
Abstract
Purpose of Review
The aim of this review is to provide a comprehensive update on the clinical assessment, diagnosis, complications, and treatment of primary central nervous system vasculitis (PCNSV).
Recent Findings
The developments in neuroimaging, molecular testing, and cerebral biopsy have enhanced clinical assessment and decision making, providing novel insights to prevent misdiagnosis increasing diagnostic certainty. Advances in imaging techniques visualizing the wall of intracranial vessels have improved the possibility to distinguish inflammatory from non-inflammatory vascular lesions. Large recent studies have revealed a more varied histopathological pictures and disclosed an association with amyloid angiopathy. Unfortunately, therapy remains largely empiric.
Summary
PCNSV is a heterogeneous group of disorders encompassing different clinical subsets that may differ in terms of prognosis and therapy. Recent evidence has described a more benign course, with good response to therapy. New diagnostic techniques will play soon a pivotal role in the appropriate diagnosis and prompt management of PCNSV.
Similar content being viewed by others


Introduction
Vasculitis is a group of clinical and pathological diseases characterized by infiltration of inflammatory cells with reactive damage to blood vessel walls of different size, from large arteries to capillaries and venules. Both loss of vessel integrity, leading to rupture and bleeding, and decrease in lumen diameter, may result in downstream ischemia, necrosis, and subsequent tissue damage. Blood vessels themselves can be damaged, resulting in permanent stenosis, aneurysmal changes, or rupture [1].
The revised 2012 Chapel Hill Consensus Conference (CHCC) [1] guides the categorization of different types of both primary and secondary vasculitis based on caliber of the vessels and based on primary localization or secondary with systemic involvement (Table 1). Classification of primary vasculitis is now by the size of the affected vessel, including large vessel, medium vessel, and small vessel vasculitis. Small vessel vasculitis affects intraparenchymal arteries, arterioles, capillaries, veins, and venules; some are related to antineutrophil cytoplasmic antibody (ANCA), while others are related to immune complex deposition. Separately there is the variable vessel vasculitis that are classified without a predominant vessel size and caliber which involve arteries, veins, and capillaries. Secondary vasculitis is the vasculitis associated with systemic diseases, mainly connective tissue disorders and the vasculitis associated with a specific cause, such as substance abuse and infections. Finally, vascular inflammation restricted to a specific organ or system, such as the vasculitis of the central nervous system (CNS) and peripheral nervous system (PNS), and immunoglobulin (Ig) G4-related disease which represent the so-called single-organ vasculitis (SOV). SOV involves arteries or veins of any size, without features of a systemic vasculitis, and includes the granulomatous angiitis of the brain and similar terms of primary angiitis of the CNS (PACNS; adult or childhood) primary CNS vasculitis (PCNSV), isolated or non-systemic PNS vasculitis (NSPNSV), and isolated aortitis (IgG4-related). See Table 1 for further classification and characterization of different vasculitis.
Although extensive literature and classifications exist, many neurologists still have a limited knowledge of vasculitis, partially because other specialists often diagnose and manage most of these patients. However, both CNS and PNS are frequently a major target of these disorders, including their early stages, when the clinical manifestations are often non-specific, thus causing underestimation of their actual prevalence. The typical CNS manifestation of vasculitis is a stroke, either because of SOV or neurological consequence of a systemic vasculitis. Other common CNS presentations include visual loss, encephalopathy, seizures, headache, venous thrombosis, and hemorrhagic stroke [2••], whereas the involvement of PNS usually presents as peripheral neuropathy of different types (polyneuropathy, mononeuropathy, or multiplex mononeuropathy, and cranial neuropathy) and neuromuscular disease [3, 4]. Overall, differential diagnosis of vasculitis is challenging, and neurologists are often on the frontline of the diagnosis and treatment of these patients.
In this scenario, the management of patients with nervous system vasculitis is based on 7 main principles. Principle 1: vasculitis are often severe diseases with potential permanent disability due to tissue ischemia and infarction and a possible fatal outcome that requires prompt recognition and therapy. Principle 2: some forms of vasculitis are associated with severe neurological complications that significantly affect patient’s prognosis. Principle 3: clinical course of vasculitis can range from fulminant to very mild forms and may fluctuate in clinical signs. Principle 4: there is a likelihood of excess morbidity and mortality due to missing diagnosis and therefore undertreatment. Principle 5: histopathological confirmation of vasculitis is essential for accurate diagnosis, as well as a correct methodology for achieving it. Principle 6: empiric trials with immunosuppressive and immunomodulating therapy should never be considered as a substitute for a confirmed diagnosis of vasculitis. Principle 7: treatments are initially guided toward stabilization of the blood–brain barrier (BBB), followed by maintenance immunosuppressive therapy directed both at the humoral and cell-mediated autoimmune and inflammatory mechanisms. Physicians should be able to early diagnose and timely treat these patients in order improve prognosis and functional outcome [1, 5, 6••].
In this review, we provide a timely update on CNS vasculitis and a practical guide for clinical neurologists in disentangling their differential diagnosis, laboratory evaluation, histological findings, and treatment of CNS vasculitis. The vasculitis affecting the PNS will be included in the companion review.
Methods and Results
To provide a comprehensive update, a PubMed-based (MEDLINE) literature review was carried out to identify all pertinent studies within the last five years (January 2016–May 2021). The following search terms were used: primary and secondary vasculitis, central nervous system, biopsy, autopsy, neuropathology, histology, morphology, encephalopathy, ischemic stroke, intracerebral hemorrhage, gliosis, necrotizing encephalopathy, encephalitis, myelitis, and inflammation.
A total of 863 articles, including those listed in the references of the retrieved studies, were screened. Then, we excluded the following items: papers different from original articles, preclinical or animal studies, non-English written papers, and any other publication that did not comply with the goal of the present review.
Overview on CNS Vasculitis Pathogenesis
The dual role of the BBB is known as a coating structure and a functional component in cerebrovascular angiogenesis and exchanges between the brain and blood compartment, although the latter function is still not completely understood. What we certainly know is that the BBB integrity is of critical importance, since its disruption is both a predisposing and precipitating factor of a variety of primary and secondary neurological diseases, including vasculitis [7,8,9]. Also, genetic background plays a crucial role as an additional predisposing factor to vasculitis [10]. Accordingly, the association between vasculitis and some human leukocyte antigen (HLA) loci that are very highly polymorphic, such as the HLA-B involved in the binding of the antigen and interactions with receptors on CD8 T cells and natural killer (NK) cells, is well known [10]. Another example is the association between PR3-ANCA vasculitis and the genes SERPINA1 (encoding alpha-1 antitrypsin), PRTN (encoding PR3), and some HLA loci, including the HLA-DP4 and HLA-DQ [11]. However, because most of vasculitis are rare diseases with a complex genetic risk conferred by hundreds of loci with a low independent effect, a genetic heritability can only account for a small proportion of cases. Indeed, only few genetic associations have been published on neurological complications of vasculitis, and these have not been replicated in independent studies [10].
Both the immune system and inflammatory responses play a pivotal role in the etiopathogenesis of CNS vasculitis, although some aspects remain unclear yet. Mechanisms of vascular damage can basically be summarized as follows [12]: (1) immune complex-mediated; (2) cell-mediated; and (3) ANCA-mediated: cytoplasmic (c-ANCA), perinuclear (p-ANCA), and atypical ANCA. The immune complex is responsible for complement activation, neutrophil chemotaxis and subsequent phagocytosis, and secretion of neutrophil granular products with vascular damage [13]. The second mechanism is mediated by cytotoxic CD8 + T cells which release interferon-γ (INFγ) that recruits and activates macrophages [13]. Finally, in the third case, inflammation and related damage is due to specific antibodies against the cytoplasm of neutrophils [14, 15]. Figure 1 summarizes the main pathogenetic mechanisms of vasculitis.

Main pathogenetic mechanisms of vasculitis and blood–brain barrier damage responsible for morphological and pathological organ change, ischemic and hemorrhagic stroke, and encephalopathy. ANCA indicates anti-neutrophil cytoplasmic antibodies; BBB, brain-blood barrier; IFN-γ, interferon-γ; ROS, radical oxygen species
Clinical Features and Diagnostic Approach
CNS vasculitis can be primary or secondary [16]. More broadly, CNS vasculitis can be divided into PACNS, where the inflammation is restricted to the vessels of the brain and/or spinal cord, and secondary CNS vasculitis, when the involvement of the brain is associated with other systemic disorders also affecting skin, kidney, lungs, sinuses, cardiovascular system, or joints. A primary vasculitis may occur without any identified underlying cause, whereas other forms of vasculitis can be secondary to a malignancy, drugs of abuse, or medications. In addition, cerebral vasculitis may be secondary to an infectious disease or a non-infectious inflammatory disorder that may be restricted to the CNS or be part of a multiorgan disorder. Since a SOV affecting only the brain is a relatively uncommon condition and because the neurological symptoms are usually non-specific, an accurate diagnosis is often challenging. Moreover, the results of laboratory tests and instrumental exams are often not precise enough to diagnose a CNS vasculitis. As a result, a cerebral vasculitis can be both over- and underdiagnosed.
Differential diagnosis of a cerebral vasculitis is also broad. The diagnosis of primary (isolated) vasculitis of the CNS can be very difficult because of the absence of symptoms or signs in other organs or systems. On the other hand, systemic diseases leading to cerebral vasculitis often show a rather stereotyped constellation of clinical symptoms and specific serologic testing abnormalities that make them less difficult to diagnose. Among them, there is also systemic vasculitis from non-autoimmune disorders which include viral infections (e.g., HIV, hepatitis C virus-associated cryoglobulinemic vasculitis, Coronavirus, Herpes virus, etc.), bacterial and fungal infections, drugs (e.g., hydralazine-associated microscopic polyangiitis, levamisole), and some cancers. Of note, also the new SARS-CoV2 virus (COVID-19) was recently identified as a viral cause of cerebral vasculitis due to an endotheliitis [17, 18], and an extensive lymphoplasmacytic perivascular inflammation, as well as a lymphocytic vasculitis [17].
CNS vasculitis secondary to connective tissue autoimmune diseases and systemic vasculitis of small, medium, and large vessels are listed in Table 1. In this context, it is known that many systemic diseases can be complicated by neurological manifestations through immune-mediated mechanisms or a direct spread to the CNS via the bloodstream through the BBB. The main complications are of neurovascular origin and are caused by endothelial damage, increased coagulability, cascade of chemical mediators, and abnormal inflammatory response. All these mechanisms eventually produce ischemic or hemorrhagic strokes that can affect both intracerebral and subependymal areas, resulting in significant disability. The parenchymal involvement is typically characterized by degeneration, necrosis, demyelination, and reactive gliosis.
In systemic viral infections, the main neurological complications are thrombosis, ischemic and hemorrhagic stroke, meningeal hemorrhage, encephalitis, and acute myelitis, characterized by demyelination, reactive gliosis, and edema [19,20,21,22,23], as well as in bacterial systemic diseases [24], complications of cancer [25], and in some immune-based diseases [26]. Of note, these conditions are included among the differential diagnoses between secondary and primary vasculitis of the nervous system and other systemic dysmetabolic diseases that can lead to stroke or degenerative diseases of the nervous system [2••, 27, 28] (Table 2). When the same mechanisms act systemically toward thromboembolic damage or vasoconstriction at the level of the brain, a determining role of the immune system responsible for secondary vasculitis and stimulation of different types of parenchymal cells can be hypothesized. Therefore, in case of systemic vasculitis, IgG, IgM, IgA, ANCA, C3-C4-C5, and fibrinogen on routinary biopsy specimen are a valuable aid in diagnosis and typing of these conditions. In Table 3 we summarized our complete blood laboratory workup.
Compared to diffuse vasculitis secondarily affecting the brain, PACNS is much less frequent, but they are often fatal, have an unclear etiopathogenesis, and typically involve the CNS without any other systemic involvement [29, 30]. Moreover, unlike systemic and secondary vasculitis, SOV of the brain shows normal blood tests or non-specific findings, such as anemia, leukocytosis, and increased inflammatory markers, and the same holds true for the analysis of cerebrospinal fluid (CSF). CSF findings usually include a mild lymphomonocytic pleocytosis or protein elevation in more than 90% of patients [2••, 31, 32]. CSF tests in suspected PACNS should search for systemic inflammation including specific antibodies but must also exclude important differential diagnoses. In Table 4 we summarized our complete CSF laboratory workup.
Neuroradiological studies are also important for a correct management and demonstrate cerebral involvement: brain MRI studies (with T1- and T2-weighted imaging, FLAIR, gradient-echo, diffusion weighted imaging, apparent diffusion coefficient, high-resolution T1-weighted imaging after contrast injection with fat suppression and flow compensation to evaluate possible wall enhancement in cerebral arteries, gadolinium-enhanced imaging), spinal cord MRI studies (with T1- and T2-weighted imaging of the entire spinal cord, as well as T1-and T2-weighted axial imaging and gadolinium-enhanced imaging of cord lesions), and vascular imaging studies (conventional digital subtraction angiography [DSA], CTA, and MRA) are the gold standard for the demonstration of vessel stenoses or aneurysms. They should be obtained at presentation, at neurological deteriorations, and during follow-up and should be systematically performed, analyzed, and compared. MRI studies should be also assessed for appropriate clinical correlations, both at presentation and during follow-up. In cerebral vasculitis, both ischemic and hemorrhagic lesions of different ages as well as findings of focal or diffuse inflammation are observed.
However, in SOV cases, frequently only the brain biopsy, although invasive, can provide evidence of diagnosis and exclude other conditions. Nevertheless, a negative biopsy does not fully exclude a PACNS due to the segmental and transmural characteristic of vessel inflammation [27, 30, 33, 34], also considering that PACNS mainly affects small- and medium-sized arteries (serving cortical and juxtacortical areas), spinal cord, and meninges. Among the complications of the vessel itself, aneurysms can develop, as well as stenosis and thrombosis, along with related histological findings [35,36,37,38,39,40,41]. As stated, unlike systemic or secondary forms, PACNS vasculitis does not have a clear etiopathogenesis. This is also due to the paucity of pathological data and the limitations of biopsy (Table 5) [6••, 42,43,44,45,46,47,48,49,50,51, 52•]. Although the role of infectious agents has been hypothesized, given the prevalent granulomatous findings observed histologically, the autoimmune pathway remains the most convincing hypothesis. Literature also indicates a higher rate of ischemic events than intracranial hemorrhages in PACNS, as it occurs in secondary vasculitis [35,36,37,38,39,40,41]. Overall, these manifestations can be explained by direct damage to the vessel, along with the inflammatory phenomena, increase in coagulability due to pro-inflammatory cytokines on endothelial cells, and changes in vasomotor tone. However, unlike systemic vasculitis, the immunological mechanisms that may be involved in PACNS are still not known. Indeed, while direct endothelial damage mediated by antibodies or an indirect damage from the immune system and mediators of inflammation are relevant and known in secondary vasculitis, as well as the response induced by the deposition of immune complexes in the vascular walls with complement and coagulation activation, this evidence is currently lacking in PACNS, although the occurrence of p-ANCA antibodies in CNS vasculitis has been described [53]. What is probably known is that CNS vasculitis is histologically independent entities from systemic vasculitis and, thus, is likely not part of a spectrum within the temporal evolution of the same vasculitis process [6••]. Lastly, a doubtful but intriguing point regards the presence of amyloid in some histological types of vasculitis. This might merely represent the product of vasculitis from chronic alteration of vascular permeability and deposit in damaged walls, but, on the other hand, this can trigger a further inflammatory/autoimmune response and immune-mediated CD20 + lymphocyte subpopulation activity [54•]. However, the main granulomatous nature of the histological lesions suggests the possibility of a T-cell-mediated immunological response (T helper-mediated) in response to cytokines produced by dendritic cells, macrophages, and natural killer (NK) cells. The T-cell-mediated response induces the production and release of mediators (such as IFN-γ, TNF-α, TNF-β, IL-2–6-7–8) that could promote the vasculitis process through a proinflammatory action, a stimulation and production of nitric oxide, and the expression of HLA class I and class II (HLA-I and HLA-II) on endothelial cells, ependymal cells, microglia, and perivascular cells [55].
Clinical Assessment Tools and Diagnostic Criteria
Current therapy has transformed the prognosis of the PANCS, and this improvement in prognosis highlighted the need for better methods of monitoring disease activity and recording the damage that occurs during the disease. While imaging and laboratory testing including histology are important aspects of diagnosis, they are of limited value in assessing response to therapy or subsequent disease course. Unfortunately, there are not well-validated clinical indices that record disease activity, damage, and the extent of disease in PANCS. Clinical assessment remains the gold standard for evaluating disease progress but requires regular training to ensure standardization. Developing standardized and validated clinical methods to quantify disease activity and damage can allow a detailed assessment of the patient’s response to therapy and provide an essential tool for insuring uniformity of patient monitoring for clinical management as well as an essential tool in conducting clinical trials and studies in PANCS (Table 6). The development of biomarkers in future may produce a more accurate description of disease and identify potential targets for therapy as well as predictors of response to drugs. However, the majority of available biomarkers is not specific for vasculitis of the nervous system [57••].
The diagnosis of PANCS is often difficult. There are neither specific clinical features nor a classical clinical course, and no blood or imaging investigations that can confirm the diagnosis. The actual diagnostic criteria are usually based on clinical experience and the evidence from published works. Here we describe a diagnostic approach based on the importance of obtaining tissue, and dividing cases into ‘definite’ PANCS, when tissue proof is available, and ‘possible,’ when it is not. Our suggested diagnostic criteria in adults and children are summarized in Table 7.
Adults
Definite PACNS requires a confirmation through the brain biopsy with specific histopathological patterns [10]. Probable PACNS is so-called when brain biopsy is lacking, and its hypothesis is supported by Digital Subtraction Angiography (DSA) and/or MRI/intracranial MR-Angiography (MRA) and/or intracranial CT-Angiography (CTA) and/or CSF analysis which may reveal lymphocytic pleocytosis and elevated levels of protein [58]. The imaging study of the CNS vessels should demonstrate alternating areas of stenosis, ectasia, or both in more than one vascular bed, unexplained by intracranial arteriosclerosis, reversible cerebral vasoconstriction syndromes (RVCS), or other diseases that may mimic PACNS [7, 25, 28, 37].
Children
In pediatric subjects (cPACNS), the diagnostic criteria [47, 59, 60] mandate a newly acquired neurological deficit with histological (definite PACNS) or angiographic (probable PACNS) evidence of CNS vasculitis in the absence of a systemic condition that could explain these findings. Specific subset of angiographically defined cPACNS (large- and medium-sized vessels) are reported:
Principles of Management and Therapy
-
1. Vasculitis is often severe diseases with potential permanent disability due to tissue ischemia and infarction and a possible fatal outcome that require prompt recognition and therapy
Vasculitis typically has an insidious onset with slow progression, though rarely occurs in acute forms. The multifocal characteristic of the lesions clinically determines non-specific and not always superimposable symptomatology in different patients. Indeed, the clinical presentation is often variable in the absence of pathognomonic signs and symptoms, although headache (60%), cognitive disturbances (50%), focal neurological deficits (mostly stroke or hemorrhage), convulsions, encephalopathy [61,62,63], and isolated myelopathies have been most described [56].
Overall, however, symptoms and signs can be within a wide spectrum, with focal lesion symptoms less frequent than those caused by multifocal lesions due to segmental or diffuse vessel involvement. For these reasons, the diagnosis of CNS vasculitis or a PACNS is rather complex, also given the lack of validated criteria, and requires a multidisciplinary approach that includes clinical features, laboratory investigations, imaging findings, and biopsy in selected cases.
-
2. Some forms of vasculitis are associated with severe neurological complications that significantly affects patient’s prognosis
Regardless of the primary or secondary form, vasculitis is a determining factor for neurological complications. The subtle inflammatory process that typically affects vessels within different regions of the CNS or PNS is responsible for severe and often unpredictable complications, sometimes with poor prognosis. Brain complications from both systemic and primary vasculitis are basically represented by acute vascular events. Thrombosis, ischemic stroke, and hemorrhagic lesions, with tissue degeneration and inflammatory involvement of other structures (such as leptomeninges), are responsible for several morphological and functional deficits, causing death in severely affected cases. These neurovascular conditions, though common, remain non-specific, thus opening a wide spectrum of differential diagnoses.
-
3. Clinical course of vasculitis can range from fulminant to very mild forms and may fluctuate in clinical signs
As a rule, clinical presentation of both primary and secondary vasculitis of the brain is very heterogeneous, including focal or multifocal deficits. Neurological symptoms depend on the affected vascular district, the extent of the process, and the duration of the disease. Therefore, symptoms can be non-specific, subtle, or overt, and the course may be subacute or chronic, up to some striking manifestations, often including neurological deficits from vascular and non-vascular complications [61].
Patients with PACNS typically present with a gradual and mild headache, often associated with signs and symptoms of cognitive impairment. Only later they can develop focal neurological deficits. The clinical course may be rapidly progressive over few days or weeks, or can be insidious over many months, with prolonged periods of stability. Conversely, patients with granulomatous angiitis present with headache, cognitive changes, and increased CSF protein content, with or without pleocytosis [62, 63]. Hemiparesis, quadriparesis, and lethargy can frequently develop and need prompt diagnosis due to the poor prognosis of their natural history.
-
4. There is a likelihood of excess morbidity and mortality due to missing diagnosis and therefore undertreatment
When a striking event of stroke or hemorrhage does not occur, the symptomatic spectrum is particularly difficult to assess and the diagnosis of vasculitis with cerebral involvement is very challenging. This is in part due to the insidious onset of symptoms and broad differential diagnosis. Diagnosis is especially challenging in SOV which do not present with the same serologic findings or constellation of clinical symptoms as seen with systemic vasculitis. Failure or delay in the diagnosis and treatment is responsible for an excess of morbidity and mortality. In these cases, the neurological damage can be by permanent.
-
5. Histopathological confirmation of vasculitis is essential for accurate diagnosis, as well as a correct methodology for achieving it
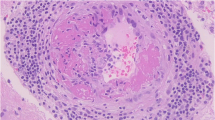
Brain biopsy remains the gold standard in diagnosing PACNS [44, 56]. Of note, the biopsy is not only useful for confirming a vasculitis, but also for excluding other disorders that can mimic a vasculitis process [2••, 37]. Since small vessels, arterioles, and capillaries are involved in primary CNS vasculitis, transmural and segmental signs of inflammation are the main diagnostic histological findings. A morphological diagnosis of vasculitis, indeed, requires the occurrence of transmural vasculocentric inflammation with vascular wall damage, with or without fibrinoid necrosis [56]. The morphological evaluation of the damaged vessel wall, with the assessment of lymphocyte populations, multinucleated giant cells, and transmural fibrinoid necrosis are all key elements for the diagnosis of a cerebral vasculitis. The next step is the typing of the PACNS and the exclusion of differential diagnoses, which basically include secondary vasculitis (such as those due to degenerative, infectious, and tumor lesions) [62]. Some authors have also confirmed the results of the biopsy in vivo with the results obtained from autopsies [45, 52•, 64]. In both cases, PACNS histologically presents as granulomatous, necrotizing, or lymphocytic, although mixed morphologic types often occur. The histological findings of vasculitis are often associated with evidence of ischemic stroke due to stenosis or total obstruction of the damaged vessel and alterations in coagulation, as well as of intraparenchymal/submeningeal hemorrhage when the damaged vessel ruptures, or of a post-thrombotic consequence as it happens in ischemic or hemorrhagic stroke due to other diseases. Histological patterns of this vasculitis are reported below [45]. Granulomatous vasculitis is the most common pattern with infiltration of the vascular wall from T lymphocytes and macrophages, which subsequently organize into granulomas with giant cells (Fig. 2A). Damage to the wall with fragmentation of the elastic fibers and reworking with proliferation of the intima causes subsequent fibrosis and vessel occlusion. Differential diagnoses include all granulomatous diseases that produce sarcoid-like granulomas, including sarcoidosis [65]. In these cases, the exclusive localization of the brain and the absence of systemic manifestations or laboratory data may be helpful. The deposition of amyloid can also be associated with the granulomatous histotype [43, 45, 66]. Lymphocytic vasculitis is characterized by predominantly lymphocytic inflammation, with a small amount of plasma cells affecting the entire thickness of the vascular wall (Fig. 2B,C,D). Indeed, some authors describe a clear vasculocentric transmural lymphocytic infiltration involving the vessels with CD3-positive (CD4 + and CD8 +) and CD20-positive lymphocytes, along with other minor elements such as macrophages (as in our case series). The main differential diagnosis of this histotype is leukodystrophy [42,43,44,45, 47, 48, 50, 52•, 56, 59, 60]. Necrotizing vasculitis is less common and is characterized by transmural fibrinoid necrosis that mainly affects small muscular arteries with rupture of the internal elastic lamina and rupture of the vessel or aneurysmal dilations. In addition, an inflammatory lymphomonocyte infiltrate may be present (Fig. 1E,F) [43, 45, 46, 49, 67]. Histological findings are similar to those observed in polyarteritis nodosa with transmural fibrinoid necrosis and may indicate the occurrence of similar underlying pathological mechanisms [5, 30]. Fibrinoid necrosis of the vessel wall may also predispose to vessel rupture, thus resulting in intracranial hemorrhage. A vasculocentric inflammatory infiltrate is considered diagnostic to primary vasculitis when other morphological aspects and clinical signs indicative of other pathologies are absent. Indeed, these infiltrates can be observed also in association with other infectious and neoplastic diseases, as well as in primary and secondary vasculopathies. For instance, the differential diagnosis with lymphomas needs to evaluate the atypia of lymphoid cells and the expression of specific markers. Similarly, the necrotizing finding can also be present in some infectious diseases with both parenchymal and vascular involvement, such as atypical mycobacteriosis [68, 69]. Finally, although the brain biopsy has a consolidated role in the oncological pathology of the CNS, it is worth to mention that despite the standardization brain biopsy, this approach is still poorly practiced and affected by some limitations, especially in non-neoplastic diseases [70]. First, the brain biopsy is a highly specialistic invasive procedure that can be complicated by intracerebral hemorrhage, seizures, transient altered mental status, cerebral infarction, and CSF leak also when performed by trained operators and in specialized centers. Second, the diagnostic accuracy of brain biopsy remains far from being pathognomonic, and the histological diagnosis is generally limited from the characteristics of segmentary and multifocality of most of vasculitis processes. Biopsy may also show negative or non-specific findings, which may be due to prolonged time to biopsy, non-lesioned biopsy, prior steroid treatment, or inadequate specimen sampling. Also, a guided biopsy does not always provide an adequate and comprehensive sample of the vascular lesion, and, on the other hand, a negative biopsy does not fully rule out a diagnosis of vasculitis. Finally, the pathologist’s training and expertise in the processing of samples and search for specific markers is of crucial importance, as well as the collaboration with neurosurgeons in the choice, timing, and modality of sampling. The diagnosis of some vasculitis requires specific methods, such as immunofluorescence for the identification of IgG, IgM, IgG, complement, and fibrinogen. The risk of negative neurological outcomes, the poor negative predictive value of the biopsy, and reliance on the clinician’s judgment make the diagnosis of vasculitis with a brain biopsy challenging. A correct indication posed by clinicians, together with an accurate medical history and clinical examination, simplifies and directs the pathologist’s evaluation toward the method of choice and, hopefully, a definitive diagnosis. The motto is that “no biopsy of any organ is foreign or independent of the rest of the body.” Fig. 2 shows the main vasculitis and immunohistochemical histological patterns. Immunohistochemistry with anti-CD3, anti-CD8, and anti-CD68 was performed on paraffinized brain samples using the detection method described in the literature [71, 72].
-
6. Empiric trials with immunosuppressive and immunomodulating therapy should never be considered as a substitute for a confirmed diagnosis of vasculitis

Main anatomopathological findings from authors’ autoptic studies. A: Granulomatous pattern: arrows indicate CD-68 + macrophages arranged around the vessel and with initial aggregation in a sarcoid-like granuloma (× 40 magnification). B-D: Lymphocytic pattern: arrows indicate T lymphocytes (CD3 +) around vessels with different caliber (B and D: CD3 + × 40 magnification; C: CD8 + × 20 magnification). E–F: Necrotizing pattern: the black box indicates a particular of degeneration and necrosis of the vessel wall with the presence of lymphocytes (H&E stain × 40). Images from G. Mansueto’s case studies
Physicians treating vasculitis must choose the sequence and combination of available immunosuppressant and immunomodulating therapies to induce and sustain remission and treat relapses, recognizing the possible beneficial and adverse effects. The standard of care for the treatment of vasculitis, notably ANCA-associated vasculitis (AAV), has been evolving in response to many factors [73]. First, the steady influx of data from multicenter, national, and international collaborative, evidence-based randomized clinical trials (RCT) and observational cohorts in adult vasculitis. Second, while many vasculitis still lacks well-validated measures of disease activity or state for use in clinical trials, there have nonetheless been advances in standardized approaches to conducting clinical trials as advocated by the EULAR and its collaborators worldwide [74]. Third, the influence of gene-wide association studies (GWAS) and biobank data such as the UK Biobank to elucidate risk gene loci, single nucleotide polymorphism (SNP) and human HLA polymorphisms in disease clusters and population cohorts [75]. Such inherited and environmental factors, gene–gene interactions, epigenetic factors, and other influences upon the immunopathogenesis of vasculitis have had important theoretical importance for the performance of RCTs in vasculitis subtypes, as well as relevance for screening studies and timing of therapy.
-
7. Treatments are initially guided toward stabilization of the blood–brain barrier (BBB), followed by maintenance immunosuppressive therapy directed both at the humoral and cell-mediated autoimmune and inflammatory mechanisms
The goal of treatment should be the rapid control of the inflammatory response and the stabilization of the BBB while protecting the brain from further injuries. Methylprednisolone has been the first-line agent administered intravenously at a dose of 30 mg/kg/d to a maximum of 1 g/d for 3 to 5 days, followed by 1 to 2 mg/kg/d of oral corticosteroids (CS) to a maximum of 60 mg/d of prednisone [76]. After stabilization, the choice of immunosuppressive treatment is directed toward the primary inflammatory or vasculitis process. Induction therapy with CS and pulse cyclophosphamide cyclosporine (CYC), followed by maintenance therapy with azathioprine (AZA) or mycophenolate mofetil (MMF), has been recommended in cPACNS. In Fig. 3 is summarized our proposed treatment algorithm for adult PACNS. Children with SV-cPACNS were treated in an open-label study with CYC in doses of 500 to 750 mg/m2 as monthly infusions for 6 months, followed with maintenance therapy with AZA of 1 mg/kg/d and a target dose of 2 to 3 mg/kg/d, and MMF at titrated doses of 800 to 1200 mg/m2/d followed for up to 24 months using pediatric stroke outcome measures (PSOM). Among 19 such patients, 13 completed 24 months of follow-up, of whom 9 had a good neurological outcome by PSOM scoring, 8 experienced disease flares, and 4 achieved remissions of disease. MMF was more effective than AZA. Rituximab may be appropriate therapy at doses of 375 mg/m2 for 4 consecutive weeks or 500 mg/m2 weekly for 2 weeks in cPACNS, as was recently reported in SV-cPACNS [77]. Figure 4, panel A and B summarizes the suggested treatment algorithms for children-PACNS, in angiography-positive (large- and medium-sized vessels) and angiography negative (small-sized vessel), as well as for adult PACNS [78, 79].

The suggested treatment algorithms for adult-PACNS

Concluding Remarks and Future Perspectives
Primary CNS vasculitis is a challenging condition due to the broad clinical manifestations and variable specificity and sensitivity of both laboratory and imaging diagnostic tools, with brain biopsy to be performed whenever possible. Large vessel primary CNS vasculitis should be managed with steroids and CYC, whereas small vessel forms with steroids alone. The most important differential diagnosis is the RCVS that may occur without thunderclap headache, whereas other differential diagnoses include secondary CNS vasculitis and non-inflammatory vasculopathies. Overall, our understanding of PACNS and the characterization of its range and subsets has been advanced, although we still need to clarify methods of diagnosis and management. Biomarker and outcome investigations might identify risk factors for an aggressive course, thus leading to treatment tailored to disease severity. Effectiveness and safety profiles of intravenous pulse and oral immunosuppressive treatments, and clarification of which patients need them at disease onset, need to be established yet. Other drug types should be considered as substitutes for immunosuppressors in selected subgroups. Moreover, since primary PACNS is uncommon, an international collaborative registry and repositories of biological specimens would help with the development of standardized classification and diagnostic criteria, help to define homogeneous patient groups for randomized clinical trials, and might aid clinicians in differentiation of primary CNS vasculitis from its much more common mimickers.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, Flores-Suarez LF, Gross WL, Guillevin L, Hagen EC, et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65:1–11. https://doi.org/10.1002/art.37715.
•• Dutra LA, de Souza AW, Grinberg-Dias G, Barsottini OG, Appenzeller S. Central nervous system vasculitis in adults: An update. Autoimmun Rev. 2017;16:123–31. https://doi.org/10.1016/j.autrev.2016.12.001. A well written review on the diagnosis and management of PCNSV, secondary vasculitis of CNS and RCVS.
Beachy N, Satkowiak K, Gwathmey KG. Vasculitic Neuropathies. Semin Neurol. 2019;39:608–19. https://doi.org/10.1055/s-0039-1688990.
Gwathmey KG, Tracy JA, Dyck PJB. Peripheral Nerve Vasculitis: Classification and Disease Associations. Neurol Clin. 2019;37:303–33. https://doi.org/10.1016/j.ncl.2019.01.013.
Miller DV, Salvarani C, Hunder GG, Brown RD, Parisi JE, Christianson TJ, Giannini C. Biopsy findings in primary angiitis of the central nervous system. Am J Surg Pathol. 2009;33:35–43. https://doi.org/10.1097/PAS.0b013e318181e097.
•• Chang HB, Gao M, Zhang JN, Cao WD, Guo SL, Wang P, Cheng G, Zhao HL. Retrospective Analysis of 28 Cases Confirmed for Primary Angiitis of the Central Nervous System by Biopsy. J Stroke Cerebrovasc Dis. 2020;29: 105400. https://doi.org/10.1016/j.jstrokecerebrovasdis.2020.105400. It suggests some good sense guidelines in diagnosis and treatment PACNS patients. An early role of biopsy is suggested together with an aggressive treatment regimen.
Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015;7: a020412. https://doi.org/10.1101/cshperspect.a020412.
Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19:1584–96. https://doi.org/10.1038/nm.3407.
Younger DS. The Blood-Brain Barrier: Implications for Vasculitis. Neurol Clin. 2019;37:235–48. https://doi.org/10.1016/j.ncl.2019.01.009.
Carmona FD, Lopez-Mejias R, Marquez A, Martin J, Gonzalez-Gay MA. Genetic Basis of Vasculitides with Neurologic Involvement. Neurol Clin. 2019;37:219–34. https://doi.org/10.1016/j.ncl.2019.01.006.
Rahmattulla C, Mooyaart AL, van Hooven D, Schoones JW, Bruijn JA, Dekkers OM, Bajema IM. Genetic variants in ANCA-associated vasculitis: a meta-analysis. Ann Rheum Dis. 2016;75:1687–92. https://doi.org/10.1136/annrheumdis-2015-207601.
Shavit E, Alavi A, Sibbald RG. Vasculitis-What Do We Have to Know? A Review of Literature. Int J Low Extrem Wounds. 2018;17:218–26. https://doi.org/10.1177/1534734618804982.
Ozen S, Batu ED. Vasculitis Pathogenesis: Can We Talk About Precision Medicine? Front Immunol. 1892;2018:9. https://doi.org/10.3389/fimmu.2018.01892.
Yates M, Watts R. ANCA-associated vasculitis. Clin Med (Lond). 2017;17:60–4. https://doi.org/10.7861/clinmedicine.17-1-60.
Kallenberg CG, Heeringa P. Pathogenesis of vasculitis. Lupus. 1998;7:280–4. https://doi.org/10.1191/096120398678920109.
Younger DS. Eleven Themes in the History of Systemic and Nervous System Vasculitides. Neurol Clin. 2019;37:149–70. https://doi.org/10.1016/j.ncl.2019.01.001.
Timmons GM, Rempe T, Bevins EA, Goodwill V, Miner A, Kavanaugh A, Ritter M, Graves JS. CNS Lymphocytic Vasculitis in a Young Woman With COVID-19 Infection. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1048. https://doi.org/10.1212/NXI.0000000000001048.
Kirschenbaum D, Imbach LL, Rushing EJ, Frauenknecht KBM, Gascho D, Ineichen BV, Keller E, Kohler S, Lichtblau M, Reimann RR, et al. Intracerebral endotheliitis and microbleeds are neuropathological features of COVID-19. Neuropathol Appl Neurobiol. 2021;47:454–9. https://doi.org/10.1111/nan.12677.
Elkind MSV, Boehme AK, Smith CJ, Meisel A, Buckwalter MS. Infection as a Stroke Risk Factor and Determinant of Outcome After Stroke. Stroke. 2020;51:3156–68. https://doi.org/10.1161/STROKEAHA.120.030429.
Yachou Y, El Idrissi A, Belapasov V, Ait Benali S. Neuroinvasion, neurotropic, and neuroinflammatory events of SARS-CoV-2: understanding the neurological manifestations in COVID-19 patients. Neurol Sci. 2020;41:2657–69. https://doi.org/10.1007/s10072-020-04575-3.
Fisicaro F, Di Napoli M, Liberto A, Fanella M, Di Stasio F, Pennisi M, Bella R, Lanza G, Mansueto G. Neurological Sequelae in Patients with COVID-19: A Histopathological Perspective. Int J Environ Res Public Health. 2021;18(4):1415. https://doi.org/10.3390/ijerph18041415.
Bradshaw MJ, Venkatesan A. Herpes Simplex Virus-1 Encephalitis in Adults: Pathophysiology, Diagnosis, and Management. Neurotherapeutics. 2016;13:493–508. https://doi.org/10.1007/s13311-016-0433-7.
Mansueto G. COVID-19: Brief check through the pathologist’s eye (autopsy archive). Pathol Res Pract. 2020;216: 153195. https://doi.org/10.1016/j.prp.2020.153195.
Pagliano P, Spera AM, Ascione T, Esposito S. Infections causing stroke or stroke-like syndromes. Infection. 2020;48:323–32. https://doi.org/10.1007/s15010-020-01415-6.
Dardiotis E, Aloizou AM, Markoula S, Siokas V, Tsarouhas K, Tzanakakis G, Libra M, Kyritsis AP, Brotis AG, Aschner M, et al. Cancer-associated stroke: Pathophysiology, detection and management (Review). Int J Oncol. 2019;54:779–96. https://doi.org/10.3892/ijo.2019.4669.
Kidd DP. Neurological complications of Behcet’s syndrome. J Neurol. 2017;264:2178–83. https://doi.org/10.1007/s00415-017-8436-9.
Salvarani C, Brown RD Jr, Christianson TJH, Huston J 3rd, Morris JM, Giannini C, Hunder GG. Primary central nervous system vasculitis mimicking brain tumor: Comprehensive analysis of 13 cases from a single institutional cohort of 191 cases. J Autoimmun. 2019;97:22–8. https://doi.org/10.1016/j.jaut.2018.10.001.
de Boysson H, Boulouis G, Dequatre N, Godard S, Neel A, Arquizan C, Detante O, Bloch-Queyrat C, Zuber M, Touze E, et al. Tumor-Like Presentation of Primary Angiitis of the Central Nervous System. Stroke. 2016;47:2401–4. https://doi.org/10.1161/STROKEAHA.116.013917.
Jacob S. Primary angiitis of the central nervous system (PACNS) - a rare and serious, but treatable entity. Neurol India. 2019;67:942–4. https://doi.org/10.4103/0028-3886.263180.
Giannini C, Salvarani C, Hunder G, Brown RD. Primary central nervous system vasculitis: pathology and mechanisms. Acta Neuropathol. 2012;123:759–72. https://doi.org/10.1007/s00401-012-0973-9.
Salvarani C, Brown RD Jr, Hunder GG. Adult primary central nervous system vasculitis. Lancet. 2012;380:767–77. https://doi.org/10.1016/S0140-6736(12)60069-5.
Mandal J, Chung SA. Primary Angiitis of the Central Nervous System. Rheum Dis Clin North Am. 2017;43:503–18. https://doi.org/10.1016/j.rdc.2017.06.001.
Torres J, Loomis C, Cucchiara B, Smith M, Messe S. Diagnostic Yield and Safety of Brain Biopsy for Suspected Primary Central Nervous System Angiitis. Stroke. 2016;47:2127–9. https://doi.org/10.1161/STROKEAHA.116.013874.
Raghavan A, Wright JM, Huang Wright C, Shammassian BH, Onyewadume L, Momotaz H, Burant CJ, Sajatovic M, Carandang R, Furlan A, et al. Concordance of angiography and cerebral biopsy results for suspected primary central nervous system vasculitis: A multi-center retrospective review. Clin Neurol Neurosurg. 2019;185:105482. https://doi.org/10.1016/j.clineuro.2019.105482.
Peng LJ, Qian HR, Mao LL, Xia DY, Qi XK. A clinical analysis of 5 patients with infratentorial primary angiitis of central nervous system. Zhonghua Nei Ke Za Zhi. 2017;56:284–9. https://doi.org/10.3760/cma.j.issn.0578-1426.2017.04.009.
Soun JE, Song JW, Romero JM, Schaefer PW. Central Nervous System Vasculopathies. Radiol Clin North Am. 2019;57:1117–31. https://doi.org/10.1016/j.rcl.2019.07.005.
Cho TA, Jones A. CNS vasculopathies: Challenging mimickers of primary angiitis of the central nervous system. Best Pract Res Clin Rheumatol. 2020;34: 101569. https://doi.org/10.1016/j.berh.2020.101569.
Kalashnikova LA, Dobrynina LA, Legenko MS. Primary central nervous system vasculitis. Zh Nevrol Psikhiatr Im S S Korsakova. 2019;119:113–23. https://doi.org/10.17116/jnevro2019119081113.
Lucke M, Hajj-Ali RA. Advances in primary angiitis of the central nervous system. Curr Cardiol Rep. 2014;16:533. https://doi.org/10.1007/s11886-014-0533-0.
Stanley E, Murphy S, Kavanagh E. Letter by Stanley et al Regarding Article, “Primary Angiitis of the Central Nervous System Magnetic Resonance Imaging Spectrum of Parenchymal, Meningeal, and Vascular Lesions at Baseline”. Stroke. 2017;48: e178. https://doi.org/10.1161/STROKEAHA.117.017518.
Boulouis G, de Boysson H, Naggara O. Response by Boulouis et al to Letter Regarding Article, “Primary Angiitis of the Central Nervous System: Magnetic Resonance Imaging Spectrum of Parenchymal, Meningeal, and Vascular Lesions at Baseline”. Stroke. 2017;48: e179. https://doi.org/10.1161/STROKEAHA.117.017609.
Wang LJ, Kong DZ, Guo ZN, Zhang FL, Zhou HW, Yang Y. Study on the Clinical, Imaging, and Pathological Characteristics of 18 Cases with Primary Central Nervous System Vasculitis. J Stroke Cerebrovasc Dis. 2019;28:920–8. https://doi.org/10.1016/j.jstrokecerebrovasdis.2018.12.007.
Sundaram S, Menon D, Khatri P, Sreedharan SE, Jayadevan ER, Sarma P, Pagnoux C, Sylaja PN. Primary angiitis of the central nervous system: Clinical profiles and outcomes of 45 patients. Neurol India. 2019;67:105–12. https://doi.org/10.4103/0028-3886.253578.
Caputi L, Erbetta A, Marucci G, Pareyson D, Eoli M, Servida M, Parati E, Salsano E. Biopsy-proven primary angiitis of the central nervous system mimicking leukodystrophy: A case report and review of the literature. J Clin Neurosci. 2019;64:42–4. https://doi.org/10.1016/j.jocn.2019.03.021.
Salvarani C, Brown RD Jr, Christianson T, Miller DV, Giannini C, Huston J 3rd, Hunder GG. An update of the Mayo Clinic cohort of patients with adult primary central nervous system vasculitis: description of 163 patients. Medicine (Baltimore). 2015;94: e738. https://doi.org/10.1097/MD.0000000000000738.
Takatsu H, Komatsu T, Fukasawa N, Fukuda T, Iguchi Y. Spontaneously changing MRI findings of primary central nervous system vasculitis: A case report. J Clin Neurosci. 2021;83:125–7. https://doi.org/10.1016/j.jocn.2020.11.012.
Denny AM, Das SK. A case of central nervous system vasculitis presenting as a mass-like lesion. Childs Nerv Syst. 2019;35:1223–6. https://doi.org/10.1007/s00381-018-04034-7.
Wilson N, Pohl D, Michaud J, Doja A, Miller E. MRI and clinicopathological correlation of childhood primary central nervous system angiitis. Clin Radiol. 2016;71:1160–7. https://doi.org/10.1016/j.crad.2016.07.013.
Spence S, Ng D, Casault C. Atypical presentation of fulminant primary central nervous system angiitis. J Neuroimmunol. 2019;330:1–4. https://doi.org/10.1016/j.jneuroim.2019.01.019.
Benson CE, Knezevic A, Lynch SC. Primary Central Nervous System Vasculitis With Optic Nerve Involvement. J Neuroophthalmol. 2016;36:174–7. https://doi.org/10.1097/WNO.0000000000000328.
Han X, Pang Z, Wang Z, Xu S, Lin Y. A case of primary angiitis of the central nervous system presenting with diffuse cerebral microbleeds and recurrent intracranial hemorrhage. Neurol Sci. 2019;40:417–9. https://doi.org/10.1007/s10072-018-3595-8.
• Van Rooij JL, Rutgers DR, Spliet WG, Frijns CJ. Vessel wall enhancement on MRI in the diagnosis of primary central nervous system vasculitis. Int J Stroke. 2018;13:NP24–7. https://doi.org/10.1177/1747493018789276. The use of MRI imaging in differential diagnosis.
Harland TA, Seinfeld J, Cava LF, Neumann RT, Roark C, Kumpe D, Case D. Anti-neutrophil cytoplasmic antibody associated central nervous system vasculitis with brain and spinal cord subarachnoid hemorrhage: A rare case report and review of the literature. J Clin Neurosci. 2019;62:253–5. https://doi.org/10.1016/j.jocn.2018.12.001.
• Strunk D, Schulte-Mecklenbeck A, Golombeck KS, Meyer Zu Horste G, Melzer N, Beuker C, Schmidt A, Wiendl H, Meuth SG, Gross CC, et al. Immune cell profiling in the cerebrospinal fluid of patients with primary angiitis of the central nervous system reflects the heterogeneity of the disease. J Neuroimmunol. 2018;321:109–16. https://doi.org/10.1016/j.jneuroim.2018.06.004. The biological basis for understanding cerebrospinal fluid resulta in pazients with PACNS.
Kraemer M, Becker J, Horn PA, Schwitalla JC, Keyvani K, Metz I, Wegner C, Bruck W, Schlamann M, Heinemann FM, et al. Association of primary central nervous system vasculitis with the presence of specific human leucocyte antigen gene variant. Clin Neurol Neurosurg. 2017;160:137–41. https://doi.org/10.1016/j.clineuro.2017.06.009.
Suthiphosuwan S, Bharatha A, Hsu CC, Lin AW, Maloney JA, Munoz DG, Palmer CA, Osborn AG. Tumefactive Primary Central Nervous System Vasculitis: Imaging Findings of a Rare and Underrecognized Neuroinflammatory Disease. AJNR Am J Neuroradiol. 2020;41:2075–81. https://doi.org/10.3174/ajnr.A6736.
•• Strunk D, Schmidt-Pogoda A, Beuker C, Milles LS, Korsukewitz C, Meuth SG, Minnerup J. Biomarkers in Vasculitides of the Nervous System. Front Neurol. 2019;10:591. https://doi.org/10.3389/fneur.2019.00591. Inflammatory markers, antibodies, cerebrospinal fluid analysis, imaging, and biopsy, are all insufficient to meet all current challenges in diagnosis of PACNS. We suggest the use of biomarkers as an approach to extend current knowledge and, ultimately, improve patient management.
Ruland T, Wolbert J, Gottschalk MG, Konig S, Schulte-Mecklenbeck A, Minnerup J, Meuth SG, Gross CC, Wiendl H, Meyer Zu Horste G. Cerebrospinal Fluid Concentrations of Neuronal Proteins Are Reduced in Primary Angiitis of the Central Nervous System. Front Neurol. 2018;9:407. https://doi.org/10.3389/fneur.2018.00407.
Deng J, Fang F, Wang XH, Ge M, He LJ, Zhang N. Small vessel-childhood primary angiitis of the central nervous system: a case report and literature review. Zhonghua Er Ke Za Zhi. 2018;56:142–7. https://doi.org/10.3760/cma.j.issn.0578-1310.2018.02.014.
Elbers J, Halliday W, Hawkins C, Hutchinson C, Benseler SM. Brain biopsy in children with primary small-vessel central nervous system vasculitis. Ann Neurol. 2010;68:602–10. https://doi.org/10.1002/ana.22075.
Smitka M, Bruck N, Engellandt K, Hahn G, Knoefler R, von der Hagen M. Clinical Perspective on Primary Angiitis of the Central Nervous System in Childhood (cPACNS). Front Pediatr. 2020;8:281. https://doi.org/10.3389/fped.2020.00281.
Byram K, Hajj-Ali RA, Calabrese L. CNS Vasculitis: an Approach to Differential Diagnosis and Management. Curr Rheumatol Rep. 2018;20:37. https://doi.org/10.1007/s11926-018-0747-z.
Younger DS. Granulomatous Angiitis: Twenty Years Later. Neurol Clin. 2019;37:267–77. https://doi.org/10.1016/j.ncl.2019.01.011.
Powers WJ. Primary angiitis of the central nervous system: diagnostic criteria. Neurol Clin. 2015;33:515–26. https://doi.org/10.1016/j.ncl.2014.12.004.
Saygin D, Jones S, Sundaram P, Calabrese LH, Messner W, Tavee JO, Hajj-Ali RA. Differentiation between neurosarcoidosis and primary central nervous system vasculitis based on demographic, cerebrospinal and imaging features. Clin Exp Rheumatol. 2020;38(Suppl 124):135–8.
de Boysson H, Zuber M, Naggara O, Neau JP, Gray F, Bousser MG, Crassard I, Touze E, Couraud PO, Kerschen P, et al. Primary angiitis of the central nervous system: description of the first fifty-two adults enrolled in the French cohort of patients with primary vasculitis of the central nervous system. Arthritis Rheumatol. 2014;66:1315–26. https://doi.org/10.1002/art.38340.
Mizuno Y, Shigeto H, Yamada T, Maeda N, Suzuki SO, Kira J. A case of primary central nervous system vasculitis diagnosed by second brain biopsy and treated successfully. Rinsho Shinkeigaku. 2016;56:186–90. https://doi.org/10.5692/clinicalneurol.cn-000847.
Mansueto G, Di Vito A, Belluomo C, Murino P, Natella V, Camastra C, Presta I, Malara N, de Rosa G, Donato G, et al. A case of intravascular large B cell lymphoma: New clinical and immunohistochemical findings. Neuropathology. 2016;36:496–503. https://doi.org/10.1111/neup.12300.
Russo CV, Sacca F, Paternoster M, Buonomo AR, Gentile I, Scotto R, Brescia Morra V, Mansueto G. Post-mortem diagnosis of invasive pulmonary aspergillosis after alemtuzumab treatment for multiple sclerosis. Mult Scler. 2020;26:123–6. https://doi.org/10.1177/1352458518813110.
Noronha C, Figueiredo G, Pinheiro C, Carvalho E, Calheiros A, Pires MM, Taipa R. Brain biopsy in suspected non-neoplastic neurological disease. Acta Neurochir (Wien). 2019;161:1139–47. https://doi.org/10.1007/s00701-019-03910-8.
Mansueto G, Costa D, Capasso E, Varavallo F, Brunitto G, Caserta R, Esposito S, Niola M, Sardu C, Marfella R, et al. The dating of thrombus organization in cases of pulmonary embolism: an autopsy study. BMC Cardiovasc Disord. 2019;19:250. https://doi.org/10.1186/s12872-019-1219-8.
Verze P, Somma A, Imbimbo C, Mansueto G, Mirone V, Insabato L. Melanotic schwannoma: a case of renal origin. Clin Genitourin Cancer. 2014;12:e37-41. https://doi.org/10.1016/j.suronc.2009.06.003.
Hoffman GS. L52. Vasculitis treatment: is it time to change the standard of care for ANCA-associated vasculitis? Presse Med. 2013;42:643–50. https://doi.org/10.1016/j.lpm.2013.01.047.
Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, Zlotogorski A, Berkun Y, Press JJ, Mukamel M, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. 2014;370:921–31. https://doi.org/10.1056/NEJMoa1307362.
Schrader ML, Hochman JS, Bulkley BH. The heart in polyarteritis nodosa: a clinicopathologic study. Am Heart J. 1985;109:1353–9. https://doi.org/10.1016/0002-8703(85)90365-5.
Beuker C, Schmidt A, Strunk D, Sporns PB, Wiendl H, Meuth SG, Minnerup J. Primary angiitis of the central nervous system: diagnosis and treatment. Ther Adv Neurol Disord. 2018;11:1756286418785071. https://doi.org/10.1177/1756286418785071.
Keenan P, Brunner J, Quan AS, Smitka M, Hahn G, Pain CE, Hafner R, Speth F, Gerstl L, Hedrich CM. Diagnosis and Treatment of Small Vessel Childhood Primary Angiitis of the Central Nervous System (sv-cPACNS): An International Survey. Front Pediatr. 2021;9: 756612. https://doi.org/10.3389/fped.2021.756612.
Kim G, Chitnis T. Child Neurology: Primary angiitis of the CNS. Neurology. 2017;89:e268–71. https://doi.org/10.1212/WNL.0000000000004718.
Cellucci T, Benseler SM. Central nervous system vasculitis in children. Curr Opin Rheumatol. 2010;22:590–7. https://doi.org/10.1097/BOR.0b013e32833c723d.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Gelsomina Mansueto, Giuseppe Lanza, Francesco Fisicaro, Danielle Alaouieh, Emily Hong, Sara Girolami, Marco Montella, Alessandro Feola, and Mario Di Napoli each declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Neurology of Systemic Diseases
Rights and permissions
About this article

Cite this article
Mansueto, G., Lanza, G., Fisicaro, F. et al. Central and Peripheral Nervous System Complications of Vasculitis Syndromes From Pathology to Bedside: Part 1—Central Nervous System. Curr Neurol Neurosci Rep 22, 47–69 (2022). https://doi.org/10.1007/s11910-022-01172-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11910-022-01172-z