function mutations; however, despite several studies, their prognostic significance remains unclear.
Additional Sex Comb-Like 1 (ASXL1)
ASXL1 loss-of-function mutations occur in ~ 5–11% of AML cases [32]. Although their mechanisms of action are not fully known, it is likely that they function, at least in part, through a loss of PRC2 (polycomb repressive complex 2) function allowing derepression of genes such as HOXA cluster genes [33]. ASXL1 mutations are five times more common in older (> 60 years) patients, are frequently associated with t(8;21), wild-type NPM1, wild-type FLT3, and mutated CEBPA, and are considered adverse prognostic factors according to European Leukemia Net (ELN) criteria.
Isocitrate Dehydrogenase (IDH)
IDH 1 and 2 gene mutations are neomorphic gain-of-function mutations that cause an alteration of normal function, allowing the mutant enzyme the novel function of further converting α-ketoglutarate into 2-hydroxyglutarate (2-HG). This “oncometabolite” inhibits the function of TET2 and other dioxygenase enzymes causing effects on DNA and histone methylation and thus epigenetic regulation [34]. IDH1 mutations more commonly affect the highly conserved arginine (R) residue at codon 132 (R132) and have been identified in 7% of AML patients. IDH2 mutations are identified in a further 9% of cases and cluster at codons R140 and R172 [1]. Interestingly IDH2 mutations are prognostically distinct, whereas R140 mutations are often associated with NPM1 and predict more favorable outcomes, and R172 mutations seem to represent a distinct genomic subgroup with mutual exclusivity with NPM1 and indicate poorer prognosis [9••].
Cohesin Complex
Cohesin is a large ring-shaped multi-protein complex consisting of four major subunits: SMC1A, SMC3, RAD21, and STAG1/2. Cohesin plays a role not only in mediating sister chromatid cohesion during mitosis, where it coordinates ordered chromosome separation and prevents mitotic catastrophe, but is also involved in DNA damage repair and the regulation of gene expression through coordinating interaction between distal and proximal cis-regulatory elements. Mutations in Cohesin subunits affect around 10% of AML patient [35] and typically co-occur with NPM1, DNMT3A, TET2, or RUNX1 mutations [36]. Cohesin mutations or knockdown of Cohesin subunits impair hematopoietic differentiation and enforce stem cell programs in both human and mouse hematopoiesis. Furthermore, studies have demonstrated alterations of chromatin accessibility upon depletion of Cohesin function and have further linked AML development with a requirement of Cohesin function for dynamic gene expression during erythroid differentiation and interaction with ETS transcription factors [37, 38].
RNA Splicing Factor Mutations
A number of RNA splicing factors are mutated in AML. The most commonly mutated genes include SF3B1, U2AF1, SRSF2, and ZRSR2 [39]. Often considered founding events, splicing factor gene mutations are frequently found in preleukemic conditions such as MDS. In newly diagnosed AML patients, spliceosome gene mutations are now considered pathognomonic of secondary AML developing from preceding MDS [40]. These mutations are likely to alter splicing and subsequent translation of critical genes and are generally associated with poorer responses.
Transcription Factor Mutations
Runt-Related Transcription Factor (RUNX1)
The RUNX1 gene is a partner in the t(8;21) fusion gene in CBF leukemia and is also affected by recurrent gene mutations in AML [41]. RUNX1 mutations are found in 5–13% of AML cases and are commonly associated with trisomy 13, trisomy 21, absence of NPM1, and CN-AML [42]. In sharp contrast with the favorable prognostic effect of gene fusions involving RUNX1, RUNX1 mutations are associated with resistance to standard induction therapy and with inferior overall survival, for both younger and older patients, and are an adverse risk factor in the 2017 ELN guidelines [43].
CCAAT Enhancer Binding Protein α (CEBPA)
CEBPA is a transcription factor that plays a key role in hematopoiesis and is a master regulator of myeloid differentiation [44]. Mutations occur in 6 to 10% of AML cases [45] and are identified in both amino and carboxy-terminal regions, with the latter resulting in a truncated protein that is unable to dimerize and bind DNA [46]. Only bi-allelic, not single, CEBPA mutations predict an increased CR rate and favorable survival. CEBPA mutations can also be inherited through the germline, with this subset of patients often going on to develop AML with the acquisition of additional mutations that include but are not exclusive to the other CEBPA allele [47].
Tumor Suppressor Gene Mutations
TP53
Mutations in the tumor suppressor TP53 are identified in 8% of AML cases and are associated with complex karyotype, therapy-related AML, chemo-resistance, high relapse rate, and poor survival [48]. These mutations also confer an adverse risk in the European Leukemia Net (ELN) guidelines and patients that carry a TP53 mutation are poorly served by current therapeutic strategies.
The Evolution of AML from a Latent Preleukemic State
More recently, AML-associated mutations have been found in healthy aging individuals, a condition named age-related clonal hematopoiesis (ARCH) or clonal hematopoiesis of indeterminate potential (CHIP), and its incidence is as high as 10% in individuals over 65 years of age. These individuals have a higher risk of hematological malignancies and cardiovascular disease [49, 50]. The acknowledgment that AML has a period of latent preleukemia of at least several years in many cases has been a major step forward in understanding the evolution and kinetics of this aggressive disease. Most cases of ARCH involve mutations in epigenetic regulators such as DNMT3A, ASXL1, and TET2, whereas FLT3 and NPM1 mutations are never observed, suggesting that these are later cooperating events. Interestingly, despite the fact that ARCH is very prevalent in the general aging population, AML remains a rare disease (~ 4 cases per 100,000 individuals) [51]. Currently, the rate of transformation of ARCH into AML is predicted to be between 0.5 and 1% per year [52]. Deep sequencing of historical samples of patients that have gone on to develop AML has suggested that mutations in TP53, IDH1, and 2 and RNA splicing factors (SRSF2, SF3B1, and U2AF1) are associated with the highest odds of transformation. In these large cohort studies, DNMT3A and TET2 mutations appeared as commonly occurring events in both AML and control cases, but higher VAFs (> 10%), the presence of a higher number of variants (two or more), along with clonal complexity, correlated with a greater risk of AML [53•, 54•].
The recent identification of a true preleukemic state has changed our view on AML and has identified the possibility of a new treatment paradigm; prevention of the evolution of this deadly disease during its latent phase, with much research, currently devoted to this. The highest risk individuals seem to be those with a detectable TP53 clone [54•], but at the moment, there are no established strategies to eliminate such clones. IDH and splicing factor mutant clones have potential therapeutics; however, these treatments are not without toxicity, and a greater predictive capacity, less toxic therapies, and careful clinical trials are needed to justify widespread preemptive intervention.
Prognostic and Therapeutic Implications of Defining the Molecular Landscape of AML in Individual Patients
The most immediate consequence of the next-generation sequencing revolution has been to improve the risk stratification of AML patients. Indeed, in 2017 the European Leukemia Network recognized the prognostic value of some of the mutations discussed above and updated their risk stratification criteria (Table 1) [55].
Several studies continue to suggest that incorporating a broader list of gene mutations than indicated by ELN 2017 could further refine risk stratification, but more importantly, it is becoming clear that the prognostic effect of a given mutation depends on the pattern of co-occurring mutations. For instance, Papaemmanuil et al. described how the negative effect of a FLT3 ITD in patients with an NPM1 mutation is much more pronounced when DNMT3A is also mutated. Likewise, in patients with an NPM1 mutation, the presence of a RAS mutation improved survival more in the presence than in the absence of a DNMT3A mutation. Analogously, the adverse effect of an MLL aberration noted in European Leukemia Net (ELN) 2017 depended on the presence of FLT3 TKD mutations, and although European Leukemia Net (ELN) 2017 regarded mutations in IDH2 or in DNMT3A as having no prognostic effect, prognosis became considerably worse when both IDH2 and DNMT3A mutations co-occurred in a large series of patients [9••]. However, to take full prognostic advantage of the genetic complexity in AML, where there are usually thought to be between 3 and 5 driver mutations per patient, will require large patient numbers and international collaboration as is planned by large consortia such as the HARMONY alliance (https://www.harmony-alliance.eu).
In addition to the presence of genetic abnormalities at diagnosis, the continued presence of particular gene mutations during or after treatment carries prognostic information for certain genetically defined AML subtypes. In NPM1-mutated AML, for instance, detection of mutant NPM1 transcripts by sensitive quantitative RT-PCR after 2 cycles of chemotherapy had an 86% cumulative incidence of relapse vs 34% for NPM1 negative patients [56•]. Minimal residual disease (MRD) detection, either by genetics or by multiparameter flow cytometry (MPFC), has therefore assumed a role in risk-adapting post-remission therapy, which had previously been based solely on pretreatment variables. Interest has more recently turned to establishing if a variety of mutations identified through NGS are persistent after treatment and the potential prognostic implications. It is becoming apparent that while detection of ARCH mutations (DNMT3A, TET2, ASXL1) has no prognostic implication and simply underlies the advantage of these clones over wild type in repopulating the marrow after chemotherapy, persistence of non-ARCH mutations is associated with higher cumulative incidence of relapse and shorter survival and relapse-free survival [57], albeit that the resolution of NGS (10-2-10-3) is more limited than either MPFC or PCR.
Finally, the research-based advances described above are translating into clinical practice leading to a very much overdue update in the treatment strategies available to combat AML, and as a result, within the last 2 years, the US Food and Drug Administration (FDA) approved eight novel therapies for patients with AML, many of which are also becoming available in Europe (Fig. 1). Midostaurin, a FLT3 inhibitor, was one of the first novel therapies to enter clinical practice and is currently recommended in combination with chemotherapy and as a single agent for maintenance therapy in patients with mutated FLT3 [58]. Similarly, the IDH1 and IDH2 inhibitors ivosidenib and enasidenib have shown promising results in clinical trials and have been approved for use in AML patients with IDH1 and IDH2 mutations, respectively [59, 60]. To further champion personalized medicine in AML, the Leukemia and Lymphoma Society is sponsoring a “BEAT AML” trial that plans to recruit 500 patients aged 60 or above with newly diagnosed AML. The trial aims firstly to assess the feasibility of enrolling patients based on mutational status. Following a genomic screening whose results should be available within 7 days of a marrow sample being taken, patients will be assigned to one of several arms based on their genetic profile. The results of this and other similar trials proposed or in set up are eagerly awaited as they will not only pave the way towards the use of precision medicine in AML but also will begin to test the efficacy of several novel targeting agents in a cohort of newly diagnosed older patients, who are the patient group with the highest mortality rates and lowest tolerance for toxic therapies.

Timing of the identification of mutations associated with AML and evolution of therapeutic strategies. More widespread use of sequencing technologies has enriched the landscape of mutations that are associated with AML. An enhanced ability to diagnose and prognosticate is now translating into an increased understanding of therapeutic vulnerabilities and the development of new therapies. Between 2017 and 2018, the FDA has approved eight novel drugs for the treatment of AML and for the first time in almost 40 years, and AML patients can benefit from a more individualized treatment approach
Conclusions
The advent of rapid and affordable genome sequencing has revolutionized our approach to the classification, prediction, and prognostication of acute myeloid leukemia and has greatly improved our scientific understanding of the pathophysiology of this deadly disease. This has initially translated into an improved ability to determine which patients will benefit most from allogeneic stem cell transplantation in first remission, but now, thanks to a growing therapeutic armamentarium, is at last leading to a long overdue clinical progress in AML therapy and to the promise of individualized approaches to improve outcomes in this deadly disease.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127(1):29–41. https://doi.org/10.1182/blood-2015-07-604496.
Rowley JD, Golomb HM, Dougherty C. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet. 1977;1(8010):549–50. https://doi.org/10.1016/s0140-6736(77)91415-5.
Lindgren V, Rowley JD. Comparable complex rearrangements involving 8;21 and 9;22 translocations in leukaemia. Nature. 1977;266(5604):744–5. https://doi.org/10.1038/266744a0.
Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89. https://doi.org/10.1056/NEJMoa1112304.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405. https://doi.org/10.1182/blood-2016-03-643544.
Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood. 1998;92:2322–33.
Grimwade D, Hills RK. Independent prognostic factors for AML outcome. Hematol Am Soc Hematol Educ Program. 2009:385–95. https://doi.org/10.1182/asheducation-2009.1.385.
Grimwade D, Biondi A, Mozziconacci MJ, Hagemeijer A, Berger R, Neat M, et al. Characterization of acute promyelocytic leukemia cases lacking the classic t(15;17): results of the European Working Party. Groupe Français de Cytogénétique Hématologique, Groupe de Français d'Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1 European Community-Concerted Action "Molecular Cytogenetic Diagnosis in Haematological Malignancies". Blood. 2000;96(4):1297–308.
•• Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374(23):2209–21. https://doi.org/10.1056/NEJMoa1516192In this large prospective cohort study the pattern of co-mutation in AML has been described in details. In addition to inform on the evolution of AML, this study has also identified new genomic categories and informed prognostic stratification.
DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17. https://doi.org/10.1182/blood-2018-08-868752.
Grimwade D, Mrózek K. Diagnostic and prognostic value of cytogenetics in acute myeloid leukemia. Hematol Oncol Clin North Am. 2011;25(6):1135–61. https://doi.org/10.1016/j.hoc.2011.09.018.
Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–18. https://doi.org/10.1056/NEJMoa074306.
Marcucci G, Haferlach T, Dohner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011;29:475–86. https://doi.org/10.1200/JCO.2010.30.2554.
Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456(7218):66–72. https://doi.org/10.1038/nature07485.
Falini B, Nicoletti I, Martelli MF, Mecucci C. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features. Blood. 2007;109(3):874–85.
Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352(3):254–66.
Vassiliou GS, Cooper JL, Rad R, Li J, Rice S, Uren A, et al. Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat Genet. 2011;43(5):470–5. https://doi.org/10.1038/ng.796.
Saultz JN, Garzon R. Acute myeloid leukemia: a concise review. J Clin Med. 2016;5(3). https://doi.org/10.3390/jcm5030033.
Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99:310–8.
Kayser S, Schlenk RF, Londono MC, Breitenbuecher F, Wittke K, Du J, et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood. 2009;114:2386–92.
Medinger M, Passweg JR. Acute myeloid leukaemia genomics. Br J Haematol. 2017;179(4):530–42. https://doi.org/10.1111/bjh.14823.
Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74. https://doi.org/10.1056/NEJMoa1301689.
Shlush LI, Zandi S, Mitchell A, Chen WC, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506(7488):328–33. https://doi.org/10.1038/nature13038.
Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiro BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci U S A. 2014;111(7):2548–53. https://doi.org/10.1073/pnas.1324297111.
Gallipoli P, Huntly BJP. Prognostic models turn the heat(IT)up on FLT3ITD -mutated AML. Clin Cancer Res. 2019;25(2):460–2. https://doi.org/10.1158/1078-0432.CCR-18-3146.
Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–33. https://doi.org/10.1056/NEJMoa1005143.
Yang L, Rau R, Goodell MA. DNMT3A in haematological malignancies. Nat Rev Cancer. 2015;15(3):152–65. https://doi.org/10.1038/nrc3895.
Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011;44(1):23–31. https://doi.org/10.1038/ng.1009.
Sehgal AR, Gimotty PA, Zhao J, Hsu JM, et al. DNMT3A mutational status affects the results of dose-escalated induction therapy in acute myelogenous leukemia. Clin Cancer Res. 2015;21:1614–20.
Chou WC, Chou SC, Liu CY, Chen CY, Hou HA, Kuo YY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118:3803–10.
Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrózek K, et al. ASXL1 mutations identify a high risk subgroup of older patients with primary cytogenetically normal AML within the ELN favorable genetic category. Blood. 2011;118:6920–9.
Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediatedgene repression. Cancer Cell. 2012;22(2):180–93. https://doi.org/10.1016/j.ccr.2012.06.032.
Inoue D, Kitaura J, Togami K, Nishimura K, Enomoto Y, et al. Myelodysplastic syndromes are induced by histone methylation-altering ASXL1 mutations. J Clin Invest. 2013;123(11):4627–40.
Molenaar RJ, Maciejewski JP, Wilmink JW, van Noorden CJF. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene. 2018;37(15):1949–60. https://doi.org/10.1038/s41388-017-0077-z.
Tsai CH, Hou HA, Tang JL, Kuo YY, Chiu YC, Lin CC, et al. Prognostic impacts and dynamic changes of cohesin complex gene mutations in de novo acute myeloid leukemia. Blood Cancer J. 2017;7(12):663. https://doi.org/10.1038/s41408-017-0022-y.
Leeke B, Marsman J, O'Sullivan JM, Horsfield JA. Cohesin mutations in myeloid malignancies: underlying mechanisms. Exp Hematol Oncol. 2014;3:13. https://doi.org/10.1186/2162-3619-3-13eCollection 2014. Review.
Mazumdar C, Shen Y, Xavy S, Zhao F, Reinisch A, Li R, et al. Leukemia associated cohesin mutants dominantly enforce stem cell programs and impair human hematopoietic progenitor differentiation. Cell Stem Cell. 2015;17(6):675–88. https://doi.org/10.1016/j.stem.2015.09.017.
Sasca D, Yun H, Giotopoulos G, Szybinski J, Evan T, Wilson NK, et al. Cohesin-dependent regulation of gene expression during differentiation is lost in cohesin-mutated myeloid malignancies. Blood. 2019. https://doi.org/10.1182/blood.2019001553.
Lindsley RC, Mar BG, Mazzola E, Grauman PV, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125:1367–76.
Cazzola M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and its clinical relevance. Blood. 2013;122:4021–34.
Meyers S, Downing JR, Hiebert SW. Identification of AML-1 and the (8;21) translocation protein (AML-1/ETO) as sequence-specific DNA-binding proteins: the runt homology domain is required for DNA binding and protein-protein interactions. Mol Cell Biol. 1993;13:6336–45.
Marcucci G, Haferlach T, Dohner H. Molecular genetic of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011;29:475–86.
Mendler JH, Maharry K, Radmacher MD, Mrozek K, Becker H, Metzeler KH. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J Clin Oncol. 2012;30:3109–18.
Koschmieder S, Halmos B, Levantini E, Tenen DG. Dysregulation of the C/EBPalpha differentiation pathway in human cancer. J Clin Oncol. 2009;27(4):619–28. https://doi.org/10.1200/JCO.2008.17.9812Review.
Mrózek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007;109(2):431–48 Review.
Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet. 2001;27(3):263–70.
Smith ML, Hills RK, Grimwade D. Independent prognostic variables in acute myeloid leukaemia. Blood Rev. 2011;25(1):39–51. https://doi.org/10.1016/j.blre.2010.10.002Review.
Devillier R, Mansat-De Mas V, Gelsi-Boyer V, et al. Role of ASXL1 and TP53 mutations in the molecular classification and prognosis of acute myeloid leukemias with myelodysplasia-related changes. Oncotarget. 2015;6(10):8388–96.
Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–87.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–98.
Deschler B, Lübbert M. Acute myeloid leukemia: epidemiology and etiology. Cancer. 2006;107(9):2099–107 Review.
Steensma DP. Clinical consequences of clonal hematopoiesis of indeterminate potential. Blood Adv. 2018;2(22):3404–10. https://doi.org/10.1182/bloodadvances.2018020222Review.
• Desai P, Mencia-Trinchant N, Savenkov O, Simon MS, Cheang G, Lee S, et al. Somatic mutations precede acute myeloid leukemia years before diagnosis. Nat Med. 2018;24(7):1015–23. https://doi.org/10.1038/s41591-018-0081-zBy analyzing a large group of individuals the two studies above identify features that can distinguish benign ARCH from preleukaemia, supporting the hypothesis that individuals at high risk of AML can be identified before overt disease develops.
• Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, Niemeyer E, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018;559(7714):400–4. https://doi.org/10.1038/s41586-018-0317-6By analyzing a large group of individuals the two studies above identify features that can distinguish benign ARCH from preleukaemia, supporting the hypothesis that individuals at high risk of AML can be identified before overt disease develops.
Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–47. https://doi.org/10.1182/blood-2016-08-733196.
• Ivey A, Hills RK, Simpson MA, et al. Assessment of minimal residual disease in standard-risk AML. N Engl J Med. 2016;374:422–33 In this study, the authors show how persistence of NPM1 transcript following two cycles of chemotherapy is associated with increased risk of relapse and lower overall survival and is a seminal example of how the continued presence of particular gene mutations during or after treatment carries independednt prognostic information.
Jongen-Lavrencic M, Grob T, Hanekamp D, Kavelaars FG, Al Hinai A, Zeilemaker A, et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med. 2018;378(13):1189–99. https://doi.org/10.1056/NEJMoa1716863.
Stone RM, Mandrekar S, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukaemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454–64.
DiNardo CD, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378:2386–98.
Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130:722–31.
Basheer F, Giotopoulos G, Meduri E, Yun H, Mazan M, et al. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J Exp Med. 2019;216(4):966–81. https://doi.org/10.1084/jem.20181276.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Ludovica Marando and Brian J. P. Huntly declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Leukemia
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marando, L., Huntly, B.J.P. Molecular Landscape of Acute Myeloid Leukemia: Prognostic and Therapeutic Implications. Curr Oncol Rep 22, 61 (2020). https://doi.org/10.1007/s11912-020-00918-7
Published:
DOI: https://doi.org/10.1007/s11912-020-00918-7