
Abstract
Purpose of Review
In autonomic failure, neurogenic orthostatic hypotension (nOH) and neurogenic supine hypertension (nSH) are interrelated conditions characterized by postural blood pressure (BP) dysregulation. nOH results in a sustained BP drop upon standing, which can lead to symptoms that include lightheadedness, orthostatic dizziness, presyncope, and syncope. nSH is characterized by elevated BP when supine and, although often asymptomatic, may increase long-term cardiovascular and cerebrovascular risk. This article reviews the pathophysiology and clinical characteristics of nOH and nSH, and describes the management of patients with both nOH and nSH.
Recent Findings
Pressor medications required to treat the symptoms of nOH also increase the risk of nSH. Because nOH and nSH are hemodynamically opposed, therapies to treat one condition may exacerbate the other. The management of patients with nOH who also have nSH can be challenging and requires an individualized approach to balance the short- and long-term risks associated with these conditions.
Summary
Approaches to manage neurogenic BP dysregulation include nonpharmacologic approaches and pharmacologic treatments. A stepwise treatment approach is presented to help guide neurologists in managing patients with both nOH and nSH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The development of hemodynamic abnormalities often results as a consequence of the autonomic failure that occurs in patients with Parkinson disease (PD) and other neurodegenerative α-synucleinopathies (e.g., multiple system atrophy [MSA], pure autonomic failure) [1••]. One manifestation is neurogenic orthostatic hypotension (nOH), a sustained blood pressure (BP) drop upon standing. nOH is caused by the inability of the autonomic nervous system to maintain BP in response to standing due to baroreflex failure causing inadequate norepinephrine release with impaired vasoconstriction of peripheral and splanchnic vessels [2, 3]. Up to 70% of patients with nOH may also have neurogenic supine hypertension (nSH) [4, 5], which differs from essential hypertension in that individuals with nSH have increased BP when supine, may be normotensive when seated, and are frequently hypotensive when standing [1••, 6••].
Both nOH and nSH are associated with significant morbidity. Symptoms of nOH can include lightheadedness, orthostatic dizziness, cognitive slowing, sleepiness, presyncope, and syncope [7, 8] and may lead to an increased risk of falls, cognitive impairment, social withdrawal, and exercise intolerance [9,10,11]. nSH is commonly asymptomatic [6••]; however, it may have potential long-term cardiovascular risks including, but not limited to, left ventricular hypertrophy and stroke [12,13,14]. When managing patients with symptomatic nOH, clinicians should evaluate supine BP before and during treatment. Although concomitant management of symptomatic nOH and nSH can be inherently challenging because they are hemodynamically opposite forms of BP dysregulation and attempts to improve one condition may exacerbate the other, it is necessary for clinicians to address coexisting nOH and nSH in patients. This review considers autonomic failure pathophysiology, clinical features, BP evaluation (e.g., circadian BP variability, postural change, meals), nonpharmacologic approaches, and pharmacologic treatment strategies for the management of patients with coexisting nOH and nSH.
Pathophysiology of Blood Pressure Control Abnormalities Associated with Autonomic Nervous System Dysfunction
Both nOH and nSH occur because of autonomic nervous system (ANS) failure and the consequent inability of the cardiovascular system to adequately respond to postural change [6••, 15]. When assuming the standing position, a large blood volume shifts to the splanchnic and lower-extremity vasculature [15]. To maintain adequate perfusion of the brain and other organs, baroreceptors in the aortic arch and carotid sinus detect these changes and signal for vasoconstriction and increases in heart rate and cardiac output through a pathway involving both the central and peripheral branches of the ANS [3, 15]. The signaling for blood vessel constriction required to maintain BP in response to standing is mediated by norepinephrine release from postganglionic sympathetic nerves [3, 16]. Attenuated increases in plasma norepinephrine that occur from supine to standing positions have been demonstrated in patients with PD, MSA, and pure autonomic failure [17], and these inadequate orthostatic increases in plasma norepinephrine levels underlie the occurrence of nOH in these conditions [18]. nOH can be caused by central or peripheral degeneration and disruption of the ANS [18,19,20]. In PD and pure autonomic failure, there is both central (e.g., locus coeruleus) and peripheral neurodegeneration (i.e., postganglionic sympathetic denervation), whereas in MSA, the ANS defects are primarily central, and postganglionic sympathetic nerves remain intact [18, 21]. nOH can also be caused by other peripheral neuropathies that result in impaired autonomic reflexes (e.g., diabetes, autoimmune disorders, chemical/toxin induced) [22, 23].
Dysfunction within the ANS pathways for BP homeostasis causes an overall inability to regulate BP in response to postural change, resulting in the coexistence of nOH and nSH. The specific mechanisms contributing to nSH may differ in patients depending on neurodegeneration pathophysiology (i.e., central and/or peripheral). In patients with intact postganglionic sympathetic innervation (e.g., MSA), nSH may result when impaired baroreflex function cannot adequately compensate for norepinephrine released by the intact postganglionic sympathetic nerves [6••, 24]. However, in patients with postganglionic sympathetic denervation and low norepinephrine plasma levels (e.g., pure autonomic failure, PD), nSH may be caused by dysfunction within the baroreflex-mediated BP control pathways and hypersensitivity to residual norepinephrine release [6••, 24]. Other abnormalities in maintaining physiologic control of BP also likely contribute to the development of nSH. Arnold et al. demonstrated that plasma angiotensin II levels are elevated in autonomic failure [25], suggesting that nOH may lead to chronic activation of the renin-angiotensin system, which could contribute to the development of nSH [15, 26]. Patients with autonomic failure experience increased nocturnal sodium excretion and decreased daytime sodium excretion, and many have renal impairment that may also be related to nSH [14, 27]. The development of nSH may also involve other disruptions in the baroreflex including the vasomotor center and effector organs (e.g., heart, arterioles, veins) [28, 29], as well as increases in central blood volume due to the shifting of fluid away from the periphery.
More research is needed to better understand the distinct pathophysiology and clinical effects of BP dysregulation in autonomic failure disorders. For example, although it is known that there is poor prognosis in patients with nOH or nSH, it is unknown whether the poor prognosis is due to underlying autonomic failure, BP variability, chronic hypoperfusion, vasculature changes, or a combination of these [6••, 30]. A better understanding of the specific pathophysiology in patients with autonomic failure may result in better approaches to mitigate the long-term clinical impact of autonomic failure.
Clinical Features and Consequences of Blood Pressure Dysregulation in Patients with Autonomic Dysfunction
Neurogenic Orthostatic Hypotension
The estimated prevalence of nOH in autonomic failure disorders varies by underlying diagnosis, ranging from 30 to 40% of patients with PD, > 75% of patients with MSA, and 100% of patients with pure autonomic failure [31,32,33,34]. Orthostatic hypotension (OH) is defined as a reduction in systolic BP of ≥ 20 mmHg or diastolic BP of ≥ 10 mmHg within 3 min of standing (or ≥ 60° head-up tilt using a tilt-table) [2]; nOH is defined as OH caused by autonomic dysfunction [26]. Clinically, nOH can be distinguished from other causes of OH, such as use of certain medications and hypovolemia and/or by an inadequate cardioacceleratory response (< 15 bpm elevation of heart rate) upon standing [26]. Patients with nOH can be asymptomatic despite a substantial drop in orthostatic BP if standing BP remains within the range of cerebral perfusion pressure and cerebral autoregulation. When standing BP falls below the cerebral autoregulation threshold, symptoms emerge as a result of cerebral hypoperfusion. The lower BP threshold of cerebral autoregulation varies among individuals, but one study of patients with nOH found that most patients had symptoms if mean upright BP was < 75 mmHg at heart level or standing systolic BP < 100 mmHg [8]. For this reason, symptomatic nOH is most reflective of a standing systolic BP below that of an individual patient’s lower range of cerebral autoregulation and not the magnitude of the orthostatic drop in BP [8].
Common symptoms of nOH include lightheadedness, orthostatic dizziness, presyncope, and syncope, but patients may also report cognitive impairment, headache, impaired vision, fatigue, general weakness, shortness of breath while standing, neck and shoulder pain, angina, and falls [3, 26, 31, 35]. nOH symptoms can adversely affect patients’ daily function and quality of life. In a survey of 363 patients with nOH, most respondents (87%) indicated that nOH symptoms affected their daily activities, such as physical activities/exercise, housework, traveling, and time spent out of the house [9]. Patients also felt that nOH symptoms decreased their quality of life (59%), caused a loss of independence (42%), and drastically changed their lives (40%) [9]. In addition to the functional and psychosocial burden associated with symptomatic nOH, risk of fainting, falls, and related injuries is a major concern. Data from nOH patient cohorts indicate that falls are a frequent occurrence and present a safety threat because of injury potential [9, 36,37,38]. Patients with PD and nOH have greater fall-related and all-cause healthcare utilization (e.g., emergency department visits, hospitalizations) and greater healthcare costs (approximately 50–250% more in adjusted costs) compared with patients with PD without nOH [39, 40]. nOH has also been associated with increased mortality in patients with PD [41].
Neurogenic Supine Hypertension
In patients with autonomic failure, the estimated prevalence of nSH varies depending on the underlying cause of autonomic failure and how nSH is defined, with estimates ranging from 21 to 46% of patients with PD [8, 42, 43], up to 50% of patients with MSA, and up to 70% of patients with pure autonomic failure [5, 42, 44]. Supine hypertension is defined as a systolic BP ≥ 140 mmHg and/or diastolic BP ≥ 90 mmHg after ≥ 5 min of supine rest and can be classified as mild (140–159/90–99 mmHg), moderate (160–179/100–109 mmHg), or severe (≥ 180/≥ 110 mmHg) [1••, 6••]. Supine BP is typically measured with patients in a 30° elevated recumbent posture, but has also been reported with patients fully supine. Although nSH is usually asymptomatic, some patients may experience nonspecific symptoms such as dizziness, fatigue, headache, and blurred vision [6••, 28]. Additionally, nSH is associated with nocturnal pressure natriuresis with frequent nocturia leading to considerable sleep disruption. Nocturia also results in volume depletion overnight, exacerbating nOH symptoms in the morning hours [6••].
The pathophysiology of nSH is different than that of essential hypertension. There is limited understanding of whether the extreme but episodic BP variability in patients with autonomic failure (i.e., high BP when supine, low BP when standing) has similar morbidity risk as seen in essential hypertension (e.g., systemic vasculature damage). nSH has been associated with left ventricular hypertrophy, as well as an increased risk for end-target organ damage and premature death [12]. Nighttime systolic BP and 24-h BP (independent of BP variability) have been linked to the development of structural cardiac changes, such as increased left ventricular mass and left ventricular hypertrophy, in patients with autonomic failure compared with normotensive individuals [45]; the extent of these cardiac structural changes was similar to that observed in hypertensive patients [13, 45, 46]. nSH may also contribute to renal impairment. Significantly higher serum creatinine levels and lower estimated glomerular filtration rates were found in patients with pure autonomic failure and nSH compared with healthy controls [27].
Altered Diurnal Blood Pressure Patterns
Another clinical feature in patients with autonomic failure is blunted nocturnal “dipping” of BP levels [6••]. A normal circadian BP pattern includes nocturnal dipping of ≥ 10% from daytime values; however, patients with nSH may have nocturnal BP decreases of < 10% (“reduced dippers”), have no nocturnal BP drop (“nondippers”), or elevated nocturnal BP (“reverse dippers”) [6••, 47]. Data suggest that abnormal circadian BP patterns may be observed in approximately two-thirds or more of patients with autonomic failure disorders [48,49,50]. There are limited data regarding the clinical effects of blunted nocturnal BP dipping specifically in patients with autonomic failure. In a study of patients with PD, left ventricular mass was significantly higher in patients with reverse dipping (defined as a change in day-night BP < 0%) compared with nonreverse dippers (P = 0.005); the increases in left ventricular mass observed in reverse-dipping patients with PD were similar to those observed in patients with essential hypertension [51•]. These findings of cardiovascular risk associated with blunted dipping in an autonomic failure population are consistent with data from general population studies that showed an association between greater morbidity and mortality (i.e., increased stroke risk, heart, vasculature, and kidney damage) with altered diurnal BP patterns [52,53,54,55,56,57].
Management of neurogenic orthostatic hypotension when neurogenic supine hypertension is also present
When treating patients with nOH, the aim of management is to increase standing systolic BP into the range of cerebral autoregulation to improve symptoms and the ability to participate in daily activities, as well as to reduce the risk of falls [1••, 26, 58]. However, when significant nSH is also present, management attempts to raise standing BP will invariably raise supine BP. The likelihood and consequences of the short- and long-term risks posed by each condition, along with evaluation of comorbidities, overall life expectancy, and concurrent medications for the individual patient, should be continually assessed. Both nonpharmacologic approaches (e.g., review and adjust medications with hypotensive effects, increase fluids and salt intake, make postural changes in stages, use of compression garments) and pharmacologic pressor medications are useful in the treatment of symptomatic nOH [26, 59]. Often, effective improvement of nOH symptoms requires treatment with pressor agents (Table 1) [50, 60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83] to increase standing BP above the lower range of cerebral perfusion pressure and cerebral autoregulation [26, 59]. When treating the symptoms of nOH, consideration of the risk/benefit profile associated with individual treatments, particularly with regard to supine BP increases, is needed.
Nonpharmacologic Management of Neurogenic Orthostatic Hypotension
Nonpharmacologic management of nOH focuses on patient education, volume expansion, and mechanical increase of vascular return. Patients should be educated about standing slowly and avoiding heat, large meals, prolonged standing, the use of daytime antihypertensives, and dehydration, as well as about diurnal variations in BP and the lower BP present in morning hours [3, 26]. Volume expansion is encouraged with liquid hydration, liberal use of table salt or salt tablets, and 30° elevation of the head of the bed overnight to reduce nocturnal diuresis and improve morning BP [3, 26]. Mechanical compression of the lower extremities and abdomen with waist-high compression stockings and abdominal binders will increase blood return into the circulation [3, 26]. Additionally, patients can be educated to contract the muscles of the buttocks and legs when they stand [3]. Symptoms of nOH should be reassessed after two weeks of nonpharmacologic treatment for the need to add pharmacologic therapy.
Pharmacologic Management of Neurogenic Orthostatic Hypotension
Pharmacologic management of nOH focuses on volume expansion and vasoconstriction. The most commonly used medications are the mineralocorticoid fludrocortisone, the acetylcholinesterase inhibitor pyridostigmine, the direct α1 receptor agonist midodrine, and the norepinephrine precursor droxidopa [26]. Other less commonly used medications to raise BP include central norepinephrine reuptake inhibitors and other medications that lead to BP increases via various mechanisms [15].
The mineralocorticoid fludrocortisone is used off-label for the treatment of OH [84] including nOH (Table 1). There are some clinical data to suggest that fludrocortisone is associated with BP increases in patients with nOH (due to PD or diabetic neuropathy); however, study design (i.e., case series) and small sample size considerations limit the overall assessment of effects [67, 85, 86]. BP elevations with fludrocortisone are initially observed 4 to 14 days after treatment initiation [87•, 88], but effects on nOH symptoms have not been rigorously evaluated. In patients with coexistent nOH and nSH, the volume expansion effects of fludrocortisone may limit its use because it raises supine BP to a greater magnitude than standing BP [68, 89]. During treatment, serum electrolytes should be regularly monitored, and pedal edema is a frequent dose-limiting side effect [90, 91].
Pyridostigmine, an acetylcholinesterase inhibitor, is used off-label to treat nOH because it enhances cholinergic neurotransmission at sympathetic ganglia. This increased cholinergic tone can result in increased norepinephrine release during orthostatic stress (Table 1) [26, 80, 82]. Because of this mechanism of action, it is thought that pyridostigmine can be used to treat nOH without exacerbating nSH [26, 80,81,82]. Studies of pyridostigmine treatment have shown increases in standing BP and improvements in orthostatic symptoms without significant effects on supine BP [80, 82], although these positive effects on nOH symptoms and standing and supine BP have not been consistently demonstrated in all trials [67, 81, 83]. Some of these differences may be due to the specific patient cohorts examined because the effects of pyridostigmine likely are related to residual sympathetic function [26, 81]. Potential side effects of pyridostigmine include nausea, vomiting, diarrhea, abdominal cramps, increased salivation, excessive sweating, and urinary incontinence [26, 78].
Midodrine is approved for the treatment of symptomatic OH (all causes). Approval was based on an increase in BP 1 min after standing (Table 1) [62], although clinical benefits (e.g., improved symptoms, ability to perform daily activities) were not confirmed in the registration trials [62]. The effectiveness of midodrine for increasing standing BP and improving OH symptoms is supported by findings of several subsequent randomized clinical trials [63,64,65]. Midodrine significantly increases standing systolic BP, but its use may be limited because of the magnitude of increases in sitting and supine BP (Table 1) [63, 64, 73,74,75,76,77]. In a single-dose study, midodrine treatment showed dose-dependent effects on incidence of nSH after 10- and 20-mg doses [65]. In other longer studies (4 weeks of active treatment), rates of study discontinuation due to nSH were 5 to 6% [63, 64]. Patients should be made aware that they should not be supine for 4 h after taking a dose of midodrine. Effective dosing can be limited by high BP when seated. Frequently reported side effects include scalp paresthesia/pruritus, piloerection, and urinary retention/urgency [62].
Droxidopa was approved by the US Food and Drug Administration for the treatment of symptomatic nOH [26]. It is the only agent approved based on symptomatic improvement, with a labeled indication for the treatment of orthostatic dizziness, lightheadedness, or the “feeling that you are about to black out” in adult patients with symptomatic nOH caused by primary autonomic failure (PD, MSA, and pure autonomic failure), dopamine β-hydroxylase deficiency, and nondiabetic autonomic neuropathy (Table 1) [60].
Clinical trials have demonstrated that droxidopa improves the symptoms of nOH and their effect on daily activities and is generally well tolerated with a low risk for the development of nSH [50, 61, 92, 93]. In clinical trials, rates of supine systolic BP > 160, > 180, and > 200 mmHg were not markedly different in patients receiving droxidopa versus placebo, although these elevated BP readings occurred more frequently in the droxidopa group (Table 1) [61]. Generally similar rates of nighttime BP readings > 160, > 180, and > 200 mmHg were found on and off droxidopa treatment in another small short-term study (N = 18; 4–5 weeks “on” then 24 h “off”), and the authors concluded that droxidopa had an “acceptably low” estimated 10 to 20% risk of increasing nocturnal nSH [50]. Although these data provide important information about the risks of nSH with droxidopa, interpretation may be limited for patients with severe preexisting hypertension (seated or supine BP ≥ 180/110 mmHg), who were excluded from the clinical trials of droxidopa [61].
Comparisons of Fludrocortisone, Pyridostigmine, Midodrine, and Droxidopa
There are no direct comparison trials between fludrocortisone, midodrine, and droxidopa regarding efficacy, tolerability, or risk of nSH; pyridostigmine has been compared with fludrocortisone or midodrine in 2 small studies [67, 83]. A recent Bayesian mixed-treatment meta-analysis compared droxidopa and midodrine using clinical trial data. Both medications were associated with significant increases in standing systolic BP versus placebo (droxidopa, 6.2 mmHg [95% credible interval (CrI), 2.4–10.0]; midodrine, 17 mmHg [95% CrI, 11.4–23.0]) [76•]. This meta-analysis found that the relative risk of nSH for droxidopa was not significant (1.4 [95% CrI, 0.71–2.7] vs placebo), whereas there was a significant corresponding relative risk for midodrine (5.1 [95% CrI, 1.6–24.0] vs placebo) [76•]. To our knowledge, no similar nSH comparisons that include fludrocortisone have been reported. However, in a retrospective analysis of patients prescribed midodrine (n = 797) or fludrocortisone (n = 1324), the risk of all-cause hospitalization was greater for patients who received fludrocortisone versus midodrine (adjusted incidence-rate ratio [aIRR], 1.20; 95% confidence interval [CI], 1.02–1.40) and the hospitalization risk associated with fludrocortisone was more pronounced in patients with a history of congestive heart failure (aIRR vs midodrine, 1.42; 95% CI, 1.07–1.90) [94•].
In a crossover study of pyridostigmine and fludrocortisone in 13 patients with PD, increases in standing or supine BP from baseline were found for fludrocortisone but not pyridostigmine [67]. Neither drug improved nOH symptoms [67]. In a 3-month open-label study of pyridostigmine and midodrine, both pyridostigmine and midodrine ameliorated orthostatic BP reductions and improved nOH symptoms, but midodrine’s effects on symptoms were more pronounced [83]. In this study, significant increases in supine systolic BP (8–11 mmHg) were observed with pyridostigmine but not midodrine [83]. However, the generalizability of these findings is limited by the low doses of midodrine used (2.5–5.0 mg, 2 times daily) and the small sample sizes studied (29 patients per treatment arm).
Stepwise Treatment Approach for Managing Supine Hypertension in Patients with Neurogenic Orthostatic Hypotension
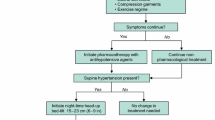
Because nSH can adversely affect patients’ morbidity risks, clinicians should identify patients with nOH who also have coexisting nSH to help optimize treatment strategies. However, because pressor agents used to treat symptoms of nOH can increase supine BP, and antihypertensive agents used to treat nSH can worsen the symptoms of nOH, an appropriate management strategy can be a clinical conundrum. A suggested treatment approach for evaluating and managing coexistent nOH and nSH based on current published evidence (clinical data and recommendations) and our clinical experience is shown in Fig. 1 [1, 26, 59].

Step 1: Awareness and Recognition of Neurogenic Supine Hypertension in Patients With Neurogenic Orthostatic Hypotension
Although some patients with nSH may report frequent nocturia or other nonspecific symptoms (e.g., headache), most often, nSH is asymptomatic [6••]. Therefore, the fundamental way to identify nSH in patients with nOH is to measure BP in a “semi-recumbent” supine position (i.e., 30° horizontal elevation of the head) because patients at risk of nSH should typically avoid being in a fully supine position, including when sleeping.
Step 2: Diagnosis of Neurogenic Supine Hypertension
The presence and magnitude of nSH can be evaluated clinically by measuring BP immediately after the patient assumes their semi-recumbent sleeping position and then after 5 min of rest in that position [6••, 26]. If required, BP assessments for nOH can also be conducted after the patient stands [26]. Because BP may fluctuate overnight [49], 24-h ambulatory BP monitoring can sometimes be useful to evaluate overnight BP and identify abnormal circadian patterns (e.g., reverse or reduced dipping) [6••, 26]. Daily BP patterns may also be assessed through a home BP log kept by the patient. Patients should be encouraged to record BP and heart rate at home (e.g., seated and standing before and after meals, in their semi-recumbent sleeping position at bedtime). These home BP logs can be particularly useful in the evaluation of nOH before implementing pharmacologic treatment for nSH and to evaluate effects after these treatments are initiated (Fig. 1, step 4).
Step 3: Nonpharmacologic Treatment of Neurogenic Supine Hypertension
There are several nonpharmacologic strategies that can be used to reduce nSH (Fig. 1) [1••, 26, 59]; information about these should be provided as part of the patient’s education about nOH.
-
Elevating the head of the bed can reduce supine BP [14, 95]. During nighttime, patients should sleep with the head of the bed elevated at least 6 to 9 inches (approximately 30°) [59]. If nSH persists, the angle of recumbence can be increasingly elevated, and some patients may need to sleep in a near-seated position (eg, in a recliner) to reduce supine hypertension [59]. A head-up sleeping position will reduce BP and natriuresis while keeping the renin-angiotensin system activated so that there will be less of a BP drop in the morning [1••, 26, 59, 96].
-
To reduce nocturnal hypertension, patients can be advised to eat a high-carbohydrate snack at bedtime, which not only lowers BP by directing blood flow to the splanchnic circulation, but also causes insulin release (which has direct vasodilator properties) [97] with resultant vasodilation [1••, 98].
-
During the daytime, patients using a pressor medication for nOH should avoid the supine position. To reduce the potential for nSH at night, the last daily dose of any nOH pressor medication should be taken at least 3 to 4 hours before bedtime [60, 62].
-
Nonpharmacologic treatments for nSH should be continued even when pharmacotherapy is implemented.
Step 4: Pharmacologic Treatment of Neurogenic Supine Hypertension and Treatment Monitoring
If nSH persists despite the use of nonpharmacologic measures, then treatment with a short-acting antihypertensive agent at bedtime may be necessary (Table 2) [1••, 25, 26, 44, 99,100,101,102,103,104,105,106]. Generally, pharmacologic management of nSH may be required for patients with severe nSH (systolic/diastolic BP ≥ 180/≥110 mmHg) and may be considered for patients with moderate nSH (BP 160–179/100–109 mmHg) based on individual patient circumstances and risk profile [6••, 26]. Short-acting agents are preferred to minimize chances that antihypertensive effects will persist into morning and worsen nOH symptoms when the patient arises for the day [1••]. Based on our clinical experience and previous published guidance, the following recommendations regarding agents and subsequent BP monitoring are provided.
-
Administer a short-acting antihypertensive agent at bedtime or in the evening after dinner (Table 2) [1••, 26, 99,100,101,102, 105, 106].
-
After therapy is initiated and at regular intervals, nOH symptoms and the patient’s home BP log should be re-evaluated (Fig. 1).
-
If symptoms of nOH emerge or are exacerbated in the morning, consider adjusting the timing of the antihypertensive agent to earlier in the evening after dinner.
Other Considerations for Treatment and Future Directions
Treatment of patients with coexisting nOH and nSH can be medically complex, and management may often require collaboration among neurology, cardiology, and other specialties with expertise in the management of autonomic disorders, especially if treatment of nSH worsens the symptoms of nOH.
Currently, much of the data on the management of coexisting nOH and nSH are from patient cohorts with a variety of autonomic failure disorders. An important area for future research may be further understanding of how to individualize treatment for both nOH and nSH based on specific underlying pathophysiology (e.g., by underlying diagnosis of central or peripheral autonomic dysfunction) and level of sympathetic activity [107]. Although some studies have indicated differential treatment effects on BP when patients are grouped by underlying diagnosis (i.e., central/preganglionic or peripheral/postganglionic ANS dysfunction) [24, 108, 109], or biomarkers thereof (eg, supine plasma norepinephrine levels as a marker of peripheral sympathetic denervation) [110], clear evidence for tailoring the management of nOH or nSH by underlying pathophysiology has not yet been identified. Additional areas for future research include a better understanding of the morbidity associated with BP dysregulation in autonomic failure populations and the potential benefits of effective simultaneous management of coexisting nOH and nSH.
Conclusions
In patients with autonomic failure, cardiovascular autonomic dysfunction frequently occurs and can manifest as coexistent nOH and nSH. Because of opposing hemodynamics, the co-management of these two opposing conditions can present a clinical challenge; however, balanced management is necessary to alleviate symptoms, improve quality of life, and mitigate associated short- and long-term risks (e.g., syncope, falls from fainting, cardiovascular complications). When treating patients with symptomatic nOH and coexisting nSH, clinicians should individualize treatment by considering the presence of nSH and the risk of increased supine BP associated with agents used to treat nOH and managing nSH with nonpharmacologic measures initially. If required, prudent use of short-acting antihypertensives can be used at night to manage nSH.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Jordan J, Fanciulli A, Tank J, Calandra-Buonaura G, Cheshire WP, Cortelli P, et al. Management of supine hypertension in patients with neurogenic orthostatic hypotension: scientific statement of the American Autonomic Society, European Federation of Autonomic Societies, and the European Society of Hypertension. J Hypertens. 2019;37(8):1541–6. https://doi.org/10.1097/hjh.0000000000002078Scientific statement that summarizes the needs and challenges of managing and treating supine hypertension in patients who also have neurogenic orthostatic hypotension.
Freeman R, Wieling W, Axelrod FB, Benditt DG, Benarroch E, Biaggioni I, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin Auton Res. 2011;21(2):69–72. https://doi.org/10.1007/s10286-011-0119-5.
Freeman R, Abuzinadah AR, Gibbons C, Jones P, Miglis MG, Sinn DI. Orthostatic hypotension: JACC state-of-the-art review. J Am Coll Cardiol. 2018;72(11):1294–309. https://doi.org/10.1016/j.jacc.2018.05.079.
Nwazue VC, Raj SR. Confounders of vasovagal syncope: orthostatic hypotension. Cardiol Clin. 2013;31(1):89–100.
Celedonio JE, Arnold AC, Dupont WD, Ramirez CE, Diedrich A, Okamoto LE, et al. Residual sympathetic tone is associated with reduced insulin sensitivity in patients with autonomic failure. Clin Auton Res. 2015;25(5):309–15.
Fanciulli A, Jordan J, Biaggioni I, Calandra-Buonaura G, Cheshire WP, Cortelli P, et al. Consensus statement on the definition of neurogenic supine hypertension in cardiovascular autonomic failure by the American Autonomic Society (AAS) and the European Federation of Autonomic Societies (EFAS): endorsed by the European Academy of Neurology (EAN) and the European Society of Hypertension (ESH). Clin Auton Res. 2018;28(4):355–62. https://doi.org/10.1007/s10286-018-0529-8Consensus statement that defines neurogenic supine hypertension and delineates its severity in patients with autonomic failure.
Novak V, Novak P, Spies JM, Low PA. Autoregulation of cerebral blood flow in orthostatic hypotension. Stroke. 1998;29(1):104–11. https://doi.org/10.1161/01.str.29.1.104.
Palma JA, Gomez-Esteban JC, Norcliffe-Kaufmann L, Martinez J, Tijero B, Berganzo K, et al. Orthostatic hypotension in Parkinson disease: how much you fall or how low you go? Mov Disord. 2015;30(5):639–45. https://doi.org/10.1002/mds.26079.
Claassen DO, Adler CH, Hewitt LA, Gibbons C. Characterization of the symptoms of neurogenic orthostatic hypotension and their impact from a survey of patients and caregivers. BMC Neurol. 2018;18(1):125. https://doi.org/10.1186/s12883-018-1129-x.
Centi J, Freeman R, Gibbons CH, Neargarder S, Canova AO, Cronin-Golomb A. Effects of orthostatic hypotension on cognition in Parkinson disease. Neurology. 2017;88(1):17–24. https://doi.org/10.1212/wnl.0000000000003452.
Stewart JM. Mechanisms of sympathetic regulation in orthostatic intolerance. J Appl Physiol (1985). 2012;113(10):1659–68. https://doi.org/10.1152/japplphysiol.00266.2012.
Palma JA, Redel-Traub G, Porciuncula A, Samaniego-Toro D, Lui YW, Norcliffe-Kaufmann L, et al. The impact of supine hypertension on target organ damage and mortality in patients with neurogenic orthostatic hypotension. Clin Auton Res. 2018;28:473.
Vagaonescu TD, Saadia D, Tuhrim S, Phillips RA, Kaufmann H. Hypertensive cardiovascular damage in patients with primary autonomic failure. Lancet. 2000;355(9205):725–6. https://doi.org/10.1016/S0140-6736(99)05320-9.
Jordan J, Biaggioni I. Diagnosis and treatment of supine hypertension in autonomic failure patients with orthostatic hypotension. J Clin Hypertens (Greenwich). 2002;4(2):139–45.
Biaggioni I. The pharmacology of autonomic failure: from hypotension to hypertension. Pharmacol Rev. 2017;69(1):53–62.
Lake CR, Ziegler MG, Kopin IJ. Use of plasma norepinephrine for evaluation of sympathetic neuronal function in man. Life Sci. 1976;18(11):1315–25.
Goldstein DS, Pechnik S, Holmes C, Eldadah B, Sharabi Y. Association between supine hypertension and orthostatic hypotension in autonomic failure. Hypertension. 2003;42(2):136–42. https://doi.org/10.1161/01.hyp.0000081216.11623.c3.
Loavenbruck A, Sandroni P. Neurogenic orthostatic hypotension: roles of norepinephrine deficiency in its causes, its treatment, and future research directions. Curr Med Res Opin. 2015;31(11):2095–104. https://doi.org/10.1185/03007995.2015.1087988.
Goldstein DS, Sharabi Y. Neurogenic orthostatic hypotension: a pathophysiological approach. Circulation. 2009;119(1):139–46. https://doi.org/10.1161/CIRCULATIONAHA.108.805887.
McDonell KE, Shibao CA, Claassen DO. Clinical relevance of orthostatic hypotension in neurodegenerative disease. Curr Neurol Neurosci Rep. 2015;15(12):78. https://doi.org/10.1007/s11910-015-0599-0.
Coon EA, Cutsforth-Gregory JK, Benarroch EE. Neuropathology of autonomic dysfunction in synucleinopathies. Mov Disord. 2018;33(3):349–58. https://doi.org/10.1002/mds.27186.
Freeman R. Clinical practice. Neurogenic orthostatic hypotension. N Engl J Med. 2008;358(6):615–24. https://doi.org/10.1056/NEJMcp074189.
Arbique D, Cheek D, Welliver M, Vongpatanasin W. Management of neurogenic orthostatic hypotension. J Am Med Dir Assoc. 2014;15(4):234–9. https://doi.org/10.1016/j.jamda.2013.10.014.
Shannon JR, Jordan J, Diedrich A, Pohar B, Black BK, Robertson D, et al. Sympathetically mediated hypertension in autonomic failure. Circulation. 2000;101(23):2710–5.
Arnold AC, Okamoto LE, Gamboa A, Black BK, Raj SR, Elijovich F, et al. Mineralocorticoid receptor activation contributes to the supine hypertension of autonomic failure. Hypertension. 2016;67(2):424–9.
Gibbons CH, Schmidt P, Biaggioni I, Frazier-Mills C, Freeman R, Isaacson S, et al. The recommendations of a consensus panel for the screening, diagnosis, and treatment of neurogenic orthostatic hypotension and associated supine hypertension. J Neurol. 2017;264(8):1567–82. https://doi.org/10.1007/s00415-016-8375-x.
Garland EM, Gamboa A, Okamoto L, Raj SR, Black BK, Davis TL, et al. Renal impairment of pure autonomic failure. Hypertension. 2009;54(5):1057–61. https://doi.org/10.1161/HYPERTENSIONAHA.109.136853.
Naschitz JE, Slobodin G, Elias N, Rosner I. The patient with supine hypertension and orthostatic hypotension: a clinical dilemma. Postgrad Med J. 2006;82(966):246–53.
Sharabi Y, Goldstein DS. Mechanisms of orthostatic hypotension and supine hypertension in Parkinson disease. J Neurol Sci. 2011;310(1-2):123–8. https://doi.org/10.1016/j.jns.2011.06.047.
Rothwell PM. Limitations of the usual blood-pressure hypothesis and importance of variability, instability, and episodic hypertension. Lancet. 2010;375(9718):938–48.
Kaufmann H, Goldstein DS. Chapter 21–Autonomic dysfunction in Parkinson disease. Handb Clin Neurol. 2013;117:259–78. https://doi.org/10.1016/B978-0-444-53491-0.00021-3.
Kaufmann H, Norcliffe-Kaufmann L, Palma JA, Biaggioni I, Low PA, Singer W, et al. Natural history of pure autonomic failure: a United States prospective cohort. Ann Neurol. 2017;81(2):287–97.
Low PA, Reich SG, Jankovic J, Shults CW, Stern MB, Novak P, et al. Natural history of multiple system atrophy in the USA: a prospective cohort study. Lancet Neurol. 2015;14(7):710–9.
Metzler M, Duerr S, Granata R, Krismer F, Robertson D, Wenning GK. Neurogenic orthostatic hypotension: pathophysiology, evaluation, and management. J Neurol. 2013;260(9):2212–9. https://doi.org/10.1007/s00415-012-6736-7.
Senard JM, Rai S, Lapeyre-Mestre M, Brefel C, Rascol O, Rascol A, et al. Prevalence of orthostatic hypotension in Parkinson's disease. J Neurol Neurosurg Psychiatry. 1997;63(5):584–9.
Craig GM. Clinical presentation of orthostatic hypotension in the elderly. Postgrad Med J. 1994;70(827):638–42.
Shaw BH, Claydon VE. The relationship between orthostatic hypotension and falling in older adults. Clin Auton Res. 2014;24(1):3–13.
François C, Shibao CA, Biaggioni I, Duhig AM, McLeod K, Ogbonnaya A, et al. Six-month use of droxidopa for neurogenic orthostatic hypotension. Mov Disord Clin Pract. 2019;6(3):235–42. https://doi.org/10.1002/mdc3.12726.
Francois C, Biaggioni I, Shibao C, Ogbonnaya A, Shih HC, Farrelly E, et al. Fall-related healthcare use and costs in neurogenic orthostatic hypotension with Parkinson’s disease. J Med Econ. 2017;20(5):525–32. https://doi.org/10.1080/13696998.2017.1284668.
Merola A, Sawyer RP, Artusi CA, Suri R, Berndt Z, Lopez-Castellanos JR, et al. Orthostatic hypotension in Parkinson disease: impact on health care utilization. Parkinsonism Relat Disord. 2017;47:45–9. https://doi.org/10.1016/j.parkreldis.2017.11.344.
Goldstein DS, Holmes C, Sharabi Y, Wu T. Survival in synucleinopathies: a prospective cohort study. Neurology. 2015;85(18):1554–61.
Fanciulli A, Gobel G, Ndayisaba JP, Granata R, Duerr S, Strano S, et al. Supine hypertension in Parkinson's disease and multiple system atrophy. Clin Auton Res. 2016;26(2):97–105. https://doi.org/10.1007/s10286-015-0336-4.
Umehara T, Matsuno H, Toyoda C, Oka H. Clinical characteristics of supine hypertension in de novo Parkinson disease. Clin Auton Res. 2016;26(1):15–21. https://doi.org/10.1007/s10286-015-0324-8.
Shannon J, Jordan J, Costa F, Robertson RM, Biaggioni I. The hypertension of autonomic failure and its treatment. Hypertension. 1997;30(5):1062–7.
Milazzo V, Maule S, Di Stefano C, Tosello F, Totaro S, Veglio F, et al. Cardiac organ damage and arterial stiffness in autonomic failure: comparison with essential hypertension. Hypertension. 2015;66(6):1168–75.
Maule S, Milan A, Grosso T, Veglio F. Left ventricular hypertrophy in patients with autonomic failure. Am J Hypertens. 2006;19(10):1049–54. https://doi.org/10.1016/j.amjhyper.2006.02.016.
Cuspidi C, Sala C, Tadic M, Gherbesi E, De Giorgi A, Grassi G, et al. Clinical and prognostic significance of a reverse dipping pattern on ambulatory monitoring: an updated review. J Clin Hypertens (Greenwich). 2017;19(7):713–21. https://doi.org/10.1111/jch.13023.
Plaschke M, Trenkwalder P, Dahlheim H, Lechner C, Trenkwalder C. Twenty-four-hour blood pressure profile and blood pressure responses to head-up tilt tests in Parkinson's disease and multiple system atrophy. J Hypertens. 1998;16(10):1433–41.
Okamoto LE, Gamboa A, Shibao C, Black BK, Diedrich A, Raj SR, et al. Nocturnal blood pressure dipping in the hypertension of autonomic failure. Hypertension. 2009;53(2):363–9.
Kaufmann H, Norcliffe-Kaufmann L, Hewitt LA, Rowse GJ, White WB. Effects of the novel norepinephrine prodrug, droxidopa, on ambulatory blood pressure in patients with neurogenic orthostatic hypotension. J Am Soc Hypertens. 2016;10(10):819–26. https://doi.org/10.1016/j.jash.2016.07.009.
Di Stefano C, Sobrero G, Milazzo V, Vallelonga F, Romagnolo A, Zibetti M, et al. Cardiac organ damage in patients with Parkinson's disease and reverse dipping. J Hypertens. 2019. https://doi.org/10.1097/hjh.0000000000002249. A study that showed patients with Parkinson disease who also had nocturnal hypertension/reverse dipping had higher left ventricular mass and increased prevalence of left ventricle hypertrophy than nonreverse patients with Parkinson disease; the magnitude of the increase in left ventricular in reverse-dipping patients was similar to that measured in patients with essential hypertension.
Staessen JA, Thijs L, Fagard R, O'Brien ET, Clement D, de Leeuw PW, et al. Predicting cardiovascular risk using conventional vs ambulatory blood pressure in older patients with systolic hypertension. Systolic Hypertension in Europe Trial Investigators. JAMA. 1999;282(6):539–46.
Ohkubo T, Hozawa A, Yamaguchi J, Kikuya M, Ohmori K, Michimata M, et al. Prognostic significance of the nocturnal decline in blood pressure in individuals with and without high 24-h blood pressure: The Ohasama Study. J Hypertens. 2002;20(11):2183–9.
Ben-Dov IZ, Kark JD, Ben-Ishay D, Mekler J, Ben-Arie L, Bursztyn M. Predictors of all-cause mortality in clinical ambulatory monitoring: unique aspects of blood pressure during sleep. Hypertension. 2007;49(6):1235–41.
Coleman CT, Stowasser M, Jenkins C, Marwick TH, Sharman JE. Central hemodynamics and cardiovascular risk in nondippers. J Clin Hypertens (Greenwich). 2011;13(8):557–62.
Routledge FS, McFetridge-Durdle JA, Dean CR. Night-time blood pressure patterns and target organ damage: a review. Can J Cardiol. 2007;23(2):132–8.
Friedman O, Logan AG. Can nocturnal hypertension predict cardiovascular risk? Integr Blood Press Control. 2009;2:25–37.
Isaacson SH, Skettini J. Neurogenic orthostatic hypotension in Parkinson's disease: evaluation, management, and emerging role of droxidopa. Vasc Health Risk Manag. 2014;10:169–76. https://doi.org/10.2147/VHRM.S53983.
Shibao C, Okamoto L, Biaggioni I. Pharmacotherapy of autonomic failure. Pharmacol Ther. 2012;134(3):279–86. https://doi.org/10.1016/j.pharmthera.2011.05.009.
Northera® (droxidopa). Full Prescribing Information, Lundbeck NA Ltd, Deerfield, IL, 2017.
Biaggioni I, Hewitt LA, Rowse GJ, Kaufmann H. Integrated analysis of droxidopa trials for neurogenic orthostatic hypotension. BMC Neurol. 2017;17(1):90. https://doi.org/10.1186/s12883-017-0867-5.
ProAmatine® (midodrine hydrochloride). Full Prescribing Information, Shire US Inc., Lexington, MA, 2017.
Low PA, Gilden JL, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA. 1997;277(13):1046–51.
Jankovic J, Gilden JL, Hiner BC, Kaufmann H, Brown DC, Coghlan CH, et al. Neurogenic orthostatic hypotension: a double-blind, placebo-controlled study with midodrine. Am J Med. 1993;95(1):38–48.
Wright RA, Kaufmann HC, Perera R, Opfer-Gehrking TL, McElligott MA, Sheng KN, et al. A double-blind, dose-response study of midodrine in neurogenic orthostatic hypotension. Neurology. 1998;51(1):120–4.
Hamitouche N, Comets E, Ribot M, Alvarez JC, Bellissant E, Laviolle B. Population pharmacokinetic-pharmacodynamic model of oral fludrocortisone and intravenous hydrocortisone in healthy volunteers. AAPS J. 2017;19(3):727–35. https://doi.org/10.1208/s12248-016-0041-9.
Schreglmann SR, Buchele F, Sommerauer M, Epprecht L, Kagi G, Hagele-Link S, et al. Pyridostigmine bromide versus fludrocortisone in the treatment of orthostatic hypotension in Parkinson's disease - a randomized controlled trial. Eur J Neurol. 2017;24(4):545–51. https://doi.org/10.1111/ene.13260.
Hakamaki T, Rajala T, Lehtonen A. Ambulatory 24-h blood pressure recordings in patients with Parkinson's disease with or without fludrocortisone. Int J Clin Pharmacol Ther. 1998;36(7):367–9.
Hussain RM, McIntosh SJ, Lawson J, Kenny RA. Fludrocortisone in the treatment of hypotensive disorders in the elderly. Heart. 1996;76(6):507–9.
Goldstein DS. L-dihydroxyphenylserine (L-DOPS): a norepinephrine prodrug. Cardiovasc Drug Rev. 2006;24(3-4):189–203. https://doi.org/10.1111/j.1527-3466.2006.00189.x.
Drugs.com. Fludrocortisone FDA Professional Drug Information. https://www.drugs.com/pro/fludrocortisone.html. Accessed February 11 2020.
Kaufmann H, Saadia D, Voustianiouk A, Goldstein DS, Holmes C, Yahr MD, et al. Norepinephrine precursor therapy in neurogenic orthostatic hypotension. Circulation. 2003;108(6):724–8. https://doi.org/10.1161/01.CIR.0000083721.49847.D7.
Arnold AC, Ramirez CE, Choi L, Okamoto LE, Gamboa A, Diedrich A, et al. Combination ergotamine and caffeine improves seated blood pressure and presyncopal symptoms in autonomic failure. Front Physiol. 2014;5:270. https://doi.org/10.3389/fphys.2014.00270.
Jordan J, Shannon JR, Biaggioni I, Norman R, Black BK, Robertson D. Contrasting actions of pressor agents in severe autonomic failure. Am J Med. 1998;105(2):116–24.
Parsaik AK, Singh B, Altayar O, Mascarenhas SS, Singh SK, Erwin PJ, et al. Midodrine for orthostatic hypotension: a systematic review and meta-analysis of clinical trials. J Gen Intern Med. 2013;28(11):1496–503. https://doi.org/10.1007/s11606-013-2520-3.
Chen JJ, Han Y, Tang J, Portillo I, Hauser RA, Dashtipour K. Standing and supine blood pressure outcomes associated with droxidopa and midodrine in patients with neurogenic orthostatic hypotension: a Bayesian meta-analysis and mixed treatment comparison of randomized trials. Ann Pharmacother. 2018;52(12):1182–94. https://doi.org/10.1177/1060028018786954A mixed-treatment meta-analysis showing that compared with placebo, the relative risk of supine hypertension was significantly higher for midodrine but not for droxidopa.
Fouad-Tarazi FM, Okabe M, Goren H. Alpha sympathomimetic treatment of autonomic insufficiency with orthostatic hypotension. Am J Med. 1995;99(6):604–10.
Mestinon® (pyridostigmine bromide). Full Prescribing Information, ICN Pharmaceuticals, Costa Mesa, CA, 2001.
Aquilonius SM, Eckernäs SA, Hartvig P, Lindström B, Osterman PO. Pharmacokinetics and oral bioavailability of pyridostigmine in man. Eur J Clin Pharmacol. 1980;18(5):423–8. https://doi.org/10.1007/bf00636797.
Singer W, Opfer-Gehrking TL, McPhee BR, Hilz MJ, Bharucha AE, Low PA. Acetylcholinesterase inhibition: a novel approach in the treatment of neurogenic orthostatic hypotension. J Neurol Neurosurg Psychiatry. 2003;74(9):1294–8.
Shibao C, Okamoto LE, Gamboa A, Yu C, Diedrich A, Raj SR, et al. Comparative efficacy of yohimbine against pyridostigmine for the treatment of orthostatic hypotension in autonomic failure. Hypertension. 2010;56(5):847–51. https://doi.org/10.1161/HYPERTENSIONAHA.110.154898.
Singer W, Sandroni P, Opfer-Gehrking TL, Suarez GA, Klein CM, Hines S, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch Neurol. 2006;63(4):513–8. https://doi.org/10.1001/archneur.63.4.noc50340.
Byun JI, Moon J, Kim DY, Shin H, Sunwoo JS, Lim JA, et al. Efficacy of single or combined midodrine and pyridostigmine in orthostatic hypotension. Neurology. 2017;89(10):1078–86. https://doi.org/10.1212/wnl.0000000000004340.
Shibao C, Grijalva CG, Lipsitz LA, Biaggioni I, Griffin MR. Early discontinuation of treatment in patients with orthostatic hypotension. Auton Neurosci. 2013;177(2):291–6. https://doi.org/10.1016/j.autneu.2013.08.064.
Campbell IW, Ewing DJ, Clarke BF. Therapeutic experience with fludrocortisone in diabetic postural hypotension. Br Med J. 1976;1(6014):872–4.
Hoehn MM. Levodopa-induced postural hypotension. Treatment with fludrocortisone. Arch Neurol. 1975;32(1):50–1.
Palma JA, Kaufmann H. Epidemiology, diagnosis, and management of neurogenic orthostatic hypotension. Mov Disord Clin Pract. 2017;4(3):298–308. https://doi.org/10.1002/mdc3.12478A review article describing the diagnosis and management of neurogenic orthostatic hypotension and providing a summary of key nonpharmacologic and pharmacologic approaches for the treatment of patients with neurogenic orthostatic hypotension.
Kaufmann H, Biaggioni I. Chapter 63 - Disorders of the autonomic nervous system. In: Fillit HM, Rockwood K, Woodhouse K, editors. Brocklehurst's Textbook of Geriatric Medicine and Gerontology. 7th ed. Philadelphia: W.B. Saunders; 2010. p. 498–510.
Pirpiris M, Sudhir K, Yeung S, Jennings G, Whitworth JA. Pressor responsiveness in corticosteroid-induced hypertension in humans. Hypertension. 1992;19(6 Pt 1):567–74. https://doi.org/10.1161/01.hyp.19.6.567.
Freeman R. Current pharmacologic treatment for orthostatic hypotension. Clin Auton Res. 2008;18(suppl 1):14–8. https://doi.org/10.1007/s10286-007-1003-1.
Handler J. Symptomatic orthostatic hypotension/supine hypertension. J Clin Hypertens (Greenwich). 2005;7(10):612–6. https://doi.org/10.1111/j.1524-6175.2005.04139.x.
Hauser RA, Isaacson S, Lisk JP, Hewitt LA, Rowse G. Droxidopa for the short-term treatment of symptomatic neurogenic orthostatic hypotension in Parkinson's disease (nOH306B). Mov Disord. 2015;30(5):646–54.
Kaufmann H, Freeman R, Biaggioni I, Low P, Pedder S, Hewitt LA, et al. Droxidopa for neurogenic orthostatic hypotension: a randomized, placebo-controlled, phase 3 trial. Neurology. 2014;83(4):328–35. https://doi.org/10.1212/WNL.0000000000000615.
Grijalva CG, Biaggioni I, Griffin MR, Shibao CA. Fludrocortisone is associated with a higher risk of all-cause hospitalizations compared with midodrine in patients with orthostatic hypotension. J Am Heart Assoc. 2017;6(10):e006848 A retrospective cohort study of patients with orthostatic hypotension showing that patients treated with fludrocortisone had a 20% greater risk of all-cause hospitalization compared with patients treated with midodrine and that those patients who also had congestive heart failure had even a greater risk (42%).
Mazza A, Ravenni R, Antonini A, Casiglia E, Rubello D, Pauletto P. Arterial hypertension, a tricky side of Parkinson's disease: physiopathology and therapeutic features. Neurol Sci. 2013;34(5):621–7. https://doi.org/10.1007/s10072-012-1251-2.
Ten Harkel AD, Van Lieshout JJ, Wieling W. Treatment of orthostatic hypotension with sleeping in the head-up tilt position, alone and in combination with fludrocortisone. J Intern Med. 1992;232(2):139–45.
Zeng G, Quon MJ. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J Clin Invest. 1996;98(4):894–8.
Kaufmann H, Norcliffe-Kaufmann L, Palma JA. Droxidopa in neurogenic orthostatic hypotension. Expert Rev Cardiovasc Ther. 2015;13(8):875–91. https://doi.org/10.1586/14779072.2015.1057504.
Cozaar (losartan potassium). Full Prescribing Information. Whitehouse Station, NJ: Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.; 2018.
Nitro-Dur (nitroglycerin patch). Full Prescribing Information. Whitehouse Station, NJ: Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc.; 2014.
Capoten (captopril). Full Prescribing Information. Spring Valley, NY: Par Pharmaceutical Companies, Inc.; 2012.
Apresoline (hydralazine). Full prescribing information, Ciba-Geigy Corporation, Summit, NJ, 1995.
Arnold AC, Okamoto LE, Gamboa A, Shibao C, Raj SR, Robertson D, et al. Angiotensin II, independent of plasma renin activity, contributes to the hypertension of autonomic failure. Hypertension. 2013;61(3):701–6. https://doi.org/10.1161/HYPERTENSIONAHA.111.00377.
Jordan J, Shannon JR, Pohar B, Paranjape SY, Robertson D, Robertson RM, et al. Contrasting effects of vasodilators on blood pressure and sodium balance in the hypertension of autonomic failure. J Am Soc Nephrol. 1999;10(1):35–42.
Procardia® (nifedipine capsule). Full prescribing information. New York, NY: Pfizer Inc; 2016.
Inspra (eplerenone). Full Prescribing Information, Pfizer, New York, NY, USA, 2008.
Baker J, Kimpinski K. Management of supine hypertension complicating neurogenic orthostatic hypotension. CNS Drugs. 2017;31(8):653–63.
Young TM, Asahina M, Watson L, Mathias CJ. Hemodynamic effects of clonidine in two contrasting models of autonomic failure: multiple system atrophy and pure autonomic failure. Mov Disord. 2006;21(5):609–15. https://doi.org/10.1002/mds.20755.
Shibao C, Raj SR, Gamboa A, Diedrich A, Choi L, Black BK, et al. Norepinephrine transporter blockade with atomoxetine induces hypertension in patients with impaired autonomic function. Hypertension. 2007;50(1):47–53. https://doi.org/10.1161/HYPERTENSIONAHA.107.089961.
Palma JA, Norcliffe-Kaufmann L, Martinez J, Kaufmann H. Supine plasma NE predicts the pressor response to droxidopa in neurogenic orthostatic hypotension. Neurology. 2018;91(16):e1539–e44.
Acknowledgements
The authors received editorial assistance from ICON plc (North Wales, PA, USA), which was supported by Lundbeck.
Funding
This work was supported by Lundbeck, who provided funding for editorial support from ICON (North Wales, PA, USA). The authors were responsible for all final content decisions during manuscript development and all authors approved the final version of the manuscript for submission.
Author information
Authors and Affiliations
Contributions
All authors were involved in the conception, drafting, reviewing, and revising of the manuscript and approved the final version submitted for publication.
Corresponding author
Ethics declarations
Conflict of Interest
SH Isaacson has served as a consultant and received honoraria for CME, research grants, and/or promotional speaker on behalf of AbbVie, Acadia, Acorda, Adamas, Addex, Affiris, Alexva, Allergan, Amarantus, Amneal, Axovant, Benevolent, Biogen, Britannia, Cadent, Cala, Cerecor, Cipla, Eli Lilly, Enterin, GE Healthcare, Global Kinetics, Impax, Impel, Intec Pharma, Ipsen, Jazz, Kyowa, Lundbeck, Merz, Michael J. Fox Foundation, Mitsubishi Tanabe, Neuralym, Neurocrine, Neuroderm, Parkinson Study Group, Pharma2B, Prilenia, Promentis, Revance, Roche, Sanofi, Sunovion, Sun Pharma, Teva, Theravance, UCB, US World Meds, and Zambon. K Dashtipour has served as an advisor, consultant, or speaker for AbbVie, Acadia, Acorda, Adamas, Allergan, Amneal, Ipsen, Kyowa Kirin, Lundbeck, Neurocrine, Revance, Sunovion, Teva, and US WorldMeds. AA Mehdirad has served as a speaker for Lundbeck, Pfizer, and Bristol-Myers Squibb. AC Peltier has received fees for consultancy, honoraria, and/or travel/lodging from Lundbeck, Alnylam and Akcea, CSL Behring, and Daiichi Sankyo Inc.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Key Points
• Neurogenic orthostatic hypotension (nOH) and neurogenic supine hypertension (nSH) are interrelated conditions associated with the inability of the cardiovascular system to appropriately adjust to postural change; patients may have coexistent nOH and nSH.
• An individualized approach for the treatment of coexisting nOH and nSH is necessary to minimize risks associated with these conditions.
• Both nonpharmacologic interventions and pharmacologic therapies are available for patients with nSH.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This Article is part of the Topical Collection on Autonomic Dysfunction
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Isaacson, S.H., Dashtipour, K., Mehdirad, A.A. et al. Management Strategies for Comorbid Supine Hypertension in Patients with Neurogenic Orthostatic Hypotension. Curr Neurol Neurosci Rep 21, 18 (2021). https://doi.org/10.1007/s11910-021-01104-3
Accepted:
Published:
DOI: https://doi.org/10.1007/s11910-021-01104-3